Structure–Function Relationships of the Repeat Domains of RTX Toxins


Abstract
:1. Introduction
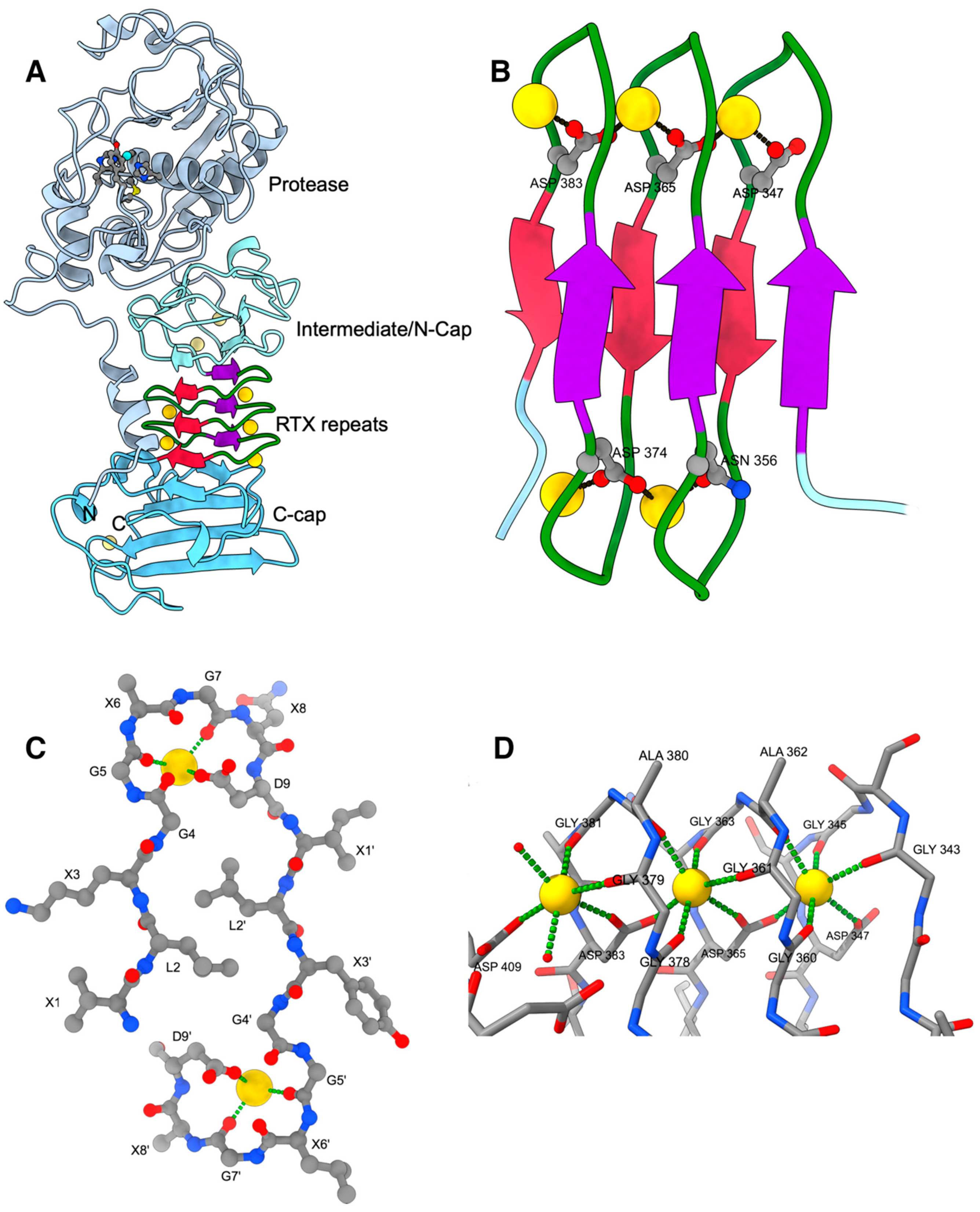
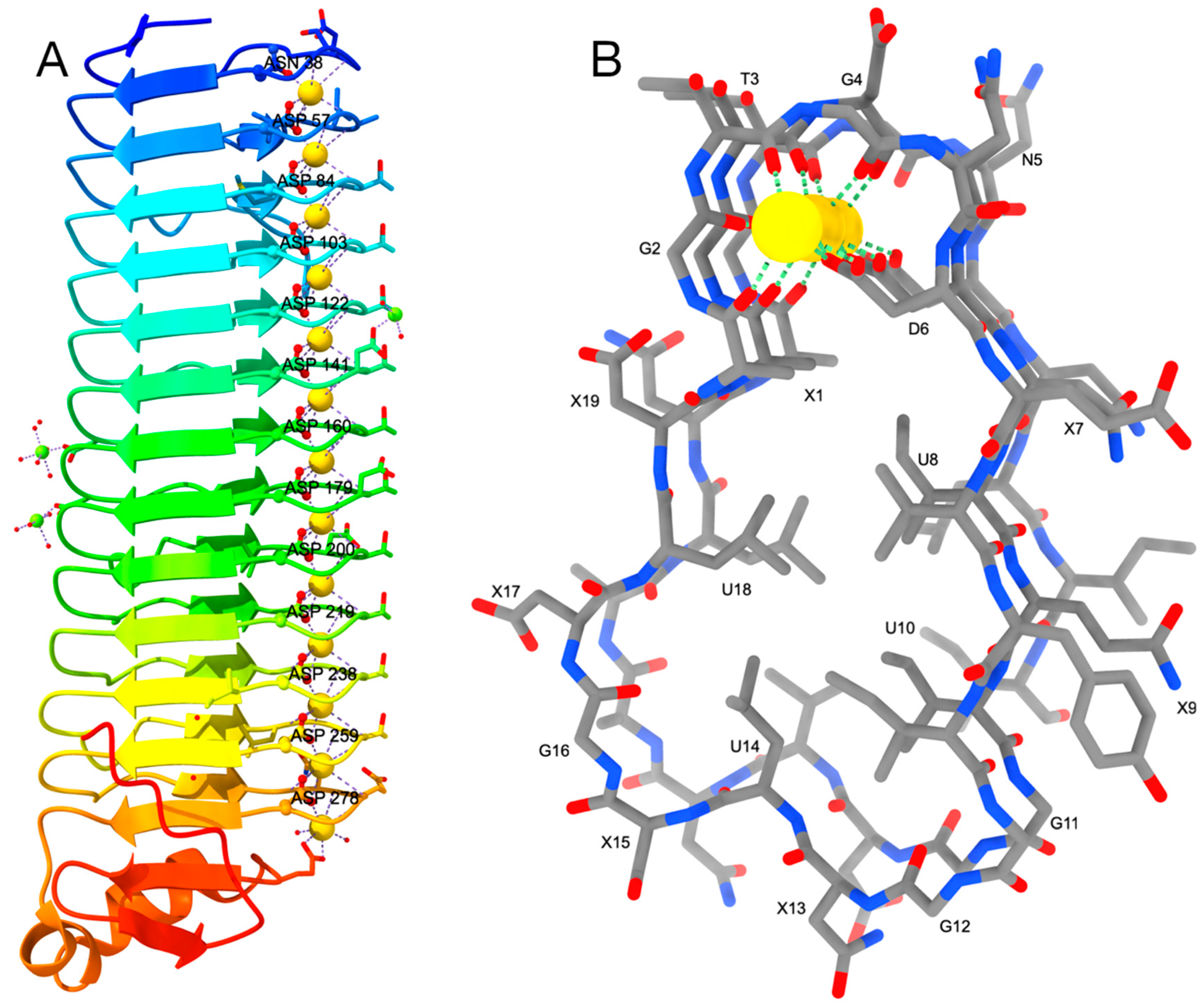
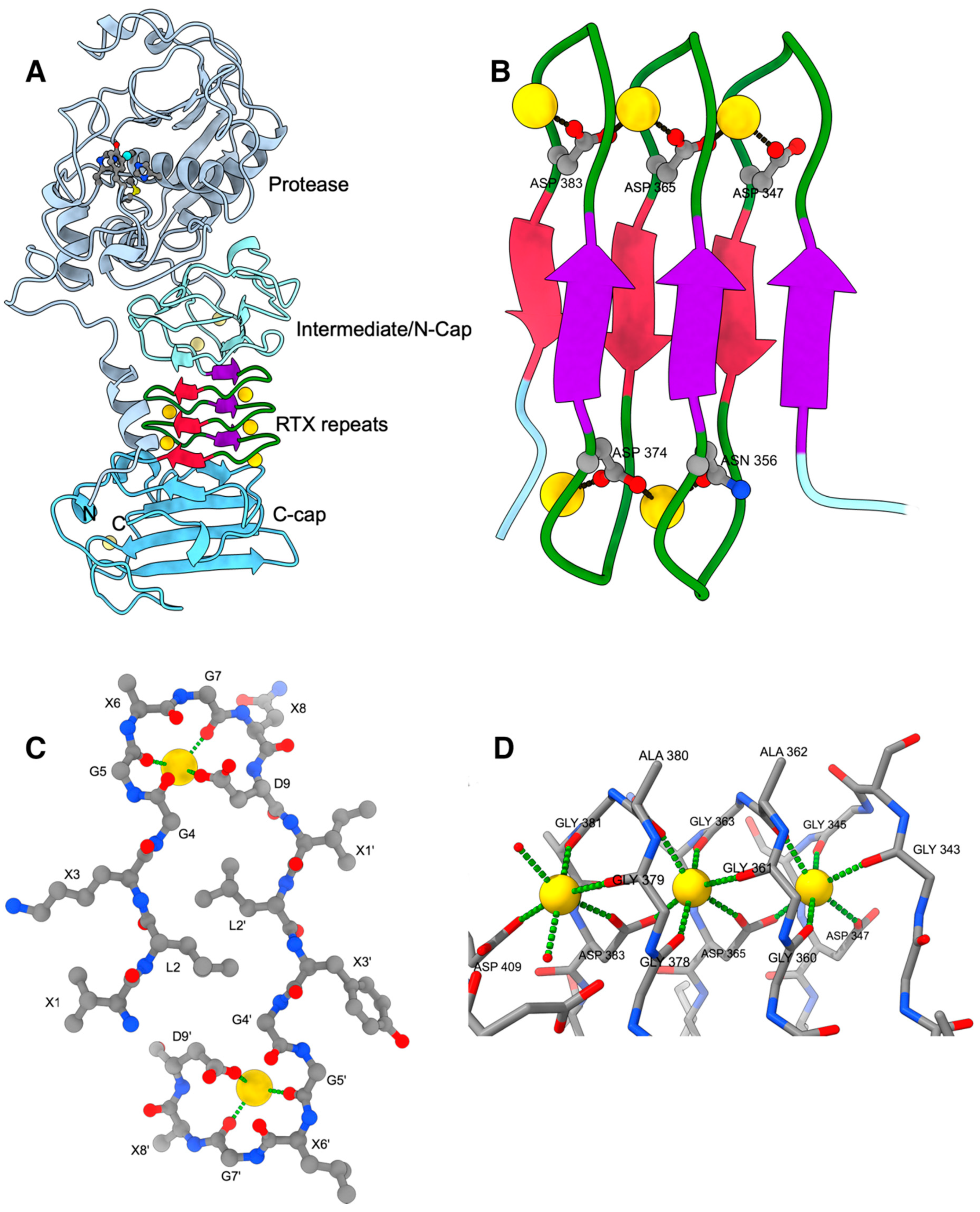
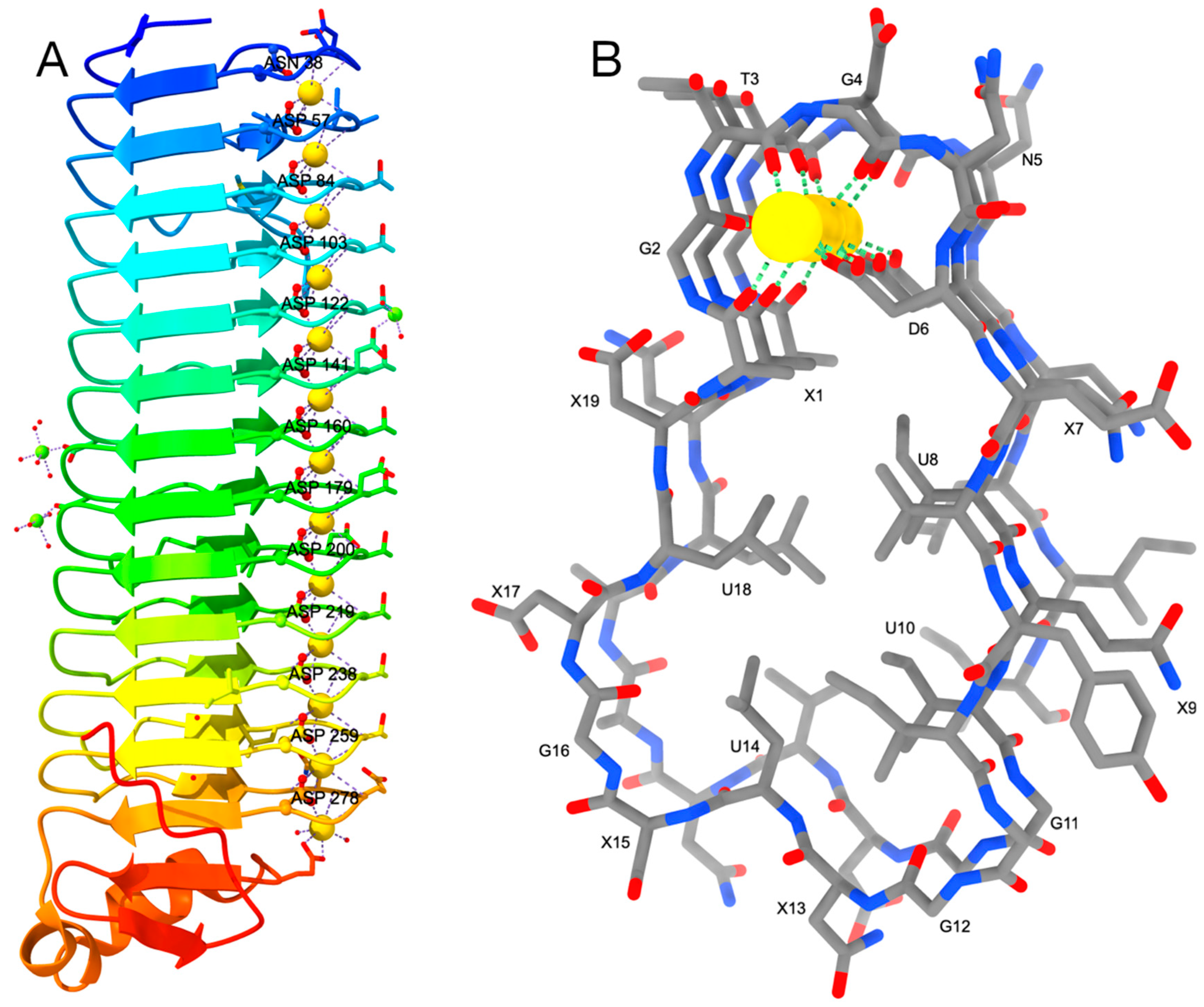
2. RTX Repeats Form a Unique Parallel β-Roll Domain Which Contains Ca2+ Ions as an Integral Part
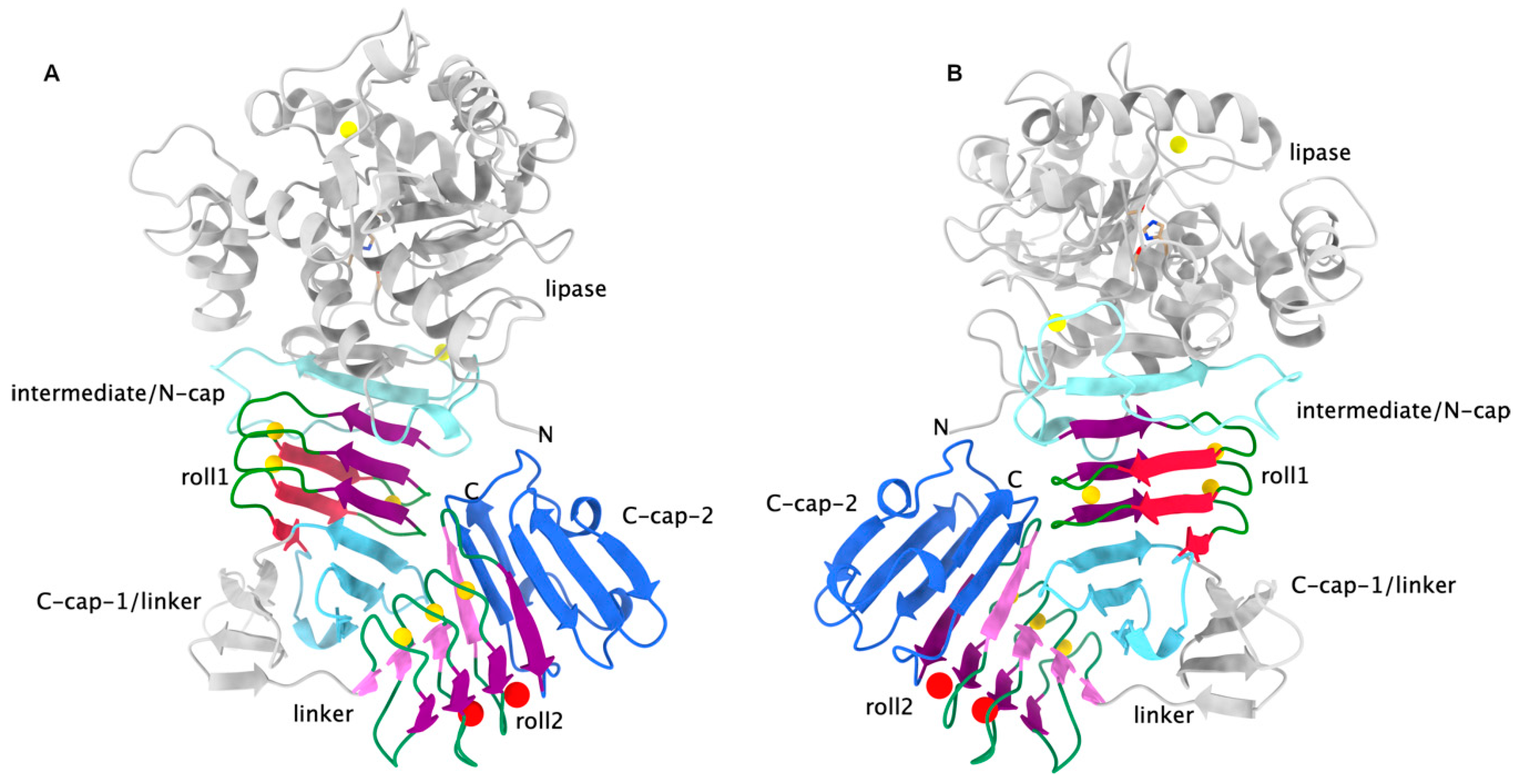
3. RTX Domains Containing more than One Block of Repeats Build a Compact Assembly that Probably Serves as a Folding Nucleus for the Functional Passenger Domains
4. Ca2+-Induced Folding of RTX Domains
5. Structure of RTX Adhesins Reveals the Presence of Canonical and Non-Canonical RTX Repeats
6. Conclusions and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Benz, R. Channel formation by RTX-toxins of pathogenic bacteria: Basis of their biological activity. Biochim. Biophys. Acta Biomembr. 2016, 1858, 526–537. [Google Scholar] [CrossRef]
- Linhartova, I.; Osicka, R.; Bumba, L.; Masin, J.; Sebo, P. Microbial Toxins; Gopalakrishnakone, P., Stiles, B., Alape-Girón, A., Dubreuil, J.D., Mandal, M., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–29. [Google Scholar]
- Welch, R.A. Pore-forming cytolysins of gram-negative bacteria. Mol. Microbiol. 1991, 5, 521–528. [Google Scholar] [CrossRef]
- Welch, R.A. RTX Toxin Structure and Function: A Story of Numerous Anomalies and Few Analogies in Toxin Biology. Curr. Top. Microbiol. Immunol. 2001, 257, 85–111. [Google Scholar]
- Linhartová, I.; Bumba, L.; Mašín, J.; Basler, M.; Osička, R.; Kamanová, J.; Procházková, K.; Adkins, I.; Hejnová-Holubová, J.; Sadílková, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Wandersman, C.; Delepelaire, P.; Letoffe, S.; Ghigo, J.M. A signal peptide-independent protein secretion pathway. Antonie Van Leeuwenhoek 1992, 61, 111–113. [Google Scholar] [CrossRef]
- Thomas, S.; Holland, I.B.; Schmitt, L. The Type 1 secretion pathway—The hemolysin system and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1629–1641. [Google Scholar] [CrossRef]
