Phobalysin: Fisheye View of Membrane Perforation, Repair, Chemotaxis and Adhesion

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hemolysins and Hemolysin Genes in Pdd
2.1. Plasmid Encoded Hemolysins
2.2. Chromosomally Encoded hlyA
3. Regulation of Hemolysin Production
3.1. Transcriptional Regulation
3.2. Secretion
4. Diversity and Molecular Epidemiology of Hemolytic Phenotypes
5. Phobalysin P
5.1. Stable Toxin-Complexes and Membrane Pores
5.2. Cytotoxicity
6. PhlyP as a Paradigm of Membrane Repair
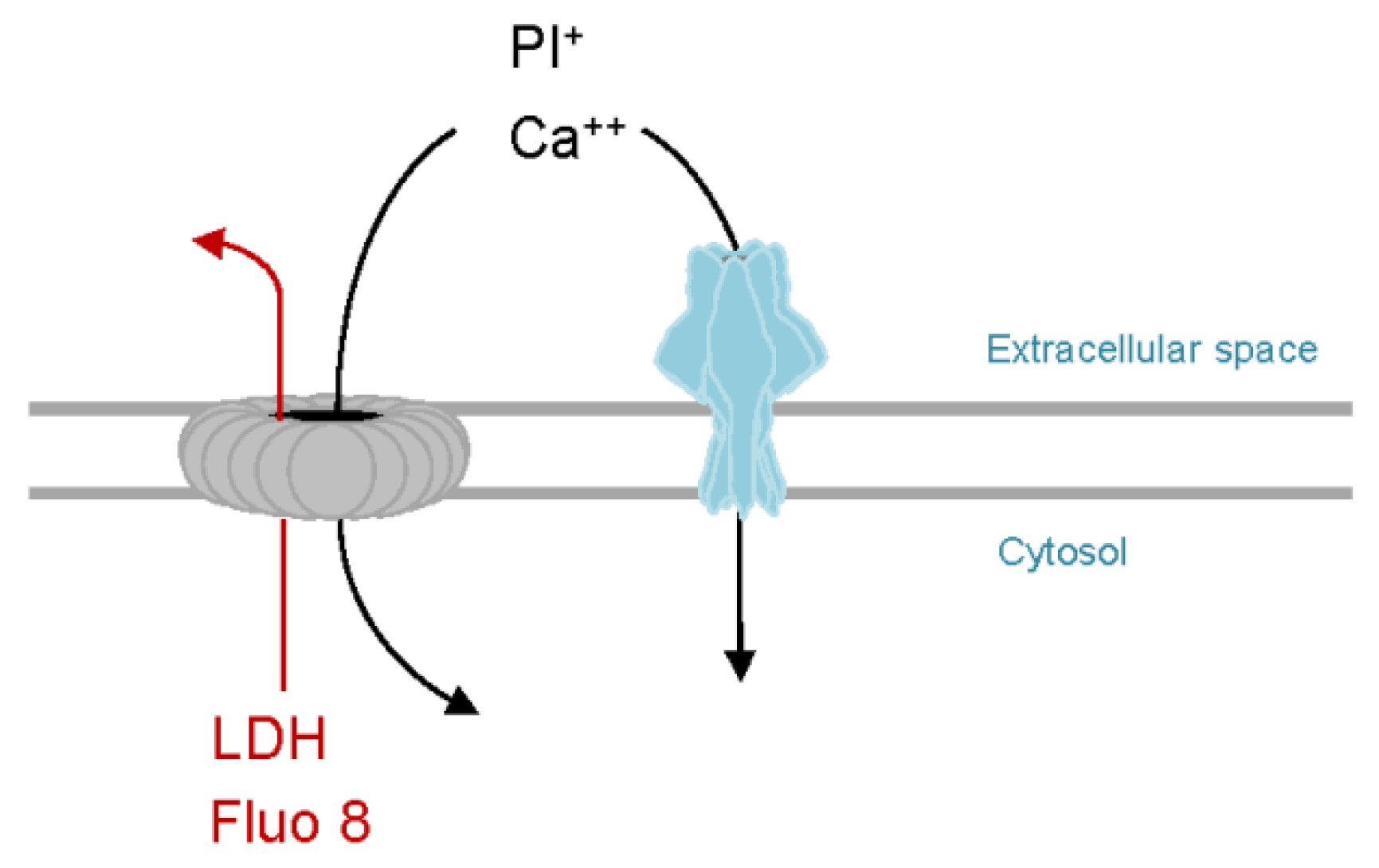
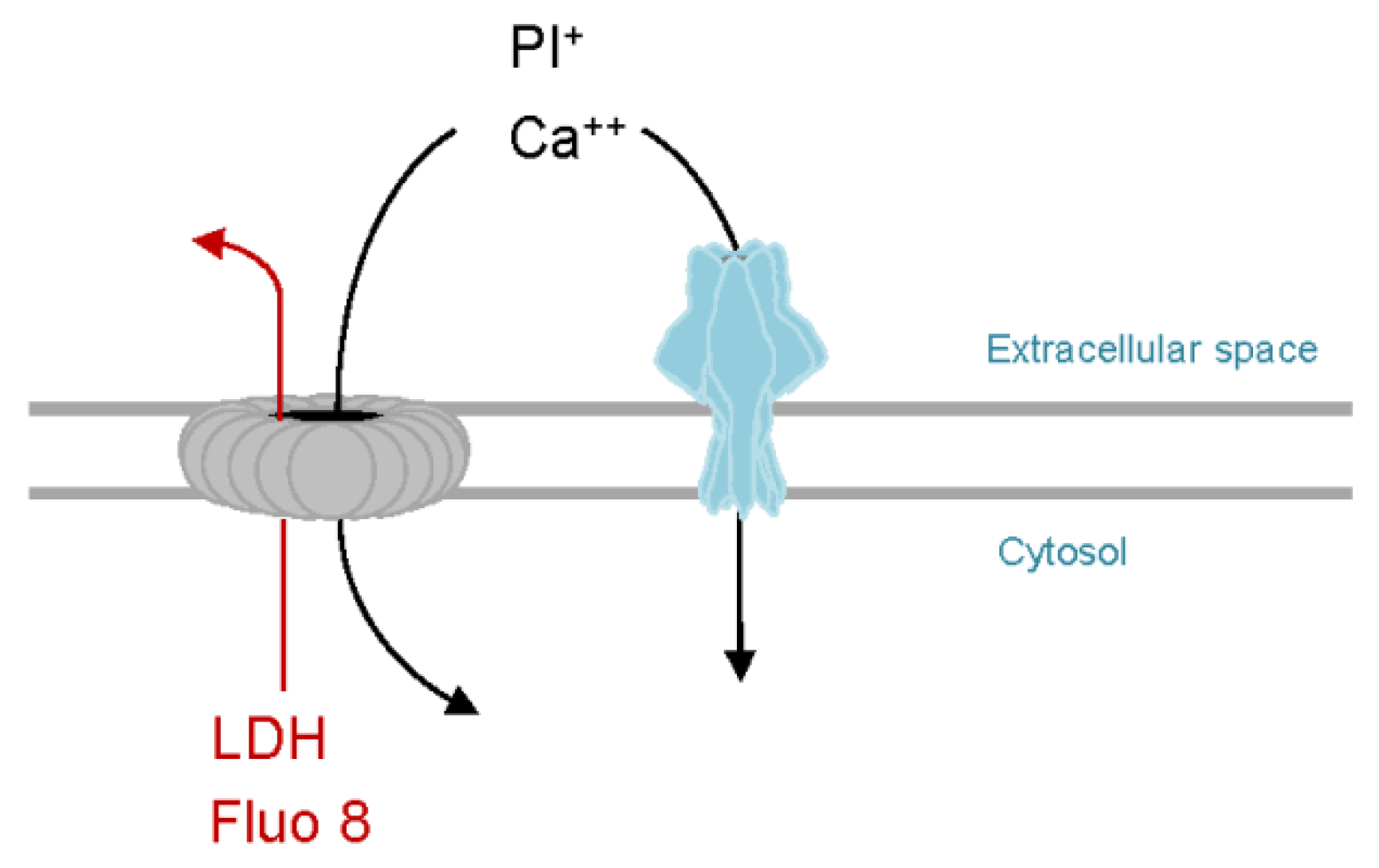
6.1. Calcium Influx-Dependent Repair (CIDRE) of Cholesterol Dependent Cytolysins (CDC) Pores
6.2. Subversion of CIDRE
6.3. Alternative Rescue Mechanisms
6.4. PhlyP—A Small β-PFT that Triggers CIDRE
6.5. Technical Advantages of PhlyP for Studying CIDRE
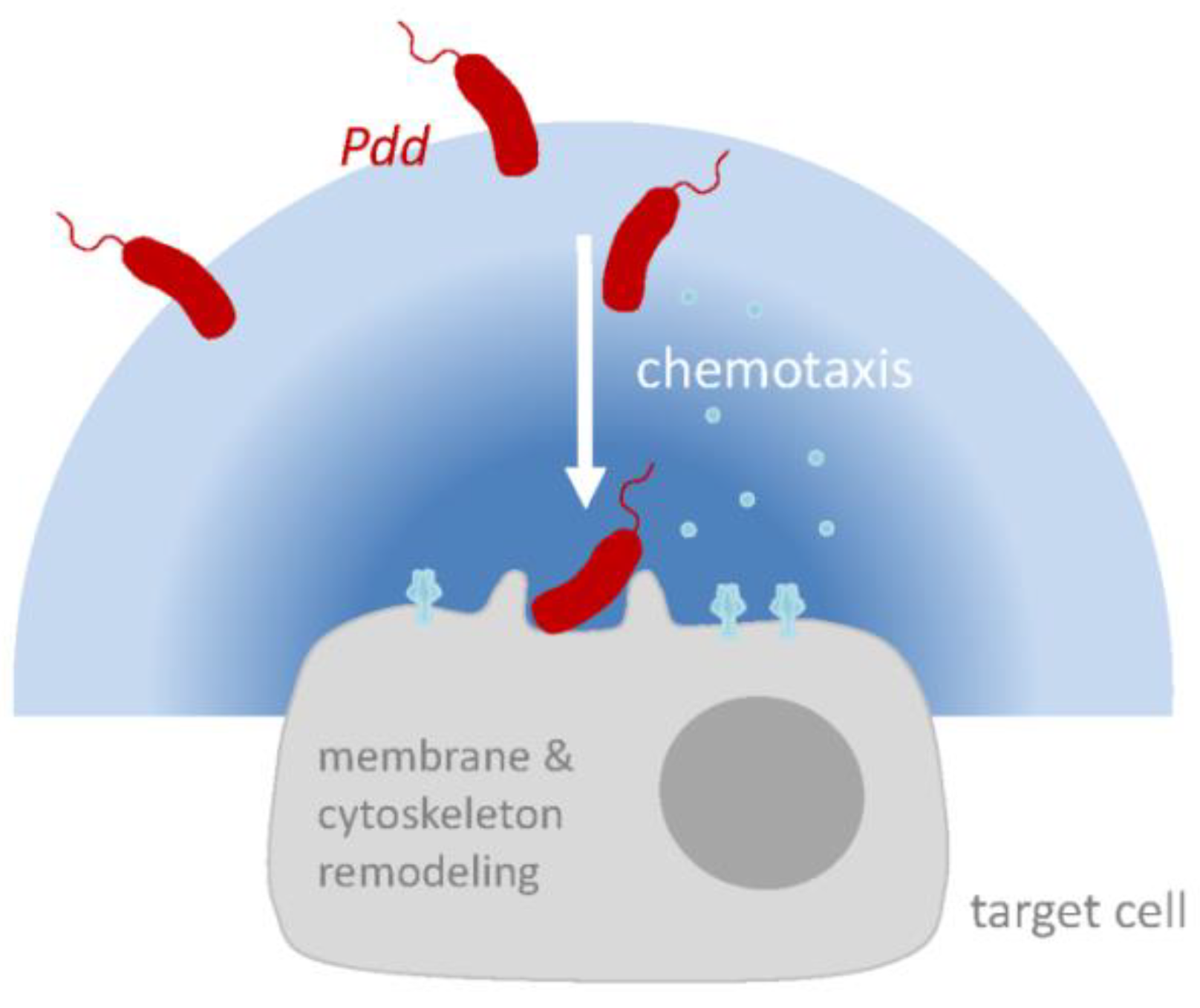
7. PhlyP Promotes the Association of Pdd with Epithelial Cells
7.1. Phlyp Enhances Adhesion
7.2. A Role of Chemotaxis Regulators
8. Perspective
8.1. Receptors
8.2. Lectin Domain (LD)
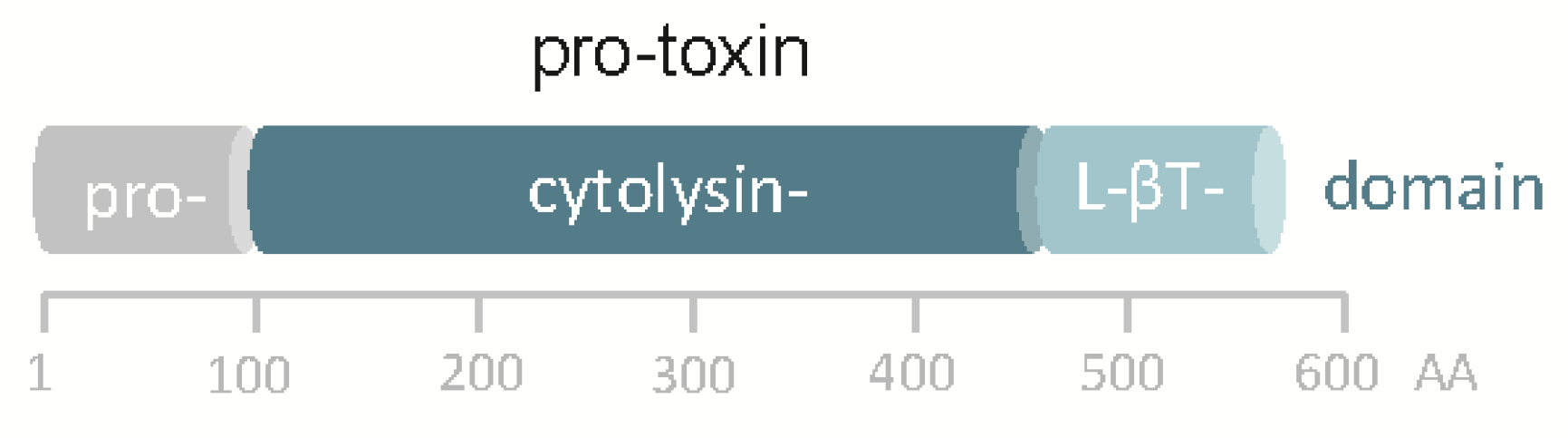
8.3. Pro-Toxin Processing
8.4. Interaction of PhlyP and Other Factors
8.5. Engineering Phobalysins
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Austin, B. Vibrios as causal agents of zoonoses. Vet. Microbiol. 2010, 140, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.G., Jr.; Miller, H.G.; Wilson, R.; Tacket, C.O.; Hollis, D.G.; Hickman, F.W.; Weaver, R.E.; Blake, P.A. Illness caused by Vibrio damsela and Vibrio hollisae. Lancet 1982, 1, 1294–1297. [Google Scholar] [CrossRef]
- Fraser, S.L.; Purcell, B.K.; Delgado, B., Jr.; Baker, A.E.; Whelen, A.C. Rapidly fatal infection due to Photobacterium (Vibrio) damsela. Clin. Infect. Dis. 1997, 25, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Goodell, K.H.; Jordan, M.R.; Graham, R.; Cassidy, C.; Nasraway, S.A. Rapidly advancing necrotizing fasciitis caused by Photobacterium (Vibrio) damsela: A hyperaggressive variant. Crit. Care Med. 2004, 32, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.; Stevens, R.P.; Konecny, P. Photobacterium damselae and Vibrio harveyi hand infection from marine exposure. Med. J. Aust. 2015, 203, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Rivas, A.J.; Lemos, M.L.; Osorio, C.R. Photobacterium damselae subsp. damselae, a bacterium pathogenic for marine animals and humans. Front. Microbiol. 2013, 4, 283. [Google Scholar] [CrossRef] [PubMed]
- Osorio, C.R.; Vences, A.; Matanza, X.M.; Terceti, M.S. Photobacterium damselae subsp. damselae, a generalist pathogen with unique virulence factors and high genetic diversity. J. Bacteriol. 2018, 200, e00002-18. [Google Scholar] [CrossRef]
- Labella, A.M.; Arahal, D.R.; Castro, D.; Lemos, M.L.; Borrego, J.J. Revisiting the genus photobacterium: Taxonomy, ecology and pathogenesis. Int. Microbiol. 2017, 20, 1–10. [Google Scholar] [CrossRef]
- Love, M.; Teebken-Fisher, D.; Hose, J.E.; Farmer, J.J., 3rd; Hickman, F.W.; Fanning, G.R. Vibrio damsela, a marine bacterium, causes skin ulcers on the damselfish chromis punctipinnis. Science 1981, 214, 1139–1140. [Google Scholar] [CrossRef]
- Aguirre, A.A.; Balazs, G.H.; Zimmerman, B.; Spraker, T.R. Evaluation of Hawaiian green turtles (Chelonia mydas) for potential pathogens associated with fibropapillomas. J. Wildl. Dis. 1994, 30, 8–15. [Google Scholar] [CrossRef]
- Liu, W.Y.; Kou, G.H.; Chen, S.N. Survey of disease of cultural shrimp in Taiwan. Chin. j. Microbiol. Immunol. 1995, 28, 59–69. [Google Scholar]
