Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases

Abstract
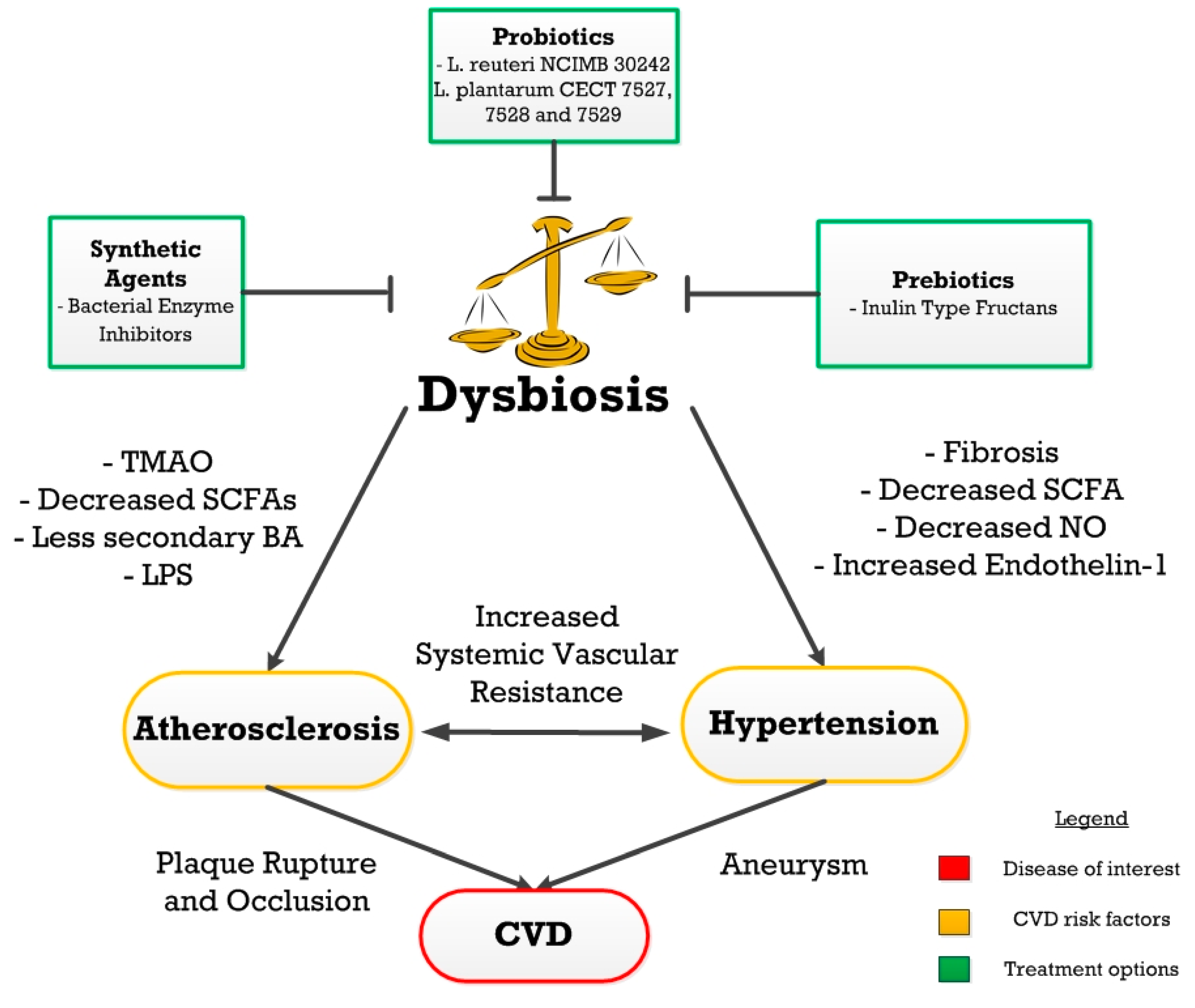
:1. Introduction
1.1. Definition and Introduction to the Gut Microbiota
1.2. Establishment, Development, and Changes to the Gut Microbiota
1.3. Homeostatic Functions
1.4. Gut Dysbiosis
2. Atherosclerosis
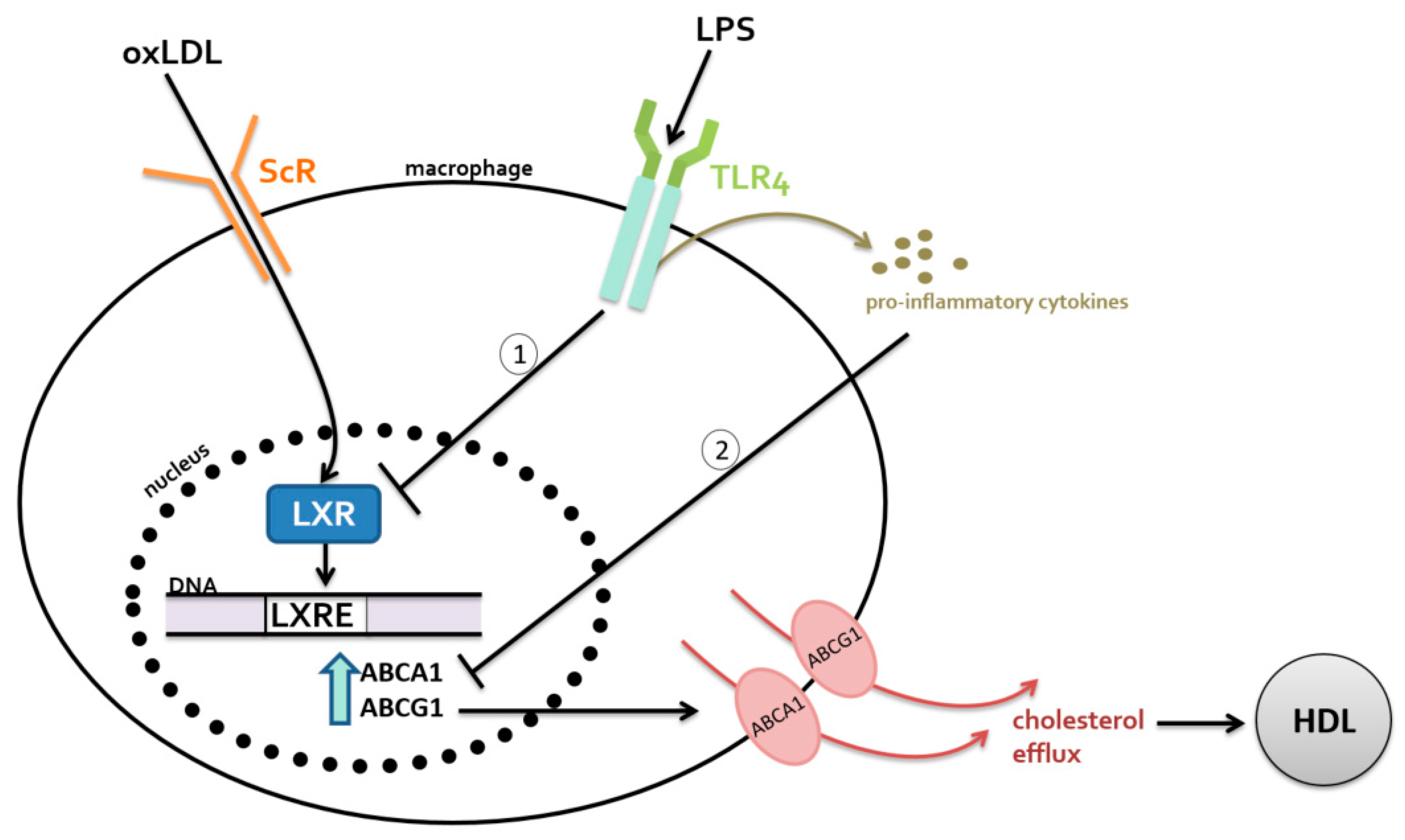
2.1. Metabolism-Independent Pathway
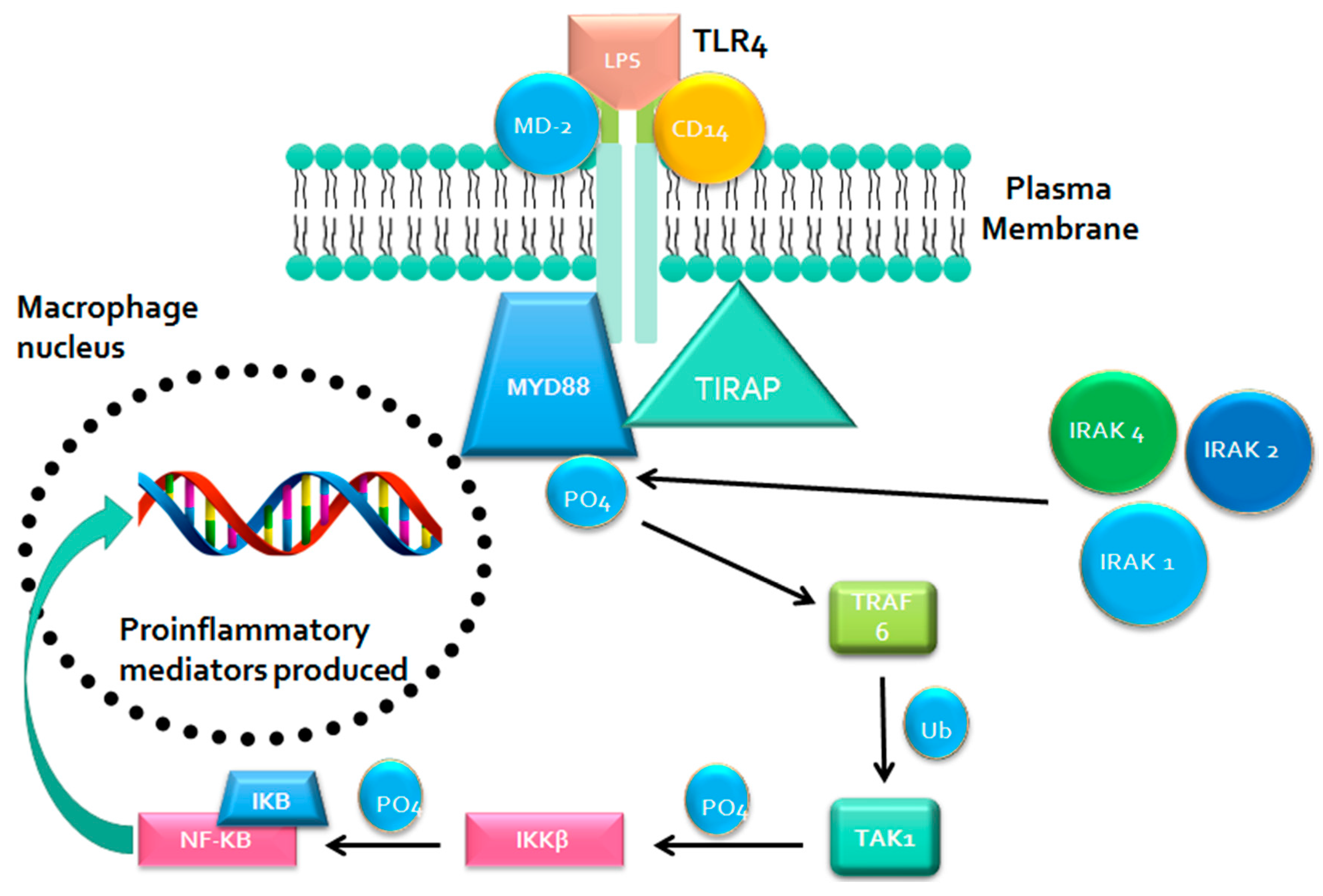
MYD88 Signaling
2.2. Metabolism-Dependent Pathway
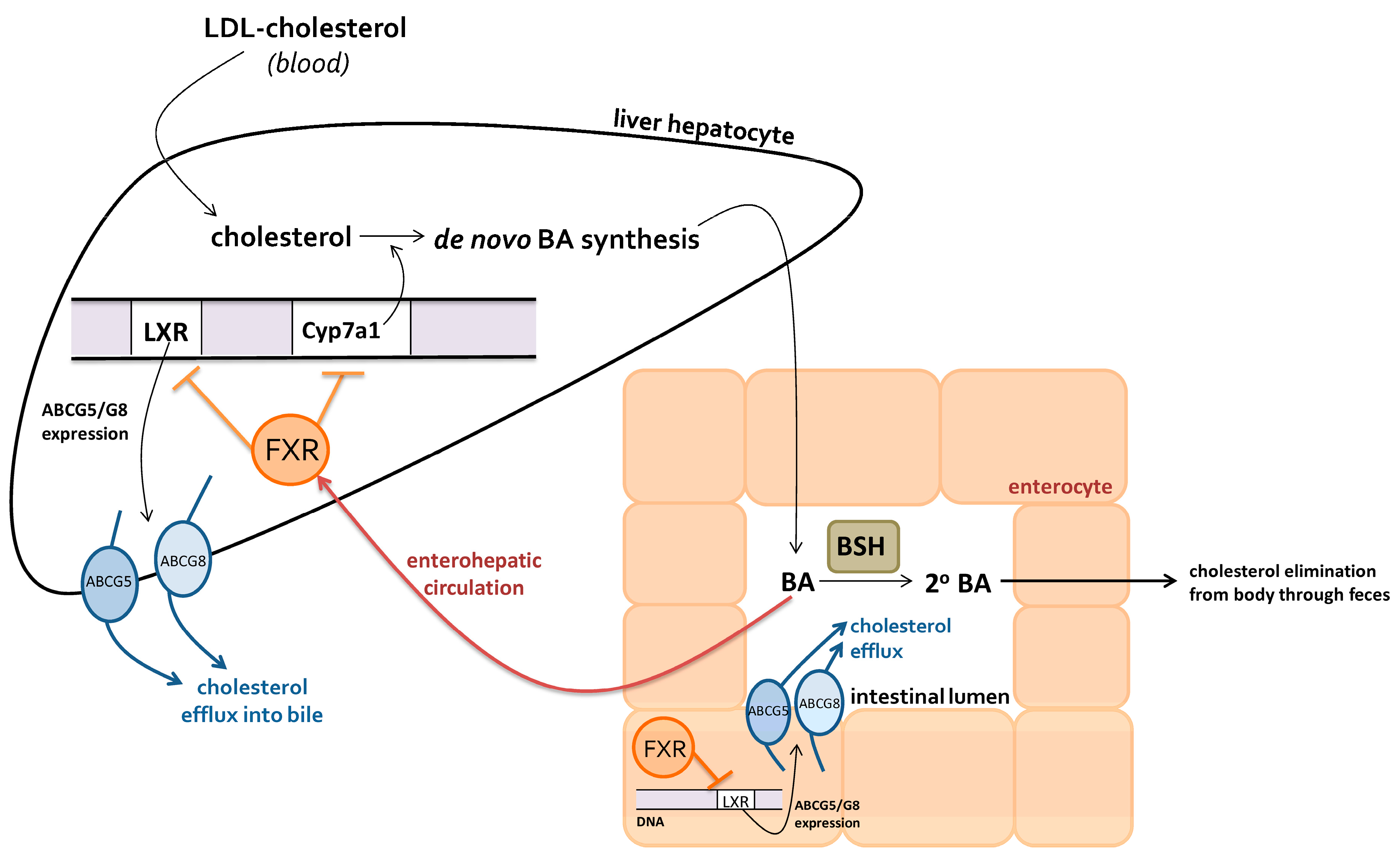
2.2.1. Bile Acids
2.2.2. Trimethylamine-N-Oxide
2.2.3. Butyrate
3. Hypertension
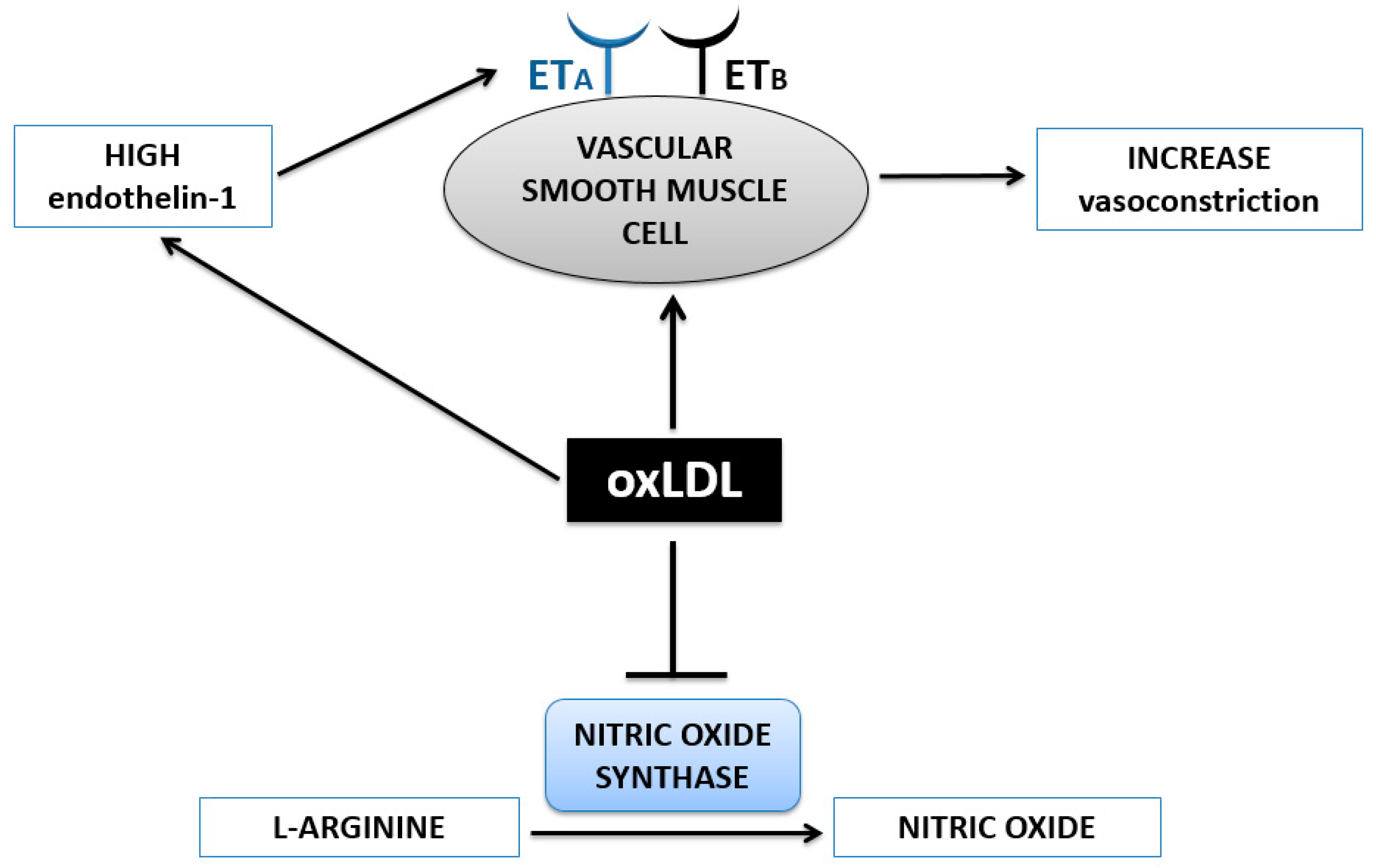
OxLDL and Vasoconstriction
4. Treatments
4.1. Dysbiosis Treatment
4.2. Prebiotics
4.3. Probiotics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Trine, N.; Nicolas, P.; Florence, L.; Takuji, Y.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Blaser, M.J.; Ley, R.E.; Knight, R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 2011, 140, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Scott, K.P.; Khan, S.; Martin, J.C.; Berry, S.H.; Stevenson, M.; Okpapi, A.; Munro, M.J.; Hold, G.L. First-Pass Meconium Samples from Healthy Term Vaginally-Delivered Neonates: An Analysis of the Microbiota. PLoS ONE 2015, 10, e0133320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.; Tremaroli, V.; Nielsen, J.; Bäckhed, F. Assessing the human gut microbiota in metabolic diseases. Diabetes 2013, 62, 3341–3349. [Google Scholar]
- Watson, J.; Jones, R.C.; Cortes, C.; Gerber, S.I.; Golash, R.G.; Price, J.; Bancroft, E.; Mascola, L.; Gorwitz, R.J.; Jernigan, D.B.; et al. Community-associated methicillin-resistant Staphylococcus aureus infection among healthy newborns—Chicago and Los Angeles County, 2004. (Reprinted from MMWR 2006, 55, 329–332). JAMA 2006, 296, 36–38. [Google Scholar]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, A.M.