- Delepelaire, P. Type I secretion in gram-negative bacteria. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1694, 149–161. [Google Scholar] [CrossRef]
- Kanonenberg, K.; Spitz, O.; Erenburg, I.N.; Beer, T.; Schmitt, L. Type I secretion system-it takes three and a substrate. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef]
- Spitz, O.; Erenburg, I.N.; Beer, T.; Kanonenberg, K.; Holland, I.B.; Schmitt, L. Type I Secretion Systems-One Mechanism for All. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Smith, T.J.; Sondermann, H.; O’Toole, G.A. Type 1 Does the Two-Step: Type 1 Secretion Substrates with a Functional Periplasmic Intermediate. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef]
- Debarbieux, L.; Wandersman, C. Folded HasA inhibits its own secretion through its ABC exporter. EMBO J. 2001, 20, 4657–4663. [Google Scholar] [CrossRef]
- Ludwig, A.; Jarchau, T.; Benz, R.; Goebel, W. The repeat domain of Escherichia coli haemolysin (HlyA) is responsible for its Ca2+-dependent binding to erythrocytes. Mol. Gen. Genet. 1988, 214, 553–561. [Google Scholar] [CrossRef]
- Létoffé, S.; Wandersman, C. Secretion of CyaA-PrtB and HlyA-PrtB fusion proteins in Escherichia coli: Involvement of the glycine-rich repeat domain of Erwinia chrysanthemi protease B. J. Bacteriol. 1992, 174, 4920–4927. [Google Scholar] [CrossRef]
- Baumann, U.; Wu, S.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: A two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 1993, 12, 3357–3364. [Google Scholar] [CrossRef]
- Hege, T.; Feltzer, R.E.; Gray, R.D.; Baumann, U. Crystal structure of a complex between Pseudomonas aeruginosa alkaline protease and its cognate inhibitor: Inhibition by a zinc-NH2 coordinative bond. J. Biol. Chem. 2001, 276, 35087–35092. [Google Scholar] [CrossRef]
- Miyatake, H.; Hata, Y.; Fujii, T.; Hamada, K.; Morihara, K.; Katsube, Y. Crystal structure of the unliganded alkaline protease from Pseudomonas aeruginosa IFO3080 and its conformational changes on ligand binding. J. Biochem. 1995, 118, 474–479. [Google Scholar] [CrossRef]