- Pedersen, K.; Skall, H.F.; Lassen-Nielsen, A.M.; Bjerrum, L.; Olesen, N.J. Photobacterium damselae subsp. damselae, an emerging pathogen in Danish rainbow trout, Oncorhynchus mykiss (Walbaum), mariculture. J. Fish. Dis. 2009, 32, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Fouz, B.; Larsen, J.L.; Nielsen, B.; Barja, J.L.; Toranzo, A.E. Characterization of vibrio-damsela strains isolated from turbot Scophthalmus maximus in Spain. Dis. Aquat. Org. 1992, 12, 155–166. [Google Scholar] [CrossRef]
- Grimes, D.J.; Stemmler, J.; Hada, H.; May, E.B.; Maneval, D.; Hetrick, F.M.; Jones, R.T.; Stoskopf, M.; Colwell, R.R. Vibrio species associated with mortality of sharks held in captivity. Microb. Ecol. 1984, 10, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Obendorf, D.L.; Carson, J.; McManus, T.J. Vibrio damsela infection in a stranded leatherback turtle (Dermochelys coriacea). J. Wildl. Dis. 1987, 23, 666–668. [Google Scholar] [CrossRef]
- Clarridge, J.E.; Zighelboim-Daum, S. Isolation and characterization of two hemolytic phenotypes of Vibrio damsela associated with a fatal wound infection. J. Clin. Microbiol. 1985, 21, 302–306. [Google Scholar] [PubMed]
- Fernandez, C.R.; Pankey, G.A. Tissue invasion by unnamed marine vibrios. JAMA 1975, 233, 1173–1176. [Google Scholar] [CrossRef]
- Yamane, K.; Asato, J.; Kawade, N.; Takahashi, H.; Kimura, B.; Arakawa, Y. Two cases of fatal necrotizing fasciitis caused by Photobacterium damsela in Japan. J. Clin. Microbiol. 2004, 42, 1370–1372. [Google Scholar] [CrossRef]
- Hundenborn, J.; Thurig, S.; Kommerell, M.; Haag, H.; Nolte, O. Severe wound infection with Photobacterium damselae ssp. damselae and Vibrio harveyi, following a laceration injury in marine environment: A case report and review of the literature. Case Rep. Med. 2013, 2013, 610632. [Google Scholar] [CrossRef]
- Austin, B.; Zhang, X.H. Vibrio harveyi: A significant pathogen of marine vertebrates and invertebrates. Lett. Appl. Microbiol. 2006, 43, 119–124. [Google Scholar] [CrossRef]
- Kreger, A.S.; Bernheimer, A.W.; Etkin, L.A.; Daniel, L.W. Phospholipase D activity of vibrio damsela cytolysin and its interaction with sheep erythrocytes. Infect. Immun. 1987, 55, 3209–3212. [Google Scholar] [PubMed]
- Kothary, M.H.; Kreger, A.S. Purification and characterization of an extracellular cytolysin produced by vibrio damsela. Infect. Immun. 1985, 49, 25–31. [Google Scholar] [PubMed]
- Kreger, A.S. Cytolytic activity and virulence of vibrio damsela. Infect. Immun. 1984, 44, 326–331. [Google Scholar] [PubMed]
- Popoff, M.R. Clostridial pore-forming toxins: Powerful virulence factors. Anaerobe 2014, 30, 220–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiu, R.; Hall, L.J. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 2018, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Bryant, A.E. Necrotizing soft-tissue infections. N. Engl. J. Med. 2017, 377, 2253–2265. [Google Scholar] [CrossRef] [PubMed]
- Baker-Austin, C.; Oliver, J.D. Vibrio vulnificus: New insights into a deadly opportunistic pathogen. Environ. Microbiol. 2018, 20, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.J.; Musser, J.M. Molecular pathogenesis of necrotizing fasciitis. Annu. Rev. Pathol. 2010, 5, 1–31. [Google Scholar] [CrossRef]
- Valerio, E.; Chaves, S.; Tenreiro, R. Diversity and impact of prokaryotic toxins on aquatic environments: A review. Toxins 2010, 2, 2359–2410. [Google Scholar] [CrossRef]
- Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins 2018, 10, 212. [Google Scholar] [CrossRef]
