Protective Effect of Genistein against Neuronal Degeneration in ApoE−/− Mice Fed a High-Fat Diet

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Diets
2.2. Analyses of Serum
2.3. Tissue Extract Preparation and Immunoblotting
2.4. Total RNA Extraction and Semiquantitative RT-PCR
2.5. Quantitative RT-PCR (qRT-PCR) Analysis
2.6. Statistical Analysis
3. Results
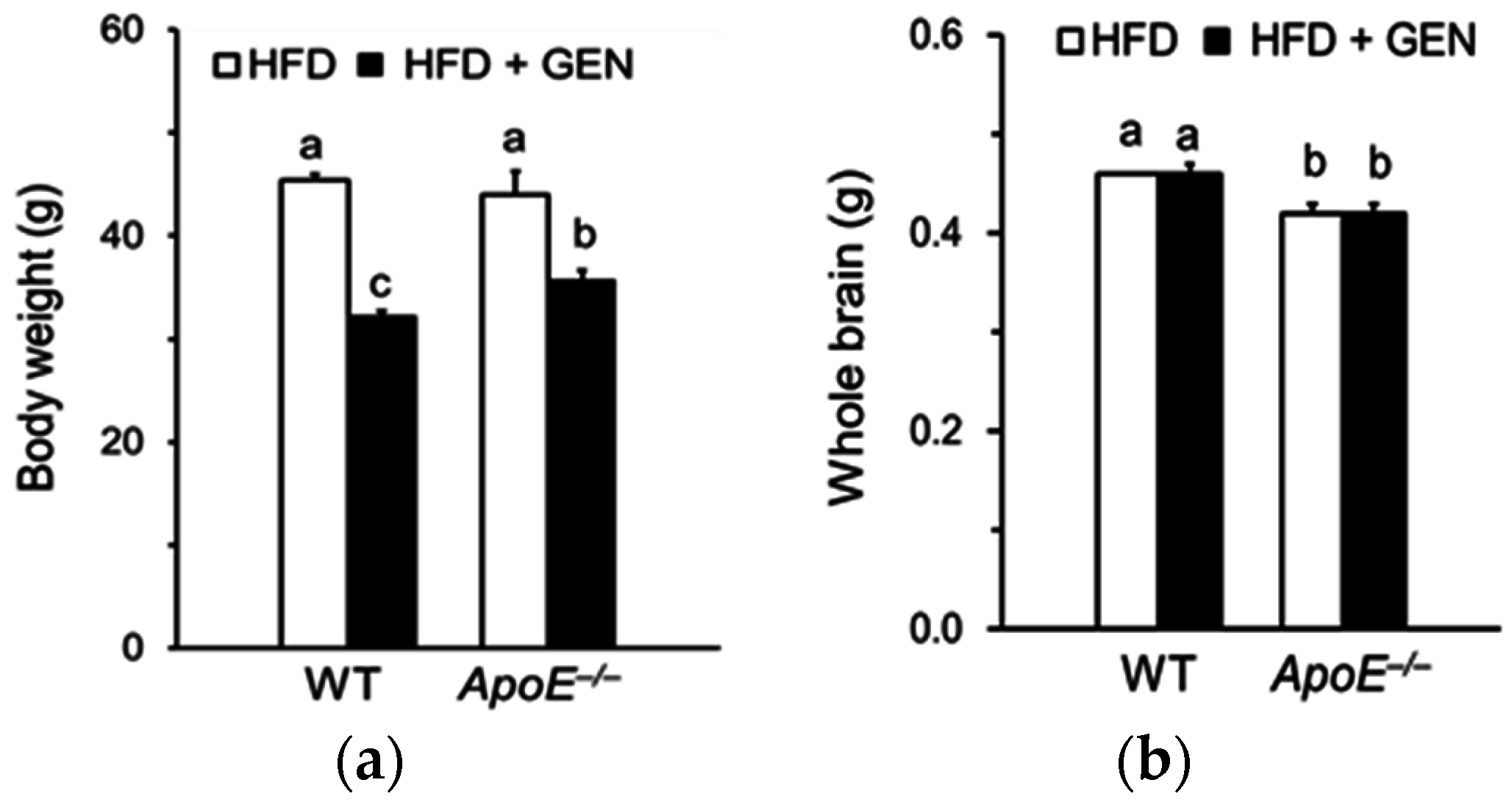
3.1. Effect of Genistein on Brain Weight in ApoE−/− Mice Fed an HFD
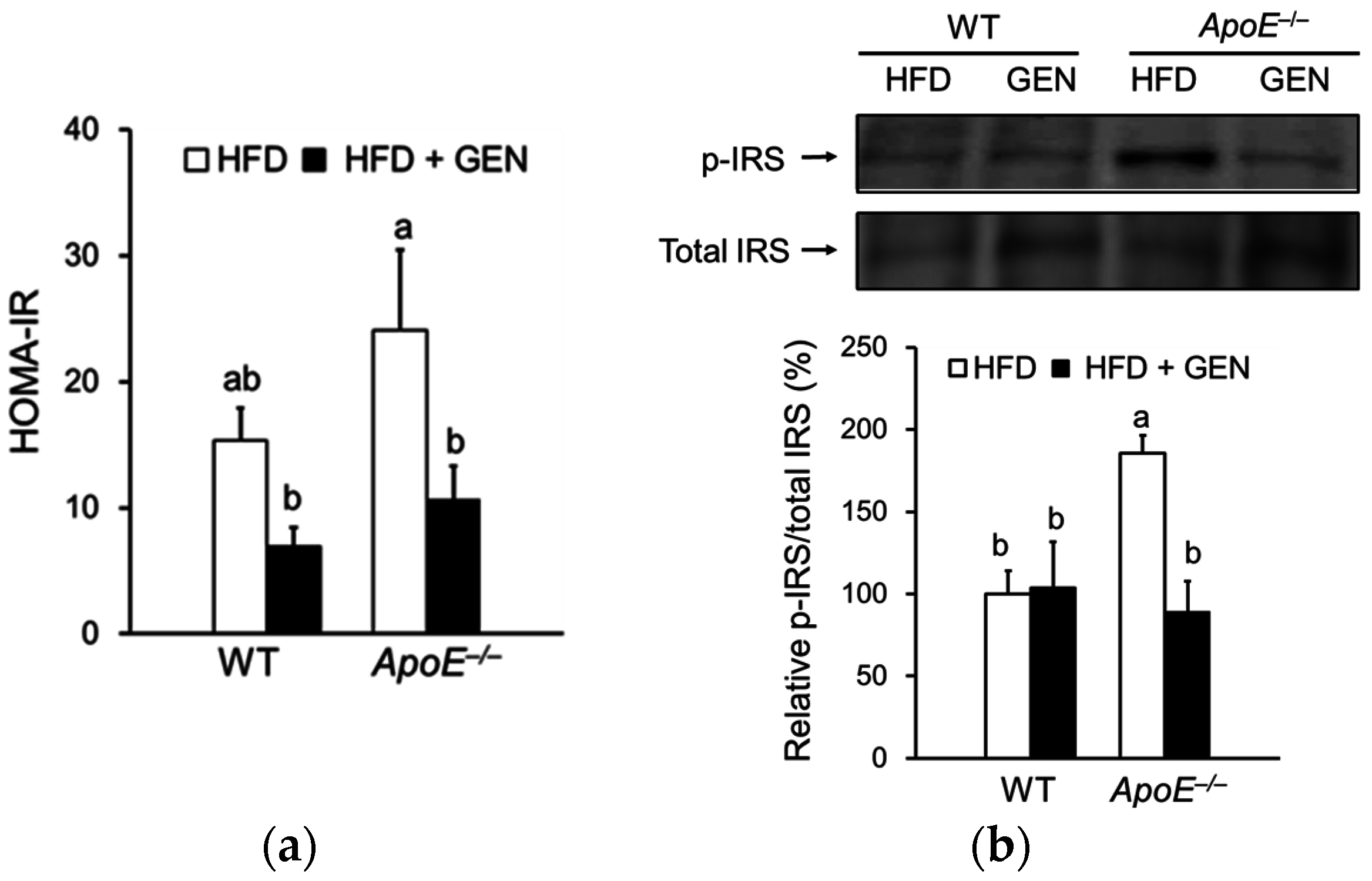
3.2. Effect of Genistein on Peripheral and Central Insulin Resistance in ApoE−/− Mice Fed an HFD
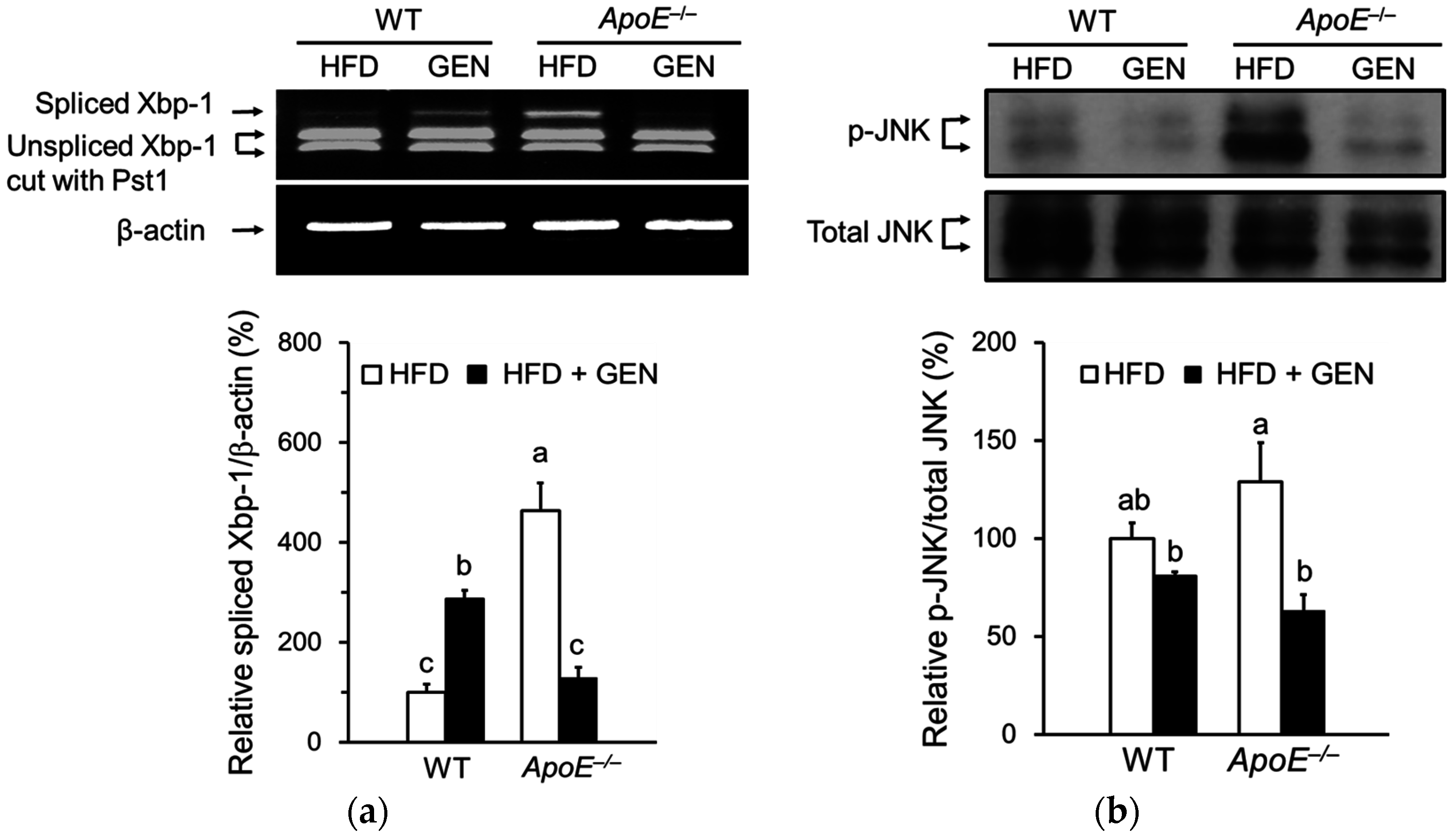
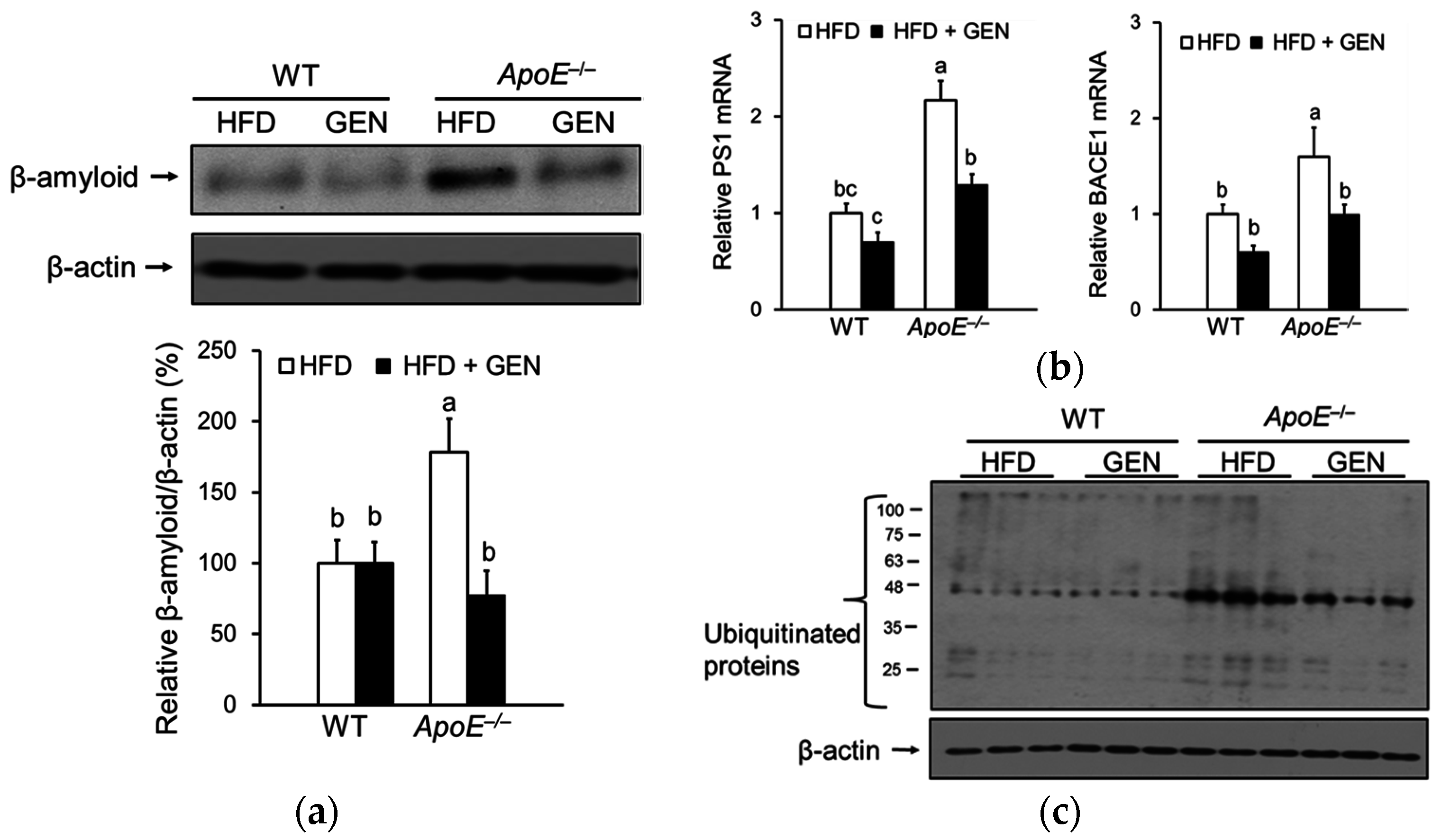
3.3. Effect of Genistein on ER Stress in the Brain of ApoE−/− Mice Fed an HFD
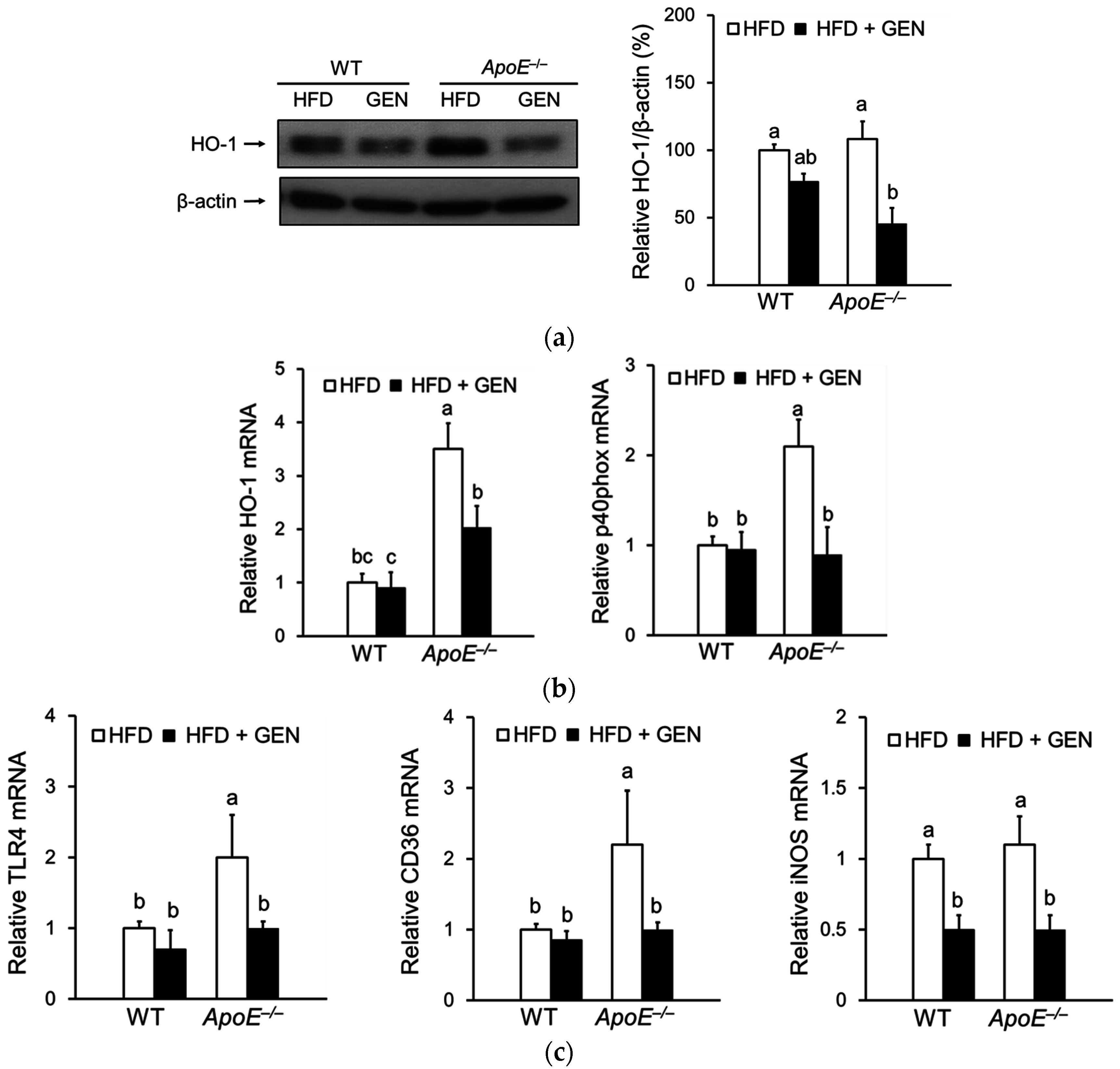
3.4. Effect of Genistein on Oxidative Stress and Inflammation in the Brain of ApoE−/− Mice Fed an HFD
3.5. Effect of Genistein on Oxidative Stress and Inflammation in the Brain of ApoE−/− Mice Fed an HFD
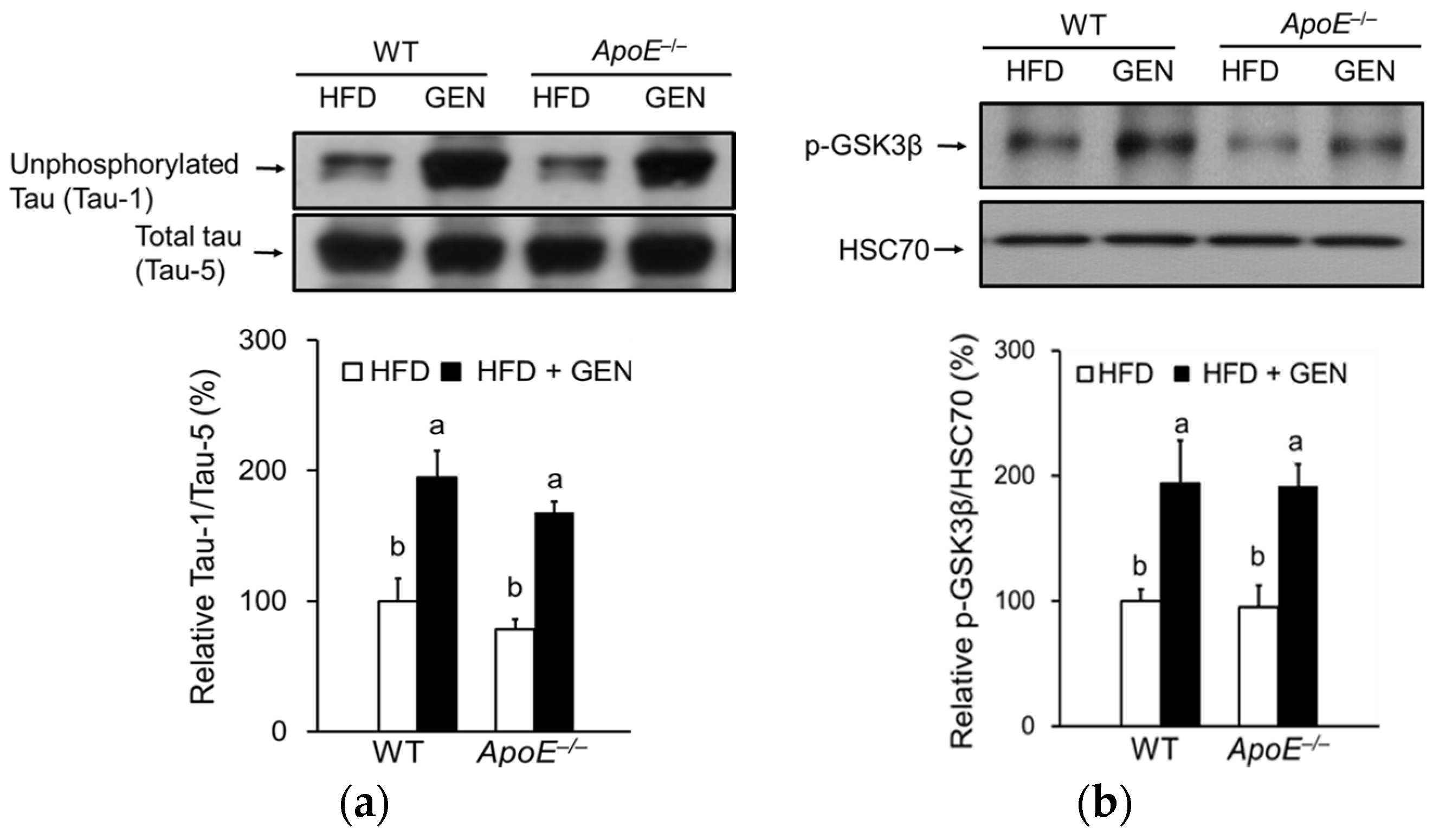
3.6. Effect of Genistein on Tau Hyperphosphorylation in the Brain of ApoE−/− Mice Fed an HFD
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Uranga, R.M.; Keller, J.N. Diet and age interactions with regards to cholesterol regulation and brain pathogenesis. Curr. Gerontol. Geriatr. Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Wang, Y. Obesity and central obesity as risk factors for incident dementia and its subtypes: A systematic review and meta-analysis. Obes. Rev. 2008, 9, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Evola, M.; Hall, A.; Wall, T.; Young, A.; Grammas, P. Oxidative stress impairs learning and memory in apoE knockout mice. Pharmacol. Biochem. Behav. 2010, 96, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Shobab, L.A.; Hsiung, G.Y.; Feldman, H.H. Cholesterol in Alzheimer’s disease. Lancet Neurol. 2005, 4, 841–852. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Ghribi, O.; Larsen, B.; Schrag, M.; Herman, M.M. High cholesterol content in neurons increases BACE, beta-amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp. Neurol. 2006, 200, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, J.H.; Choi, K.H.; Jang, Y.J.; Bae, S.S.; Choi, B.T.; Shin, H.K. Hypercholesterolemia accelerates amyloid beta-induced cognitive deficits. Int. J. Mol. Med. 2013, 31, 577–582. [Google Scholar] [PubMed]
- Leoni, V.; Solomon, A.; Kivipelto, M. Links between ApoE, brain cholesterol metabolism, tau and amyloid beta-peptide in patients with cognitive impairment. Biochem. Soc. Trans. 2010, 38, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol. 2011, 68, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J. Apolipoprotein E and Alzheimer’s disease. A role in amyloid catabolism. Ann. N. Y. Acad. Sci. 2000, 924, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Herz, J. Is apolipoprotein e required for cognitive function in humans? Implications for Alzheimer drug development. JAMA Neurol. 2014, 71, 1213–1215. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Ge, N.; Alford, M.; Veinbergs, I.; Roses, A.D. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp. Neurol. 1995, 136, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Veinbergs, I.; Mante, M.; Jung, M.W.; van Uden, E.; Masliah, E. Synaptotagmin and synaptic transmission alterations in apolipoprotein E-deficient mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 1999, 23, 519–531. [Google Scholar] [CrossRef]
- Anderson, R.; Barnes, J.C.; Bliss, T.V.; Cain, D.P.; Cambon, K.; Davies, H.A.; Errington, M.L.; Fellows, L.A.; Gray, R.A.; Hoh, T.; et al. Behavioural, physiological and morphological analysis of a line of apolipoprotein E knockout mouse. Neuroscience 1998, 85, 93–110. [Google Scholar] [CrossRef]
- Ekuni, D.; Endo, Y.; Tomofuji, T.; Azuma, T.; Irie, K.; Kasuyama, K.; Morita, M. Effects of apoE deficiency and occlusal disharmony on amyloid-beta production and spatial memory in rats. PLoS ONE 2013, 8, e74966. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Park, Y.J.; Kwon, Y.H. Genistein alleviates the development of nonalcoholic steatohepatitis in ApoE(−/−) mice fed a high-fat diet. Mol. Nutr. Food Res. 2014, 58, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Bingham, S.A.; Atkinson, C.; Liggins, J.; Bluck, L.; Coward, A. Phyto-oestrogens: Where are we now? Br. J. Nutr. 1998, 79, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Jang, Y.; Kwon, Y.H. Protective effect of isoflavones against homocysteine-mediated neuronal degeneration in SH-SY5Y cells. Amino Acids 2010, 39, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Jang, Y.-M.; Kwon, Y.H. Isoflavones prevent endoplasmic reticulum stress-mediated neuronal degeneration by inhibiting tau hyperphosphorylaltion in SH-SY5Y cells. J. Med. Food 2009, 12, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Chen, W.F.; Xie, J.X.; Wong, M.S. Neuroprotective effects of genistein on dopaminergic neurons in the mice model of Parkinson’s disease. Neurosci. Res. 2008, 60, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Yu, H.L.; Ma, W.W.; Xi, Y.D.; Zhao, X.; Yuan, L.H.; Feng, J.F.; Xiao, R. Soy isoflavone attenuates brain mitochondrial oxidative stress induced by beta-amyloid peptides 1-42 injection in lateral cerebral ventricle. J. Neurosci. Res. 2013, 91, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Audet, A.; Cusic, J.; Seeger, D.; Cochran, R.; Ghribi, O. Broad DNA repair responses in neural injury are associated with activation of the IL-6 pathway in cholesterol-fed rabbits. J. Neurochem. 