Rapid Detection of Botulinum Neurotoxins—A Review


Abstract
:1. Introduction
2. Botulism: Types and Medical Application
2.1. Human Botulism
2.2. Foodborne Botulism
2.3. Infant Botulism
2.4. Wound Botulism
2.5. Inhalation Botulism
2.6. Other Types of Intoxication
2.7. Botulism in Animals
2.8. Medical Applications of Botulinum Neurotoxin
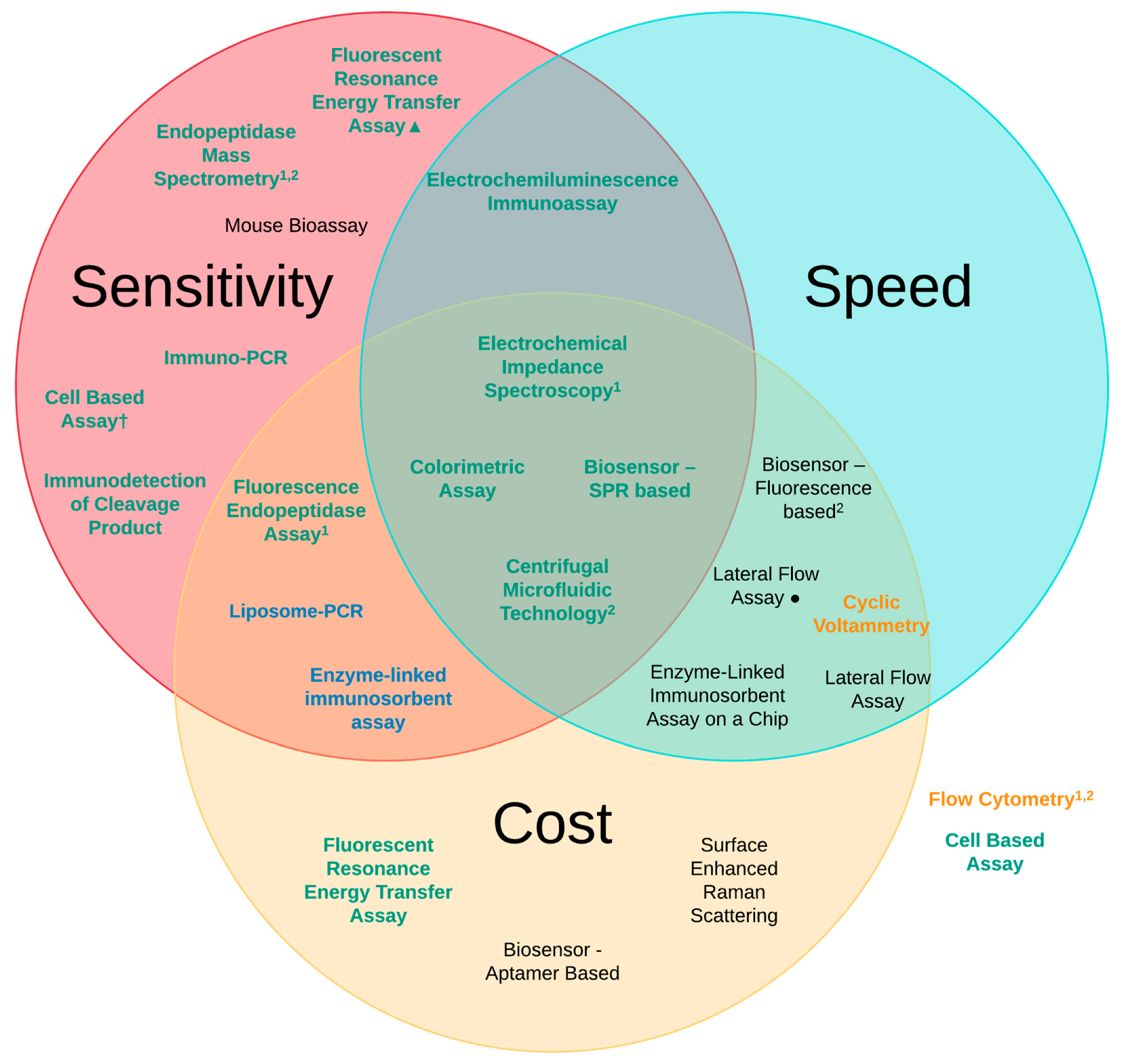