- Daniel, L.W.; King, L.; Kennedy, M. Phospholipase activity of bacterial toxins. Methods Enzymol. 1988, 165, 298–301. [Google Scholar] [PubMed]
- Cutter, D.L.; Kreger, A.S. Cloning and expression of the damselysin gene from vibrio damsela. Infect. Immun. 1990, 58, 266–268. [Google Scholar] [PubMed]
- Christie, R.; Atkins, N.E.; Munch-Petersen, E. A note on a lytic phenomenon shown by group B streptococci. Aust. J. Exp. Biol. Med. Sci. 1944, 22, 197–200. [Google Scholar] [CrossRef]
- Rivas, A.J.; Balado, M.; Lemos, M.L.; Osorio, C.R. The Photobacterium damselae subsp. damselae hemolysins damselysin and HlyA are encoded within a new virulence plasmid. Infect. Immun. 2011, 79, 4617–4627. [Google Scholar] [CrossRef] [PubMed]
- Roig, F.J.; Amaro, C. Plasmid diversity in Vibrio vulnificus biotypes. Microbiology 2009, 155, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Rivas, A.J.; von Hoven, G.; Neukirch, C.; Meyenburg, M.; Qin, Q.; Fuser, S.; Boller, K.; Lemos, M.L.; Osorio, C.R.; Husmann, M. Phobalysin, a small beta-pore-forming toxin of Photobacterium damselae subsp. damselae. Infect. Immun. 2015, 83, 4335–4348. [Google Scholar] [CrossRef]
- De, S.; Olson, R. Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. USA 2011, 108, 7385–7390. [Google Scholar] [CrossRef]
- Rivas, A.J.; Balado, M.; Lemos, M.L.; Osorio, C.R. Synergistic and additive effects of chromosomal and plasmid-encoded hemolysins contribute to hemolysis and virulence in Photobacterium damselae subsp. damselae. Infect. Immun 2013, 81, 3287–3299. [Google Scholar] [CrossRef]
- Rivas, A.J.; Labella, A.M.; Borrego, J.J.; Lemos, M.L.; Osorio, C.R. Evidence for horizontal gene transfer, gene duplication and genetic variation as driving forces of the diversity of haemolytic phenotypes in Photobacterium damselae subsp. damselae. FEMS Microbiol. Lett. 2014, 355, 152–162. [Google Scholar] [CrossRef]
- Terceti, M.S.; Rivas, A.J.; Alvarez, L.; Noia, M.; Cava, F.; Osorio, C.R. rstB Regulates expression of the Photobacterium damselae subsp. damselae major virulence factors damselysin, phobalysin P and phobalysin, C. Front. Microbiol. 2017, 8, 582. [Google Scholar] [CrossRef]
- Terceti, M.S.; Vences, A.; Matanza, X.M.; Barca, A.V.; Noia, M.; Lisboa, J.; dos Santos, N.M.S.; do Vale, A.; Osorio, C.R. The RstAB system impacts virulence, motility, cell morphology, penicillin tolerance and production of type II secretion system-dependent factors in the fish and human pathogen Photobacterium damselae subsp. damselae. Front. Microbiol. 2019, 10, 897. [Google Scholar] [CrossRef] [PubMed]
- Von Hoven, G.; Neukirch, C.; Meyenburg, M.; Schmidt, S.; Vences, A.; Osorio, C.R.; Husmann, M.; Rivas, A.J. Cytotoxin- and chemotaxis-genes cooperate to promote adhesion of Photobacterium damselae subsp. damselae. Front. Microbiol 2018, 9, 2996. [Google Scholar] [CrossRef] [PubMed]
- Rivas, A.J.; Vences, A.; Husmann, M.; Lemos, M.L.; Osorio, C.R. Photobacterium damselae subsp. damselae major virulence factors Dly, plasmid-encoded HlyA, and chromosome-encoded HlyA are secreted via the type II secretion system. Infect. Immun. 2015, 83, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Green, E.R.; Mecsas, J. Bacterial secretion systems: An overview. Microbiol. Spectr. 2016, 4, 1–19. [Google Scholar] [CrossRef]
- Sandkvist, M.; Bagdasarian, M.; Howard, S.P. Characterization of the multimeric Eps complex required for cholera toxin secretion. Int. J. Med. Microbiol. 2000, 290, 345–350. [Google Scholar] [CrossRef]