- Baumann, U. Crystal structure of the 50 kDa metallo protease from Serratia marcescens. J. Mol. Biol. 1994, 242, 244–251. [Google Scholar] [CrossRef]
- Baumann, U.; Bauer, M.; Létoffé, S.; Delepelaire, P.; Wandersman, C. Crystal structure of a complex between Serratia marcescens metallo-protease and an inhibitor from Erwinia chrysanthemi. J. Mol. Biol. 1995, 248, 653–661. [Google Scholar] [CrossRef]
- Wu, D.; Ran, T.; Wang, W.; Xu, D. Structure of a thermostable serralysin from Serratia sp. FS14 at 1.1 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 10–15. [Google Scholar] [CrossRef]
- Hamada, K.; Hata, Y.; Katsuya, Y.; Hiramatsu, H.; Fujiwara, T.; Katsube, Y. Crystal structure of Serratia protease, a zinc-dependent proteinase from Serratia sp. E-15, containing a beta-sheet coil motif at 2.0 A resolution. J. Biochem. 1996, 119, 844–851. [Google Scholar] [CrossRef]
- Hege, T.; Baumann, U. The conserved methionine residue of the metzincins: A site-directed mutagenesis study. J. Mol. Biol. 2001, 314, 181–186. [Google Scholar] [CrossRef]
- Hege, T.; Baumann, U. Protease C of Erwinia chrysanthemi: The crystal structure and role of amino acids Y228 and E189. J. Mol. Biol. 2001, 314, 187–193. [Google Scholar] [CrossRef]
- Oberholzer, A.E.; Bumann, M.; Hege, T.; Russo, S.; Baumann, U. Metzincin’s canonical methionine is responsible for the structural integrity of the zinc-binding site. Biol. Chem. 2009, 390, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Bharat, T.A.M.; Kureisaite-Ciziene, D.; Hardy, G.G.; Yu, E.W.; Devant, J.M.; Hagen, W.J.H.; Brun, Y.V.; Briggs, J.A.G.; Löwe, J. Structure of the hexagonal surface layer on Caulobacter crescentus cells. Nat. Microbiol. 2017, 2, 17059. [Google Scholar] [CrossRef]