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl. Acad. Sci. USA 2011, 108, 4653–4658. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Laparra, J.M.; Sanz, Y. Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol. Res. 2010, 61, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.W.; de Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T. The gut is still the biggest lymphoid organ in the body. Mucosal Immunol. 2008, 1, 246–247. [Google Scholar]
- Serino, M.; Blasco-Baque, V.; Nicolas, S.; Burcelin, R. Far from the eyes, close to the heart: Dysbiosis of gut microbiota and cardiovascular consequences. Curr. Cardiol. Rep. 2014, 16, 540. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26. [Google Scholar] [CrossRef] [PubMed]
- Al Khodor, S.; Reichert, B.; Shatat, I.F. The Microbiome and Blood Pressure: Can Microbes Regulate Our Blood Pressure? Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.L.; Triplett, E.W.; Gevers, D.; Xavier, R.; Insel, R.; Danska, J.; Atkinson, M.A. The intestinal microbiome in type 1 diabetes. Clin. Exp. Immunol. 2014, 177, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.J.; Prabhu, K.S.; Vijay-Kumar, M. The microbiome and obesity—An established risk for certain types of cancer. Cancer J. Sudbury Mass 2014, 20, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Gross, M. Does the gut microbiome hold clues to obesity and diabetes? Curr. Biol. 2013, 23, R359–R362. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Microbiome, inflammation, and cancer. Cancer J. Sudbury Mass 2014, 20, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, K.E.; Lynch, S.V. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe 2015, 17, 592–602. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.; Tan, J.; Macia, L.; Mackay, C.R. The nutrition-gut microbiome—Physiology axis and allergic diseases. Immunol. Rev. 2017, 278, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Emoto, T.; Yamashita, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; Mizoguchi, T. Analysis of Gut Microbiota in Coronary Artery Disease Patients: A Possible Link between Gut Microbiota and Coronary Artery Disease. J. Atheroscler. Thromb. 2016, 23, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.W. Gut microbiota and cardiometabolic outcomes: Influence of dietary patterns and their associated components. Am. J. Clin. Nutr. 2014, 100, 369S–377S. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to biomarker discovery. Mediat. Inflamm. 2012, 2012, 693083. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Hansson, G.K.; Hansson, G.K. Mechanisms of disease: Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Noncommunicable Diseases 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.-K.L.; Söderberg-Nauclér, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C.-K. Foam cells in atherosclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Shashkin, P.; Dragulev, B.; Ley, K. Macrophage differentiation to foam cells. Curr. Pharm. Des. 2005, 11, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Annema, W.; Tietge, U.J. Regulation of reverse cholesterol transport—A comprehensive appraisal of available animal studies. Nutr. Metab. 2012, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Spady, D.K. Reverse cholesterol transport and atherosclerosis regression. Circulation 1999, 100, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM Mon. J. Assoc. Physicians 2005, 98, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. An overview of reverse cholesterol transport. Eur. Heart J. 1998, 19, A31–A35. [Google Scholar] [CrossRef]