- Terceti, M.S.; Vences, A.; Matanza, X.M.; Dalsgaard, I.; Pedersen, K.; Osorio, C.R. Molecular epidemiology of Photobacterium damselae subsp. damselae outbreaks in marine rainbow trout farms reveals extensive horizontal gene transfer and high genetic diversity. Front. Microbiol. 2018, 9, 2155. [Google Scholar] [CrossRef]
- Vences, A.; Rivas, A.J.; Lemos, M.L.; Husmann, M.; Osorio, C.R. Chromosome-encoded hemolysin, phospholipase, and collagenase in plasmidless isolates of Photobacterium damselae subsp. damselae contribute to virulence for fish. Appl. Environ. Microbiol. 2017, 83, e00401-17. [Google Scholar] [CrossRef]
- Von Hoven, G.; Rivas, A.J.; Neukirch, C.; Meyenburg, M.; Qin, Q.; Parekh, S.; Hellmann, N.; Husmann, M. Repair of a bacterial small beta-barrel toxin pore depends on channel width. MBio 2017, 8, e02083-16. [Google Scholar] [CrossRef]
- Bhakdi, S.; Fussle, R.; Tranum-Jensen, J. Staphylococcal alpha-toxin: Oligomerization of hydrophilic monomers to form amphiphilic hexamers induced through contact with deoxycholate detergent micelles. Proc. Natl. Acad. Sci. USA 1981, 78, 5475–5479. [Google Scholar] [CrossRef]
- Harris, J.R.; Palmer, M. Cholesterol specificity of some heptameric beta-barrel pore-forming bacterial toxins: Structural and functional aspects. Subcell Biochem. 2010, 51, 579–596. [Google Scholar] [CrossRef]
- Woll, S.; Windoffer, R.; Leube, R.E. p38 MAPK-dependent shaping of the keratin cytoskeleton in cultured cells. J. Cell Biol. 2007, 177, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Von Hoven, G.; Neukirch, C.; Meyenburg, M.; Fuser, S.; Petrivna, M.B.; Rivas, A.J.; Ryazanov, A.; Kaufman, R.J.; Aroian, R.V.; Husmann, M. eIF2alpha confers cellular tolerance to S. aureus alpha-toxin. Front. Immunol. 2015, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Girardin, S.E. Translation inhibition and metabolic stress pathways in the host response to bacterial pathogens. Nat. Rev. Microbiol. 2013, 11, 365–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- Etxaniz, A.; Gonzalez-Bullon, D.; Martin, C.; Ostolaza, H. Membrane repair mechanisms against permeabilization by pore-forming toxins. Toxins 2018, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host. Microbe. 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Cabanes, D.; Sarmento Mesquita, F.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell Mol. Life Sci. 2019, 76, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Bouillot, S.; Reboud, E.; Huber, P. Functional consequences of calcium influx promoted by bacterial pore-forming toxins. Toxins 2018, 10, 387. [Google Scholar] [CrossRef]
- Chambers, R.; Chambers, E.L. Explorations into the Nature of the Living Cell; Harvard University Press: Cambridge, MA, USA, 1961. [Google Scholar]
- Heilbrunn, L.V. Cellular physiology and aging. Fed. Proc. 1956, 15, 948–953. [Google Scholar] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; McNeil, P.L. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J. Cell Biol. 1995, 131, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed]
- Thelestam, M.; Mollby, R. Survival of cultured cells after functional and structural disorganization of plasma membrane by bacterial haemolysins and phospholipases. Toxicon 1983, 21, 805–815. [Google Scholar] [CrossRef]