- Buchinger, E.; Knudsen, D.H.; Behrens, M.A.; Pedersen, J.S.; Aarstad, O.A.; Tøndervik, A.; Valla, S.; Skjåk-Bræk, G.; Wimmer, R.; Aachmann, F.L. Structural and functional characterization of the R-modules in alginate C-5 epimerases AlgE4 and AlgE6 from Azotobacter vinelandii. J. Biol. Chem. 2014, 289, 31382–31396. [Google Scholar] [CrossRef] [Green Version]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-Driven Folding of RTX Domain β-Rolls Ratchets Translocation of RTX Proteins through Type I Secretion Ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Meier, R.; Drepper, T.; Svensson, V.; Jaeger, K.E.; Baumann, U. A calcium-gated lid and a large beta-roll sandwich are revealed by the crystal structure of extracellular lipase from Serratia marcescens. J. Biol. Chem. 2007, 282, 31477–31483. [Google Scholar] [CrossRef] [Green Version]
- Angkawidjaja, C.; You, D.J.; Matsumura, H.; Kuwahara, K.; Koga, Y.; Takano, K.; Kanaya, S. Crystal structure of a family I.3 lipase from Pseudomonas sp. MIS38 in a closed conformation. FEBS Lett. 2007, 581, 5060–5064. [Google Scholar] [CrossRef] [Green Version]
- Aghajari, N.; Van Petegem, F.; Villeret, V.; Chessa, J.P.; Gerday, C.; Haser, R.; Van Beeumen, J. Crystal structures of a psychrophilic metalloprotease reveal new insights into catalysis by cold-adapted proteases. Proteins 2003, 50, 636–647. [Google Scholar] [CrossRef]
- Zhang, S.C.; Sun, M.; Li, T.; Wang, Q.H.; Hao, J.H.; Han, Y.; Hu, X.J.; Zhou, M.; Lin, S.X. Structure analysis of a new psychrophilic marine protease. PLoS ONE 2011, 6, e26939. [Google Scholar] [CrossRef] [Green Version]
- Garnham, C.P.; Campbell, R.L.; Davies, P.L. Anchored clathrate waters bind antifreeze proteins to ice. Proc. Natl. Acad. Sci. USA 2011, 108, 7363–7367. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Stevens, C.A.; Vance, T.D.R.; Olijve, L.L.C.; Graham, L.A.; Campbell, R.L.; Yazdi, S.R.; Escobedo, C.; Bar-Dolev, M.; Yashunsky, V.; et al. Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci. Adv. 2017, 3, e1701440. [Google Scholar] [CrossRef] [Green Version]
- Delepelaire, P.; Wandersman, C. Protein secretion in gram-negative bacteria. The extracellular metalloprotease B from Erwinia chrysanthemi contains a C-terminal secretion signal analogous to that of Escherichia coli alpha-hemolysin. J. Biol. Chem. 1990, 265, 17118–17125. [Google Scholar]