2009, 111, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Kutty, R.K.; Richey, P.L.; Yan, S.D.; Stern, D.; Chader, G.J.; Wiggert, B.; Petersen, R.B.; Perry, G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1994, 145, 42–47. [Google Scholar] [PubMed]
- Canevari, L.; Clark, J.B. Alzheimer’s disease and cholesterol: The fat connection. Neurochem. Res. 2007, 32, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Titani, K.; Ihara, Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 1995, 270, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Betts, J.C.; Loviny, T.L.; Blackstock, W.P.; Anderton, B.H. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 1998, 71, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 309–317. [Google Scholar] [PubMed]
- Li, T.; Paudel, H.K. Glycogen synthase kinase 3beta phosphorylates Alzheimer’s disease-specific ser of microtubule-associated protein tau by a sequential mechanism. Biochemistry 2006, 45, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.M.; Wu, W.M.; Hu, M.L. Soy isoflavones attenuate oxidative stress and improve parameters related to aging and Alzheimer’s disease in C57BL/6J mice treated with d-galactose. Food Chem. Toxicol. 2009, 47, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Roghani, M.; Joghataei, M.T.; Mohseni, S. Genistein inhibits aggregation of exogenous amyloid-beta1–40 and alleviates astrogliosis in the hippocampus of rats. Brain Res. 2012, 1429, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, A.E.; Franchi, S.; Panerai, A.E.; Rossi, A.; Sacerdote, P.; Colleoni, M. The soy isoflavone genistein reverses oxidative and inflammatory state, neuropathic pain, neurotrophic and vasculature deficits in diabetes mouse model. Eur. J. Pharmacol. 2011, 650, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflamm. 2016, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Holsinger, R.M.; McLean, C.A.; Laughton, K.M.; Beyreuther, K.; Evin, G.; Masters, C.L.; Li, Q.X. APP intracellular domain is increased and soluble Abeta is reduced with diet-induced hypercholesterolemia in a transgenic mouse model of Alzheimer disease. Neurobiol. Dis. 2004, 16, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Planel, E.; Tatebayashi, Y.; Miyasaka, T.; Liu, L.; Wang, L.; Herman, M.; Yu, W.H.; Luchsinger, J.A.; Wadzinski, B.; Duff, K.E.; et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 2007, 27, 13635–13648. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, D.A.; Hafez, H.S.; Hussien, H.M.; Kabapy, N.F. Non-alcoholic fatty liver induces insulin resistance and metabolic disorders with development of brain damage and dysfunction. Metab. Brain Dis. 2011, 26, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.S.; Hong, H.; Kim, C.; Mook-Jung, I. Acute ER stress regulates amyloid precursor protein processing through ubiquitin-dependent degradation. Sci. Rep. 2015, 5, 8805. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.; Sohn, H. Soy isoflavones and cognitive function. J. Nutr. Biochem. 2005, 16, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Shughrue, P.J.; Lane, M.V.; Merchenthaler, I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J. Comp. Neurol. 1997, 388, 507–525. [Google Scholar] [CrossRef]
- Coldham, N.G.; Sauer, M.J. Pharmacokinetics of [14C]Genistein in the rat: Gender-related differences, potential mechanisms of biological action, and implications for human health. Toxicol. Appl. Pharmacol. 2000, 164, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Zhang, Q.H. Genistein reduced the neural apoptosis in the brain of ovariectomised rats by modulating mitochondrial oxidative stress. Br. J. Nutr. 2010, 104, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.H.; Allred, C.D.; Allred, K.F.; Karko, K.L.; Doerge, D.R.; Helferich, W.G. Physiological concentrations of dietary genistein dose-dependently stimulate growth of estrogen-dependent human breast cancer (MCF-7) tumors implanted in athymic nude mice. J. Nutr. 2001, 131, 2957–2962. [Google Scholar] [PubMed]
- Xu, X.; Harris, K.S.; Wang, H.J.; Murphy, P.A.; Hendrich, S. Bioavailability of soybean isoflavones depends upon gut microflora in women. J. Nutr. 1995, 125, 2307–2315. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| BACE1 | GCATGATCATTGGTGGTATC | CCATCTTGAGATCTTGACCA |