- Walev, I.; Palmer, M.; Martin, E.; Jonas, D.; Weller, U.; Hohn-Bentz, H.; Husmann, M.; Bhakdi, S. Recovery of human fibroblasts from attack by the pore-forming alpha-toxin of Staphylococcus aureus. Microb. Pathog. 1994, 17, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Walev, I.; Bhakdi, S.C.; Hofmann, F.; Djonder, N.; Valeva, A.; Aktories, K.; Bhakdi, S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 2001, 98, 3185–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walev, I.; Hombach, M.; Bobkiewicz, W.; Fenske, D.; Bhakdi, S.; Husmann, M. Resealing of large transmembrane pores produced by streptolysin O in nucleated cells is accompanied by NF-kappaB activation and downstream events. FASEB J. 2002, 16, 237–239. [Google Scholar] [CrossRef]
- Keefe, D.; Shi, L.; Feske, S.; Massol, R.; Navarro, F.; Kirchhausen, T.; Lieberman, J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 2005, 23, 249–262. [Google Scholar] [CrossRef]
- Pilzer, D.; Gasser, O.; Moskovich, O.; Schifferli, J.A.; Fishelson, Z. Emission of membrane vesicles: Roles in complement resistance, immunity and cancer. Springer Semin. Immun. 2005, 27, 375–387. [Google Scholar] [CrossRef]
- Andrews, N.W.; Corrotte, M.; Castro-Gomes, T. Above the fray: Surface remodeling by secreted lysosomal enzymes leads to endocytosis-mediated plasma membrane repair. Semin. Cell Dev. Biol. 2015, 45, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Atanassoff, A.P.; Wolfmeier, H.; Schoenauer, R.; Hostettler, A.; Ring, A.; Draeger, A.; Babiychuk, E.B. Microvesicle shedding and lysosomal repair fulfill divergent cellular needs during the repair of streptolysin O-induced plasmalemmal damage. PLoS ONE 2014, 9, e89743. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.M.; von Einem, J.; Frick, M.; Miklavc, P.; Mayenburg, M.; Husmann, M.; Dietl, P.; Wittekindt, O.H. Molecular basis of early epithelial response to streptococcal exotoxin: Role of STIM1 and Orai1 proteins. Cell Microbiol. 2012, 14, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.K.; O'Riordan, M.X. More than a pore: The cellular response to cholesterol-dependent cytolysins. Toxins 2013, 5, 618–636. [Google Scholar] [CrossRef] [PubMed]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef]
- Maurer, J.; Hupp, S.; Pillich, H.; Mitchell, T.J.; Chakraborty, T.; Iliev, A.I. Missing elimination via membrane vesicle shedding contributes to the diminished calcium sensitivity of listeriolysin O. Sci. Rep. 2018, 8, 15846. [Google Scholar] [CrossRef]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Freche, B.; Ho, S.; Parton, R.G.; van der Goot, F.G. Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol. 2011, 13, 1026–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reboud, E.; Bouillot, S.; Patot, S.; Beganton, B.; Attree, I.; Huber, P. Pseudomonas aeruginosa ExlA and Serratia marcescens ShlA trigger cadherin cleavage by promoting calcium influx and ADAM10 activation. PLoS Pathog. 2017, 13, e1006579. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Contreras, M.L.; Lelkes, P.I.; Lazarovici, P. Staphylococcus aureus alpha-toxin activates phospholipases and induces a Ca2+ influx in PC12 cells. Cell Signal. 1989, 1, 387–393. [Google Scholar] [CrossRef]
- Eichstaedt, S.; Gabler, K.; Below, S.; Muller, C.; Kohler, C.; Engelmann, S.; Hildebrandt, P.; Volker, U.; Hecker, M.; Hildebrandt, J.P. Effects of Staphylococcus aureus—Hemolysin A on calcium signalling in immortalized human airway epithelial cells. Cell Calcium 2009, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, X.; Yu, L.; Xu, H. Calcium signaling in membrane repair. Semin. Cell Dev. Biol. 2015, 45, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Nagahama, M.; Seike, S.; Shirai, H.; Takagishi, T.; Kobayashi, K.; Takehara, M.; Sakurai, J. Role of P2X7 receptor in Clostridium perfringens beta-toxin-mediated cellular injury. Biochim. Biophys. Acta 2015, 1850, 2159–2167. [Google Scholar] [CrossRef]