- Delepelaire, P.; Wandersman, C. Characterization, localization and transmembrane organization of the three proteins PrtD, PrtE and PrtF necessary for protease secretion by the gram-negative bacterium Erwinia chrysanthemi. Mol. Microbiol. 1991, 5, 2427–2434. [Google Scholar] [CrossRef]
- Létoffé, S.; Delepelaire, P.; Wandersman, C. Protease secretion by Erwinia chrysanthemi: The specific secretion functions are analogous to those of Escherichia coli alpha-haemolysin. EMBO J. 1990, 9, 1375–1382. [Google Scholar] [CrossRef]
- Baumann, U. Handbook of Proteolytic Enzymes, 2nd ed.; Barrett, A.J., Rawlings, N.D., Woessner, F.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 579–581. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, 624–632. [Google Scholar] [CrossRef]
- Duong, F.; Lazdunski, A.; Cami, B.; Murgier, M. Sequence of a cluster of genes controlling synthesis and secretion of alkaline protease in Pseudomonas aeruginosa: Relationships to other secretory pathways. Gene 1992, 121, 47–54. [Google Scholar] [CrossRef]
- Létoffé, S.; Delepelaire, P.; Wandersman, C. Characterization of a protein inhibitor of extracellular proteases produced by Erwinia chrysanthemi. Mol. Microbiol. 1989, 3, 79–86. [Google Scholar] [CrossRef]
- Stöcker, W.; Grams, F.; Baumann, U.; Reinemer, P.; Gomis-Rüth, F.X.; McKay, D.B.; Bode, W. The metzincins—Topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. 1995, 4, 823–840. [Google Scholar] [CrossRef] [Green Version]
- Goddard, T.D.; Brilliant, A.A.; Skillman, T.L.; Vergenz, S.; Tyrwhitt-Drake, J.; Meng, E.C.; Ferrin, T.E. Molecular Visualization on the Holodeck. J. Mol. Biol. 2018, 430, 3982–3996. [Google Scholar] [CrossRef]
- Yoder, M.D.; Keen, N.T.; Jurnak, F. New domain motif: The structure of pectate lyase C, a secreted plant virulence factor. Science 1993, 260, 1503–1507. [Google Scholar] [CrossRef]
- Yoder, M.D.; Lietzke, S.E.; Jurnak, F. Unusual structural features in the parallel beta-helix in pectate lyases. Structure 1993, 1, 241–251. [Google Scholar] [CrossRef]