- Cuchel, M.; Rader, D.J. Macrophage reverse cholesterol transport: Key to the regression of atherosclerosis? Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Murzilli, S.; Salvatore, L.; D’Errico, I.; Petruzzelli, M.; Conca, P.; Jiang, Z.Y.; Calabresi, L.; Parini, P.; Moschetta, A. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. 2010, 12, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Harris, K.; Kassis, A.; Major, G.; Chou, C.J. Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J. Obes. 2012, 2012, 879151. [Google Scholar] [PubMed]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51–R64. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M.; Kagan, J.C. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.; Ayer, A.; Basta, S.; Banfield, B.W.; Gee, K. IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 2012, 188, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.-H.; Huang, X.-Q.; Tan, H.-M. Vascular fibrosis in atherosclerosis. Cardiovasc. Pathol. 2013, 22, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Shirwany, N.A.; Zou, M. Arterial stiffness: A brief review. Acta Pharmacol. Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R. Getting to the core of atherosclerosis. Nat. Med. 2008, 14, 1015–1016. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, A.C.; Becker, A.E. Atherosclerotic plaque rupture—Pathologic basis of plaque stability and instability. Cardiovasc. Res. 1999, 41, 334–344. [Google Scholar] [CrossRef]
- Jones, M.L.; Martoni, C.J.; Ganopolsky, J.G.; Labbé, A.; Prakash, S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin. Biol. Ther. 2014, 14, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Parent, M.; Prakash, S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase—Active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012, 107, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.I.A.; Gibson, G.R. Effects of consumption of probiotics and prebiotics on serum lipid levels in humans. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Menzel, T.; Lührs, H.; Zirlik, S.; Schauber, J.; Kudlich, T.; Gerke, T.; Gostner, A.; Neumann, M.; Melcher, R.; Scheppach, W. Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm. Bowel Dis. 2004, 10, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Segain, J.; de la Bletiere, D.R.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottiere, H.; Galmiche, J. Butyrate inhibits inflammatory responses through NFκB inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Brar, M.; Kirjavainen, P.; Chen, Y.; Peng, J.; Li, D.; Leung, F.C.; El Nezami, H. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: a pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE−/− mice. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Barton, M. Biology of the endothelium. Clin. Cardiol. 1997, 20, II-3-10. [Google Scholar] [PubMed]
- Subah Packer, C. Estrogen protection, oxidized LDL, endothelial dysfunction and vasorelaxation in cardiovascular disease: New insights into a complex issue. Cardiovasc. Res. 2007, 73, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M. Secondary endothelial dysfunction: Hypertension and heart failure. J. Mol. Cell. Cardiol. 1999, 31, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.X.; Zhou, B.; Chen, Z.; Ren, Q.; Lu, S.H.; Sawamura, T.; Han, Z.C. Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 2006, 47, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.; Lüscher, T.F. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J. Clin. Investig. 1990, 85, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Yeoh, B.S.; Vijay-Kumar, M. Gut microbiome as a novel cardiovascular therapeutic target. Curr. Opin. Pharmacol. 2016, 27, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M. Prebiotics: the concept revisited. J. Nutr. 2007, 137, 830S–837S. [Google Scholar] [PubMed]