| CD36 | TCCTCTGACATTTGCAGGTCTATC | AAAGGCATTGGCTGGAAGAA |
| HO-1 | CCTCACTGGCAGGAAATCATC | CCTCGTGGAGACGCTTTACATA |
| iNOS | CAGGAGGAGAGAGATCCGATTTA | GCATTAGCATGGAAGCAAAGA |
| p40phox | CCTGCCCACATTGCCAGCCA | AGACCGGCAGGCTCAGGAGG |
| PS1 | TGCGGCCATCATGATCAGTGTC | ATAAGCCAGGCGTGGATGAC |
| TLR4 | AGGAAGTTTCTCTGGACTAACAAGTTTAGA | AAATTGTGAGCCACATTGAGTTTC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.-J.; Ko, J.W.; Jeon, S.; Kwon, Y.H. Protective Effect of Genistein against Neuronal Degeneration in ApoE−/− Mice Fed a High-Fat Diet. Nutrients 2016, 8, 692. https://doi.org/10.3390/nu8110692
Park Y-J, Ko JW, Jeon S, Kwon YH. Protective Effect of Genistein against Neuronal Degeneration in ApoE−/− Mice Fed a High-Fat Diet. Nutrients. 2016; 8(11):692. https://doi.org/10.3390/nu8110692
Chicago/Turabian StylePark, Yoon-Jin, Je Won Ko, Sookyoung Jeon, and Young Hye Kwon. 2016. "Protective Effect of Genistein against Neuronal Degeneration in ApoE−/− Mice Fed a High-Fat Diet" Nutrients 8, no. 11: 692. https://doi.org/10.3390/nu8110692
APA StylePark, Y.-J., Ko, J. W., Jeon, S., & Kwon, Y. H. (2016). Protective Effect of Genistein against Neuronal Degeneration in ApoE−/− Mice Fed a High-Fat Diet. Nutrients, 8(11), 692. https://doi.org/10.3390/nu8110692