- Lilie, H.; Haehnel, W.; Rudolph, R.; Baumann, U. Folding of a synthetic parallel beta-roll protein. FEBS Lett. 2000, 470, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Bauche, C.; Chenal, A.; Knapp, O.; Bodenreider, C.; Benz, R.; Chaffotte, A.; Ladant, D. Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 2006, 281, 16914–16926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blenner, M.A.; Shur, O.; Szilvay, G.R.; Cropek, D.M.; Banta, S. Calcium-induced folding of a beta roll motif requires C-terminal entropic stabilization. J. Mol. Biol. 2010, 400, 244–256. [Google Scholar] [CrossRef]
- Sotomayor-Pérez, A.C.; Subrini, O.; Hessel, A.; Ladant, D.; Chenal, A. Molecular crowding stabilizes both the intrinsically disordered calcium-free state and the folded calcium-bound state of a repeat in toxin (RTX) protein. J. Am. Chem. Soc. 2013, 135, 11929–11934. [Google Scholar] [CrossRef]
- Richardson, J.S.; Richardson, D.C. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenal, A.; Guijarro, J.I.; Raynal, B.; Delepierre, M.; Ladant, D. RTX calcium binding motifs are intrinsically disordered in the absence of calcium: Implication for protein secretion. J. Biol. Chem. 2009, 284, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Chenal, A.; Karst, J.C.; Sotomayor Pérez, A.C.; Wozniak, A.K.; Baron, B.; England, P.; Ladant, D. Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys. J. 2010, 99, 3744–3753. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, D.P.; Perez, A.C.S.; Karst, J.; Cannella, S.E.; Enguéné, V.Y.N.; Hessel, A.; Raoux-Barbot, D.; Voegele, A.; Subrini, O.; Davi, M.; et al. Calcium-dependent disorder-to-order transitions are central to the secretion and folding of the CyaA toxin of Bordetella pertussis, the causative agent of whooping cough. Toxicon 2018, 149, 37–44. [Google Scholar] [CrossRef]