- Delzenne, N.M.; Neyrinck, A.M.; Bäckhed, F.; Cani, P.D. Targeting gut microbiota in obesity: Effects of prebiotics and probiotics. Nat. Rev. Endocrinol. 2011, 7, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Koutsos, A.; Tuohy, K.M.; Lovegrove, J.A. Apples and Cardiovascular Health—Is the Gut Microbiota a Core Consideration? Nutrients 2015, 7, 3959–3998. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Ohashi, Y.; Kawasumi, K.; Terada, A.; Fujisawa, T. Effect of apple intake on fecal microbiota and metabolites in humans. Anaerobe 2010, 16, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Pietro Femia, A.; Luceri, C.; Bianchini, F.; Salvadori, M.; Salvianti, F.; Pinzani, P.; Dolara, P.; Calorini, L.; Caderni, G. Marie Ménard apples with high polyphenol content and a low-fat diet reduce 1,2-dimethylhydrazine-induced colon carcinogenesis in rats: Effects on inflammation and apoptosis. Mol. Nutr. Food Res. 2012, 56, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Aprikian, O.; Duclos, V.; Guyot, S.; Besson, C.; Manach, C.; Bernalier, A.; Morand, C.; Rémésy, C.; Demigné, C. Apple pectin and a polyphenol-rich apple concentrate are more effective together than separately on cecal fermentations and plasma lipids in rats. J. Nutr. 2003, 133, 1860–1865. [Google Scholar] [PubMed]
- Watzl, B.; Girrbach, S.; Roller, M. Inulin, oligofructose and immunomodulation. Br. J. Nutr. 2005, 93, S49–S55. [Google Scholar] [CrossRef] [PubMed]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiao, G.; Yao, Y.; Guo, S.; Lu, K.; Sheng, Z. The role of Bifidobacteria in gut barrier function after thermal injury in rats. J. Trauma 2006, 61, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.E. Probiotics: Definition, sources, selection, and uses. Clin. Infect. Dis. 2008, 46, S58–S61, discussion S144–S151. [Google Scholar] [CrossRef] [PubMed]
