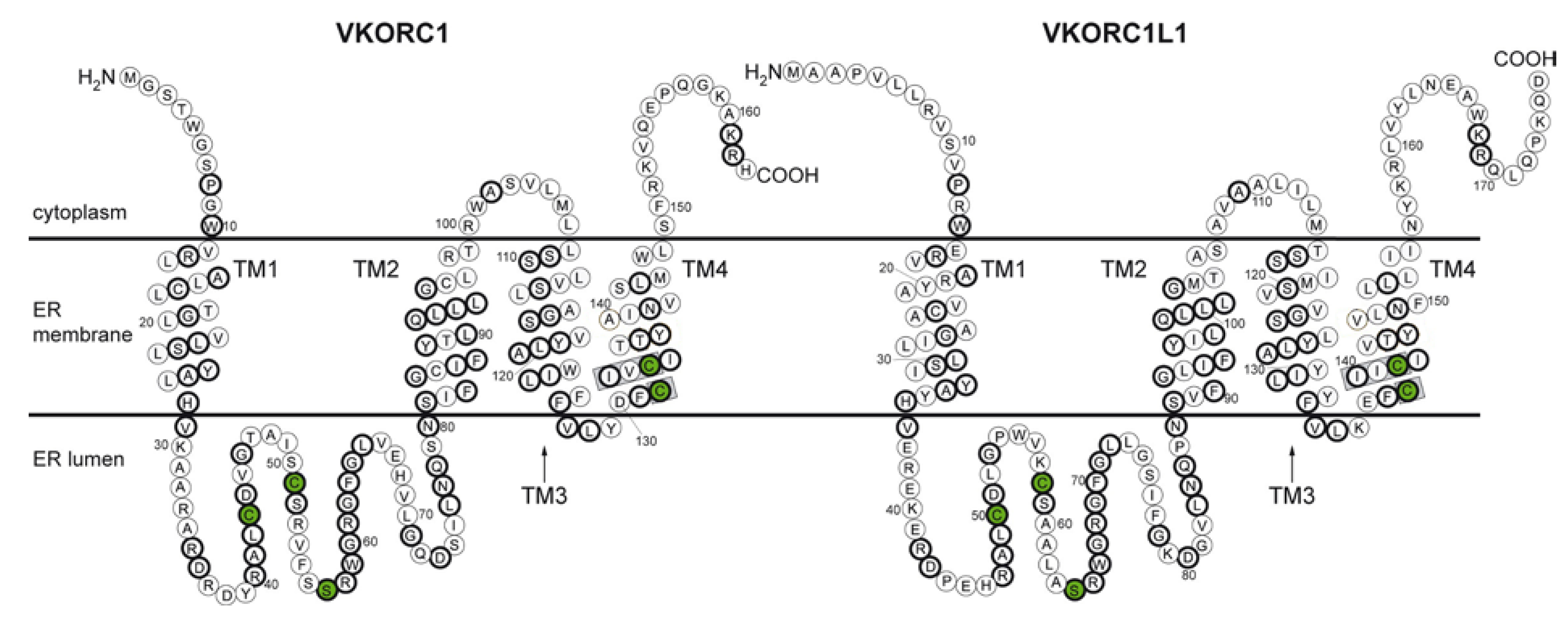
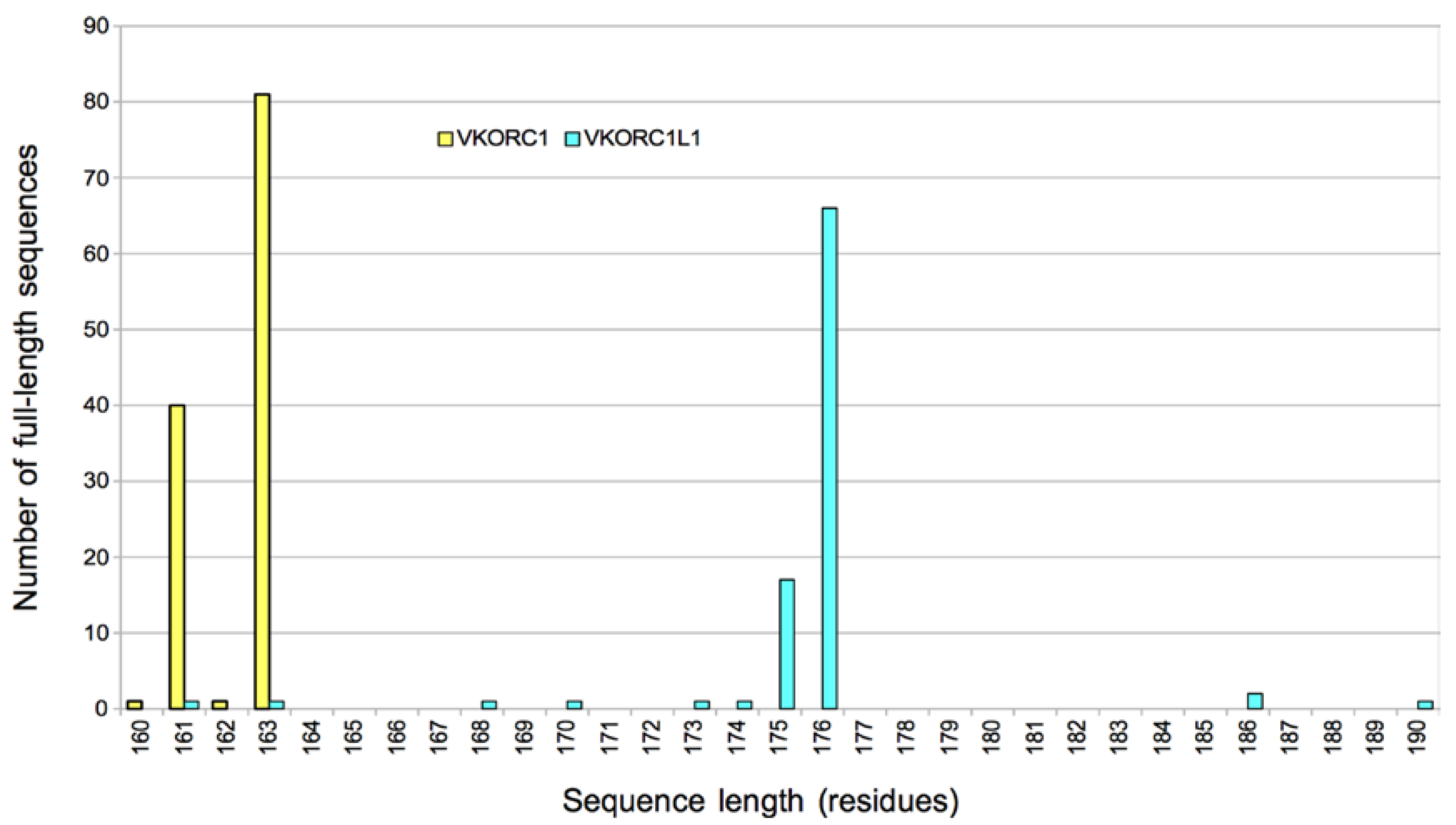
VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Oldenburg, J.; Watzka, M.; Bevans, C.G. VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases? Nutrients 2015, 7, 6250-6280. https://doi.org/10.3390/nu7085280
Oldenburg J, Watzka M, Bevans CG. VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases? Nutrients. 2015; 7(8):6250-6280. https://doi.org/10.3390/nu7085280
Chicago/Turabian StyleOldenburg, Johannes, Matthias Watzka, and Carville G. Bevans. 2015. "VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases?" Nutrients 7, no. 8: 6250-6280. https://doi.org/10.3390/nu7085280
APA StyleOldenburg, J., Watzka, M., & Bevans, C. G. (2015). VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases? Nutrients, 7(8), 6250-6280. https://doi.org/10.3390/nu7085280