- Sotomayor-Pérez, A.C.; Ladant, D.; Chenal, A. Calcium-induced folding of intrinsically disordered repeat-in-toxin (RTX) motifs via changes of protein charges and oligomerization states. J. Biol. Chem. 2011, 286, 16997–17004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotomayor-Pérez, A.C.; Ladant, D.; Chenal, A. Disorder-to-order transition in the CyaA toxin RTX domain: Implications for toxin secretion. Toxins 2014, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Szilvay, G.R.; Blenner, M.A.; Shur, O.; Cropek, D.M.; Banta, S. A FRET-based method for probing the conformational behavior of an intrinsically disordered repeat domain from Bordetella pertussis adenylate cyclase. Biochemistry 2009, 48, 11273–11282. [Google Scholar] [CrossRef]
- Zhang, L.; Conway, J.F.; Thibodeau, P.H. Calcium-induced folding and stabilization of the Pseudomonas aeruginosa alkaline protease. J. Biol. Chem. 2012, 287, 4311–4322. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.H.; Beer, T.; Smits, S.H.; Schmitt, L. In vivo quantification of the secretion rates of the hemolysin A Type I secretion system. Sci. Rep. 2016, 6, 33275. [Google Scholar] [CrossRef] [PubMed]
- Satchell, K.J. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 2011, 65, 71–90. [Google Scholar] [CrossRef]
- Guo, S.; Vance, T.D.R.; Stevens, C.A.; Voets, I.; Davies, P.L. RTX Adhesins are Key Bacterial Surface Megaproteins in the Formation of Biofilms. Trends Microbiol. 2019, 27, 453–467. [Google Scholar] [CrossRef] [PubMed]
- El-Azami-El-Idrissi, M.; Bauche, C.; Loucka, J.; Osicka, R.; Sebo, P.; Ladant, D.; Leclerc, C. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: Role of toxin acylation and identification of the main integrin interaction domain. J. Biol. Chem. 2003, 278, 38514–38521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulutoglu, B.; Dooley, K.; Szilvay, G.; Blenner, M.; Banta, S. Catch and Release: Engineered Allosterically Regulated β-Roll Peptides Enable On/Off Biomolecular Recognition. ACS Synth. Biol. 2017, 6, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Bulutoglu, B.; Banta, S. Calcium-Dependent RTX Domains in the Development of Protein Hydrogels. Gels 2019, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Protein | PDB Entry | No of Repeats XUXGGXGXD |