- Kailasapathy, K.; Chin, J. Survival and therapeutic potential of probiotic organisms with reference to Lactobacillus acidophilus and Bifidobacterium spp. Immunol. Cell Biol. 2000, 78, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, M.C.; Lajo, T.; Carrión, J.M.; Cuñé, J. Cholesterol-lowering efficacy of Lactobacillus plantarum CECT 7527, 7528 and 7529 in hypercholesterolaemic adults. Br. J. Nutr. 2013, 109, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Liu, X.M.; Zhang, Q.X.; Shen, Z.; Tian, F.W.; Zhang, H.; Sun, Z.H.; Zhang, H.P.; Chen, W. Influence of consumption of probiotics on the plasma lipid profile: A meta-analysis of randomised controlled trials. Nutr. Metab. Cardiovasc. Dis. NMCD 2011, 21, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Ishimwe, N.; Daliri, E.B.; Lee, B.H.; Fang, F.; Du, G. The perspective on cholesterol-lowering mechanisms of probiotics. Mol. Nutr. Food Res. 2015, 59, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Martoni, C.J.; Prakash, S. Cholesterol lowering and inhibition of sterol absorption by Lactobacillus reuteri NCIMB 30242: A randomized controlled trial. Eur. J. Clin. Nutr. 2012, 66, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, K.M.; Fava, F.; Viola, R. ‘The way to a man’s heart is through his gut microbiota’—Dietary pro- and prebiotics for the management of cardiovascular risk. Proc. Nutr. Soc. 2014, 73, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.; Rivière, A.; Moens, F.; Van den Abbeele, P.; Geirnaert, A.; Rogelj, I.; Leroy, F.; De Vuyst, L. Inulin-type fructan fermentation by Bifidobacteria depends on the strain rather than the species and region in the human intestine. Appl. Microbiol. Biotechnol. 2016, 100, 4097–4107. [Google Scholar] [CrossRef] [PubMed]