- Skals, M.; Praetorius, H.A. Mechanisms of cytolysin-induced cell damage—A role for auto- and paracrine signalling. Acta Physiol. 2013, 209, 95–113. [Google Scholar] [CrossRef]
- Schwiering, M.; Husmann, M.; Hellmann, N. P2X-receptor antagonists inhibit the interaction of S. aureus hemolysin a with membranes. Toxins 2017, 9, 332. [Google Scholar] [CrossRef]
- Von Hoven, G.; Qin, Q.; Neukirch, C.; Husmann, M.; Hellmann, N.S. aureus alpha-toxin: Small pore, large consequences. Biol. Chem. 2018. [Google Scholar] [CrossRef]
- Valeva, A.; Walev, I.; Gerber, A.; Klein, J.; Palmer, M.; Bhakdi, S. Staphylococcal alpha-toxin: Repair of a calcium-impermeable pore in the target cell membrane. Mol. Microbiol. 2000, 36, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kloft, N.; Neukirch, C.; Bobkiewicz, W.; Veerachato, G.; Busch, T.; von Hoven, G.; Boller, K.; Husmann, M. Pro-autophagic signal induction by bacterial pore-forming toxins. Med. Microbiol. Immunol. 2010, 199, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoven, G.; Kloft, N.; Neukirch, C.; Ebinger, S.; Bobkiewicz, W.; Weis, S.; Boller, K.; Janda, K.D.; Husmann, M. Modulation of translation and induction of autophagy by bacterial exoproducts. Med. Microbiol. Immunol. 2012, 201, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloft, N.; Neukirch, C.; von Hoven, G.; Bobkiewicz, W.; Weis, S.; Boller, K.; Husmann, M. A subunit of eukaryotic translation initiation factor 2alpha-phosphatase (CreP/PPP1R15B) regulates membrane traffic. J. Biol. Chem. 2012, 287, 35299–35317. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. eLife 2013, 2, e00926. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Rouaud, F.; Guerrera, D.; Vasileva, E.; Popov, L.M.; Kelley, W.L.; Rubinstein, E.; Carette, J.E.; Amieva, M.R.; Citi, S. A dock-and-lock mechanism clusters ADAM10 at cell-cell junctions to promote alpha-toxin cytotoxicity. Cell Rep. 2018, 25, 2132–2147. [Google Scholar] [CrossRef] [PubMed]
- Von Hoven, G.; Husmann, M. Staphylococcus aureus alpha-toxin’s close contacts ensure the kill. Trends Microbiol. 2019, 27, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Porta, H.; Cancino-Rodezno, A.; Soberon, M.; Bravo, A. Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides 2011, 32, 601–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloft, N.; Busch, T.; Neukirch, C.; Weis, S.; Boukhallouk, F.; Bobkiewicz, W.; Cibis, I.; Bhakdi, S.; Husmann, M. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem. Biophys. Res. Commun. 2009, 385, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; van der Goot, F.G.; Freche, B. Bacterial pore-forming toxins: The (w)hole story? Cell Mol. Life Sci. 2008, 65, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Shibutani, M.; Seike, S.; Yonezaki, M.; Takagishi, T.; Oda, M.; Kobayashi, K.; Sakurai, J. The p38 MAPK and JNK pathways protect host cells against Clostridium perfringens beta-toxin. Infect. Immun. 2013, 81, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Hayashi, S.; Morimitsu, S.; Sakurai, J. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 2003, 278, 36934–36941. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk-Balska, A.; Bielecki, J. Listeria monocytogenes listeriolysin O and phosphatidylinositol-specific phospholipase C affect adherence to epithelial cells. Can. J. Microbiol. 2005, 51, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Lucas, E.A.; Billington, S.J.; Carlson, P.; McGee, D.J.; Jost, B.H. Phospholipase D promotes Arcanobacterium haemolyticum adhesion via lipid raft remodeling and host cell death following bacterial invasion. BMC Microbiol. 2010, 10, 270. [Google Scholar] [CrossRef]