|---|---|---|
| Ps. aeruginosa alkaline protease APRA_PSEAE | 1KAP [15], 1JIW [16], 1AKL [17], 3VI1 | 5 |
| S. marcescens metalloprotease PRZN_SERMA | 1SAT [18], 1SMP [19], 1AF0, 4I35, 5D7W [20] | 5 |
| S. marcescens sp. Metalloprotease PRTZN_SERME | 1SRP [21] | 5 |
| Erwinia chrysanthemi metalloprotease PrtC PRTC_DICCH | 1GO8 [22], 1K7G [23], 1K7I, 1K7Q, 3HB2 [24], 3HBU, 3HBV, 3HDA | 5 |
| Caulobacter vibroides S-Layer protein A0A0H3C8J1_CAUVN | 5N97, 5N8P [25] | 4 |
| Azotobacter vinelandii mannrinan-C5-Epimerase ALGE4_AZOVI | 2AGM, 2ML1, 2ML2, 2ML3 [26] | 6 |
| Bordetella pertussis adenlyate cyclase CYAA_BORP1 | 5CVW, 5CXL [27] | 8 |
| Serratia marcescens lipase LipA Q5933_SERMA | 2QUA, 2QUB [28] | 5 + 8 |
| Ps. Fluorescence extracellular lipase Q9RBY1_9PSED | 2Z8X, 2Z8Z, 2ZJ6, 2ZJ7, 2ZVD, 3A6Z, 3A70 [29] | 5 + 8 |
| Pseudomonas sp. psychrophilic metalloprotease O69771_9PSED | 1H71 [30], 1G9K, 1O0Q, 1O0T, 1OM6, 1OM7, 1OM8, 1OMJ | 5 |
| Flaviobacterium sp. alkaline metalloprotease DOVMS8_9FLAO | 3U1R [31] | 5 |
| Marinomonas primoryensis ice-binding protein region IV A1YIY3_9GAMM | 3P4G [32] | 12 19-residue repeats |
| Marinomonas primoryensis ice-binding protein region V A1YIY3_9GAMM | 5 JUH [33] | 4 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumann, U. Structure–Function Relationships of the Repeat Domains of RTX Toxins. Toxins 2019, 11, 657. https://doi.org/10.3390/toxins11110657
Baumann U. Structure–Function Relationships of the Repeat Domains of RTX Toxins. Toxins. 2019; 11(11):657. https://doi.org/10.3390/toxins11110657
Chicago/Turabian StyleBaumann, Ulrich. 2019. "Structure–Function Relationships of the Repeat Domains of RTX Toxins" Toxins 11, no. 11: 657. https://doi.org/10.3390/toxins11110657
APA StyleBaumann, U. (2019). Structure–Function Relationships of the Repeat Domains of RTX Toxins. Toxins, 11(11), 657. https://doi.org/10.3390/toxins11110657