- Dewulf, E.M.; Cani, P.D.; Claus, S.P.; Fuentes, S.; Puylaert, P.G.B.; Neyrinck, A.M.; Bindels, L.B.; de Vos, W.M.; Gibson, G.R.; Thissen, J. Insight into the prebiotic concept: Lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut 2013, 62, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Martin, J.C.; Duncan, S.H.; Flint, H.J. Prebiotic stimulation of human colonic butyrate-producing bacteria and Bifidobacteria, in vitro. FEMS Microbiol. Ecol. 2014, 87, 30–40. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment: | Prebiotics | Probiotics |
|---|---|---|
| Definition: | Dietary constituents that fertilize and promote healthy gut microbiota composition | Beneficial live microorganisms that can colonize the human gut to develop or restore healthy gut microbiota composition |
| Examples and Effects: | Plant polyphenols Fruits and vegetables (e.g., apples): reduce inflammation [76] and total cholesterol levels [74,77], promote bifidobacteria growth [75] Dietary fructans Foods high in inulin and/or oligofructose: promote bifidobacteria growth [90], restore butyrate-producing bacterial populations [91,92] | Lactobaccillus strains L. reuteri (microencapsulated in yogurt): reduce LDL-cholesterol, serum total cholesterol, and non-HDL cholesterol [88] L. plantarum (capsules): reduce serum total cholesterol [84] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients 2017, 9, 859. https://doi.org/10.3390/nu9080859
Lau K, Srivatsav V, Rizwan A, Nashed A, Liu R, Shen R, Akhtar M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients. 2017; 9(8):859. https://doi.org/10.3390/nu9080859
Chicago/Turabian StyleLau, Kimberley, Varun Srivatsav, Ayesha Rizwan, Andrew Nashed, Rui Liu, Rui Shen, and Mahmood Akhtar. 2017. "Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases" Nutrients 9, no. 8: 859. https://doi.org/10.3390/nu9080859
APA StyleLau, K., Srivatsav, V., Rizwan, A., Nashed, A., Liu, R., Shen, R., & Akhtar, M. (2017). Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients, 9(8), 859. https://doi.org/10.3390/nu9080859