- Seitz, M.; Baums, C.G.; Neis, C.; Benga, L.; Fulde, M.; Rohde, M.; Goethe, R.; Valentin-Weigand, P. Subcytolytic effects of suilysin on interaction of Streptococcus suis with epithelial cells. Vet. Microbiol. 2013, 167, 584–591. [Google Scholar] [CrossRef]
- Vadia, S.; Arnett, E.; Haghighat, A.C.; Wilson-Kubalek, E.M.; Tweten, R.K.; Seveau, S. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog. 2011, 7, e1002356. [Google Scholar] [CrossRef]
- Gellings, P.S.; McGee, D.J. Arcanobacterium haemolyticum utilizes both phospholipase d and arcanolysin to mediate its uptake into nonphagocytic cells. Infect. Immun. 2019, 87, e00832-18. [Google Scholar] [CrossRef]
- Satchell, K.J. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 2011, 65, 71–90. [Google Scholar] [CrossRef]
- Itoh, T.; Erdmann, K.S.; Roux, A.; Habermann, B.; Werner, H.; De Camilli, P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev. Cell 2005, 9, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Kruchten, A.E.; McNiven, M.A. Dynamin as a mover and pincher during cell migration and invasion. J. Cell Sci. 2006, 119, 1683–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.G.T.; Vadia, S.; Pathak-Sharma, S.; McLaughlin, E.; Zhang, X.; Swanson, J.; Seveau, S. Host cell perforation by listeriolysin O (LLO) activates a Ca(2+)-dependent cPKC/Rac1/Arp2/3 signaling pathway that promotes Listeria monocytogenes internalization independently of membrane resealing. Mol. Biol. Cell 2018, 29, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Dons, L.; Eriksson, E.; Jin, Y.; Rottenberg, M.E.; Kristensson, K.; Larsen, C.N.; Bresciani, J.; Olsen, J.E. Role of flagellin and the two-component CheA/CheY system of Listeria monocytogenes in host cell invasion and virulence. Infect. Immun. 2004, 72, 3237–3244. [Google Scholar] [CrossRef] [PubMed]
- Fenchel, T. Microbial behavior in a heterogeneous world. Science 2002, 296, 1068–1071. [Google Scholar] [CrossRef]
- Stock, A.; Chen, T.; Welsh, D.; Stock, J. CheA protein, a central regulator of bacterial chemotaxis, belongs to a family of proteins that control gene expression in response to changing environmental conditions. Proc. Natl. Acad. Sci. USA 1988, 85, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.H.; Blackburn, N.; Larsen, J.L.; Olsen, J.E. Influences of temperature, salinity and starvation on the motility and chemotactic response of Vibrio anguillarum. Microbiology 2004, 150, 1283–1290. [Google Scholar] [CrossRef]
- Thompson, J.R.; Randa, M.A.; Marcelino, L.A.; Tomita-Mitchell, A.; Lim, E.; Polz, M.F. Diversity and dynamics of a north atlantic coastal Vibrio community. Appl. Environ. Microbiol. 2004, 70, 4103–4110. [Google Scholar] [CrossRef]
- Takemura, A.F.; Chien, D.M.; Polz, M.F. Associations and dynamics of Vibrionaceae in the environment, from the genus to the population level. Front. Microbiol. 2014, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Marco-Noales, E.; Biosca, E.G.; Amaro, C. Effects of salinity and temperature on long-term survival of the eel pathogen Vibrio vulnificus biotype 2 (serovar E). Appl. Environ. Microbiol. 1999, 65, 1117–1126. [Google Scholar]
- Fouz, B.; Toranzo, A.E.; Marco-Noales, E.; Amaro, C. Survival of fish-virulent strains of Photobacterium damselae subsp. damselae in seawater under starvation conditions. FEMS Microbiol. Lett. 1998, 168, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Carda-Dieguez, M.; Silva-Hernandez, F.X.; Hubbard, T.P.; Chao, M.C.; Waldor, M.K.; Amaro, C. Comprehensive identification of Vibrio vulnificus genes required for growth in human serum. Virulence 2018, 9, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo, D.; Lee, C.T.; Roig, F.J.; Lemos, M.L.; Hor, L.I.; Amaro, C. Host-nonspecific iron acquisition systems and virulence in the zoonotic serovar of Vibrio vulnificus. Infect. Immun. 2014, 82, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Ganguly, S.; Chatterjee, N.S.; Banerjee, K.K. Vibrio cholerae hemolysin: The beta-trefoil domain is required for folding to the native conformation. Biochem. Biophys. Rep. 2016, 8, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; Mukherjee, A.; Mazumdar, B.; Ghosh, A.N.; Banerjee, K.K. The beta-prism lectin domain of Vibrio cholerae hemolysin promotes self-assembly of the beta-pore-forming toxin by a carbohydrate-independent mechanism. J. Biol. Chem. 2014, 289, 4001–4008. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.K.; Jwa, N.S.; Lebrun, M.H.; Job, D.; Rakwal, R. Plant secretome: Unlocking secrets of the secreted proteins. Proteomics 2010, 10, 799–827. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Walev, I.; Weis, S.; Boukhallouk, F.; Wassenaar, T.M.; Endres, K.; Fahrenholz, F.; Bhakdi, S.; Zitzer, A. A cellular metalloproteinase activates Vibrio cholerae pro-cytolysin. J. Biol. Chem. 2004, 279, 25143–25148. [Google Scholar] [CrossRef]
- Koo, S.; Cheley, S.; Bayley, H. Redirecting pore assembly of staphylococcal alpha-hemolysin by protein engineering. ACS Cent. Sci. 2019, 5, 629–639. [Google Scholar] [CrossRef]
- Yang, N.; Wittrup, D. Engineered pore-forming proteins for the intracellular delivery of macromolecular therapeutics. In Proceedings of the 40th Annual Northeast Bioengineering Conference (NEBEC), Boston, MA, USA, 25–27 April 2014. [Google Scholar] [CrossRef]
- Tabata, A.; Ohkubo, Y.; Sakakura, E.; Tomoyasu, T.; Ohkura, K.; Nagamune, H. Investigation of a bacterial pore-forming chimera toxin for application as a novel drug-delivery system tool. Anticancer Res. 2012, 32, 2323–2329. [Google Scholar]
- Gurnev, P.A.; Nestorovich, E.M. Channel-forming bacterial toxins in biosensing and macromolecule delivery. Toxins 2014, 6, 2483–2540. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Oliver, J.D.; Alam, M.; Ali, A.; Waldor, M.K.; Qadri, F.; Martinez-Urtaza, J. Vibrio spp. infections. Nat. Rev. Dis. Primers 2018, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.J.; Harris, J.B.; Morris, J.G., Jr.; Calderwood, S.B.; Camilli, A. Cholera transmission: The host, pathogen and bacteriophage dynamic. Nat. Rev. Microbiol. 2009, 7, 693–702. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Hoven, G.; Rivas, A.J.; Husmann, M. Phobalysin: Fisheye View of Membrane Perforation, Repair, Chemotaxis and Adhesion. Toxins 2019, 11, 412. https://doi.org/10.3390/toxins11070412
von Hoven G, Rivas AJ, Husmann M. Phobalysin: Fisheye View of Membrane Perforation, Repair, Chemotaxis and Adhesion. Toxins. 2019; 11(7):412. https://doi.org/10.3390/toxins11070412
Chicago/Turabian Stylevon Hoven, Gisela, Amable J. Rivas, and Matthias Husmann. 2019. "Phobalysin: Fisheye View of Membrane Perforation, Repair, Chemotaxis and Adhesion" Toxins 11, no. 7: 412. https://doi.org/10.3390/toxins11070412
APA Stylevon Hoven, G., Rivas, A. J., & Husmann, M. (2019). Phobalysin: Fisheye View of Membrane Perforation, Repair, Chemotaxis and Adhesion. Toxins, 11(7), 412. https://doi.org/10.3390/toxins11070412