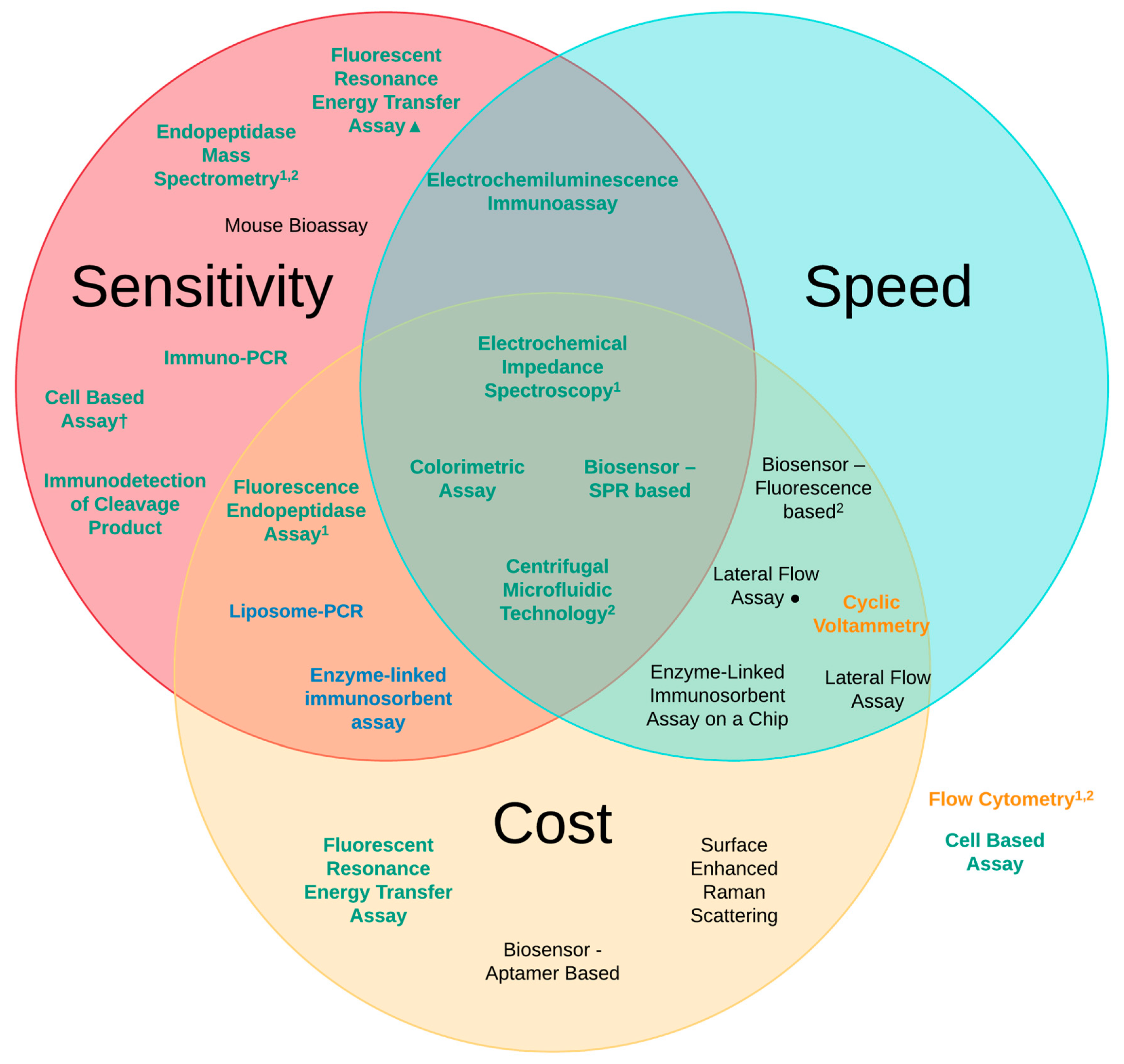
3. Methods of Detection
3.1. Mouse Bioassay
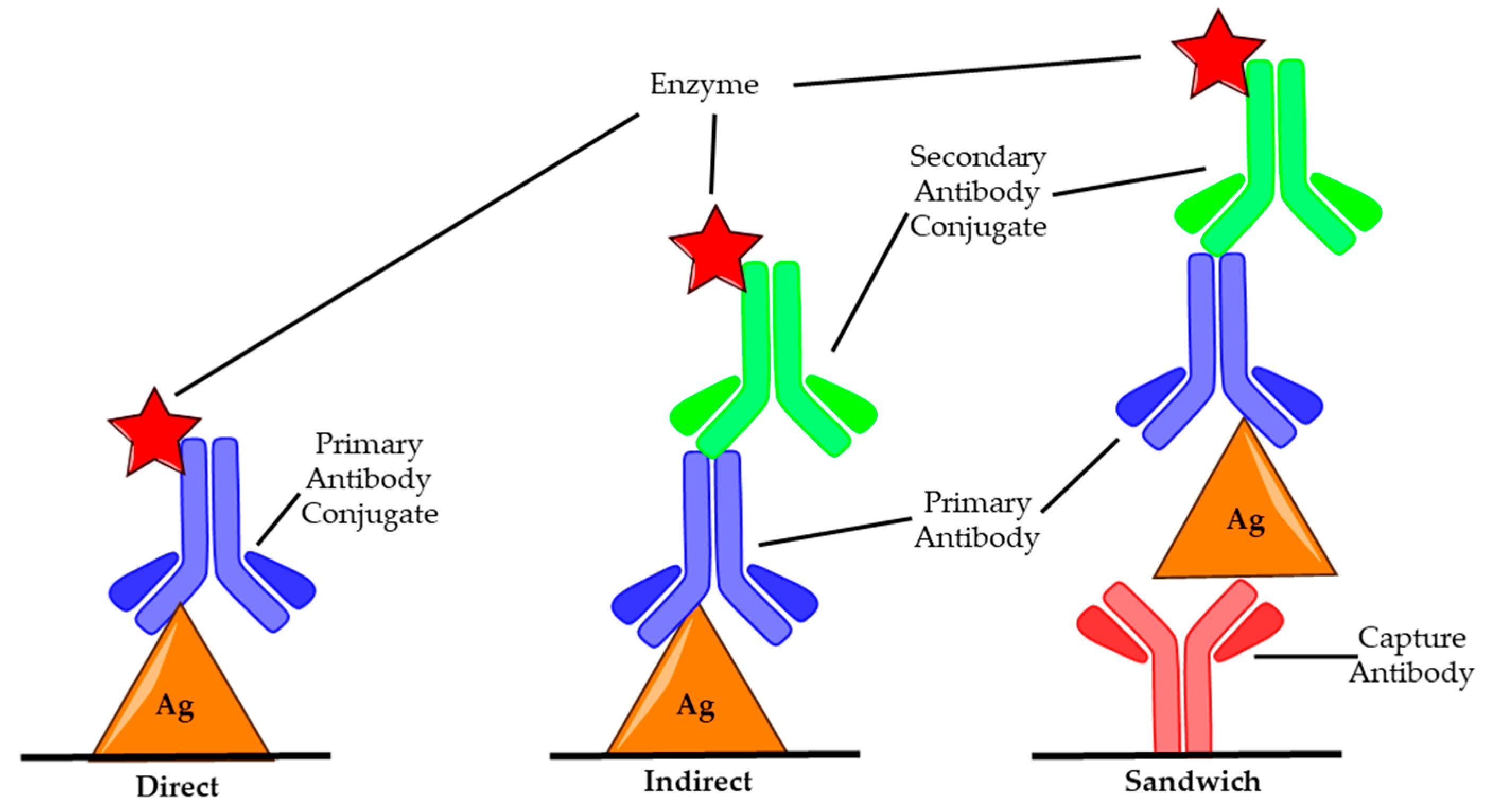
3.2. Enzyme-Linked Immunosorbent Assay
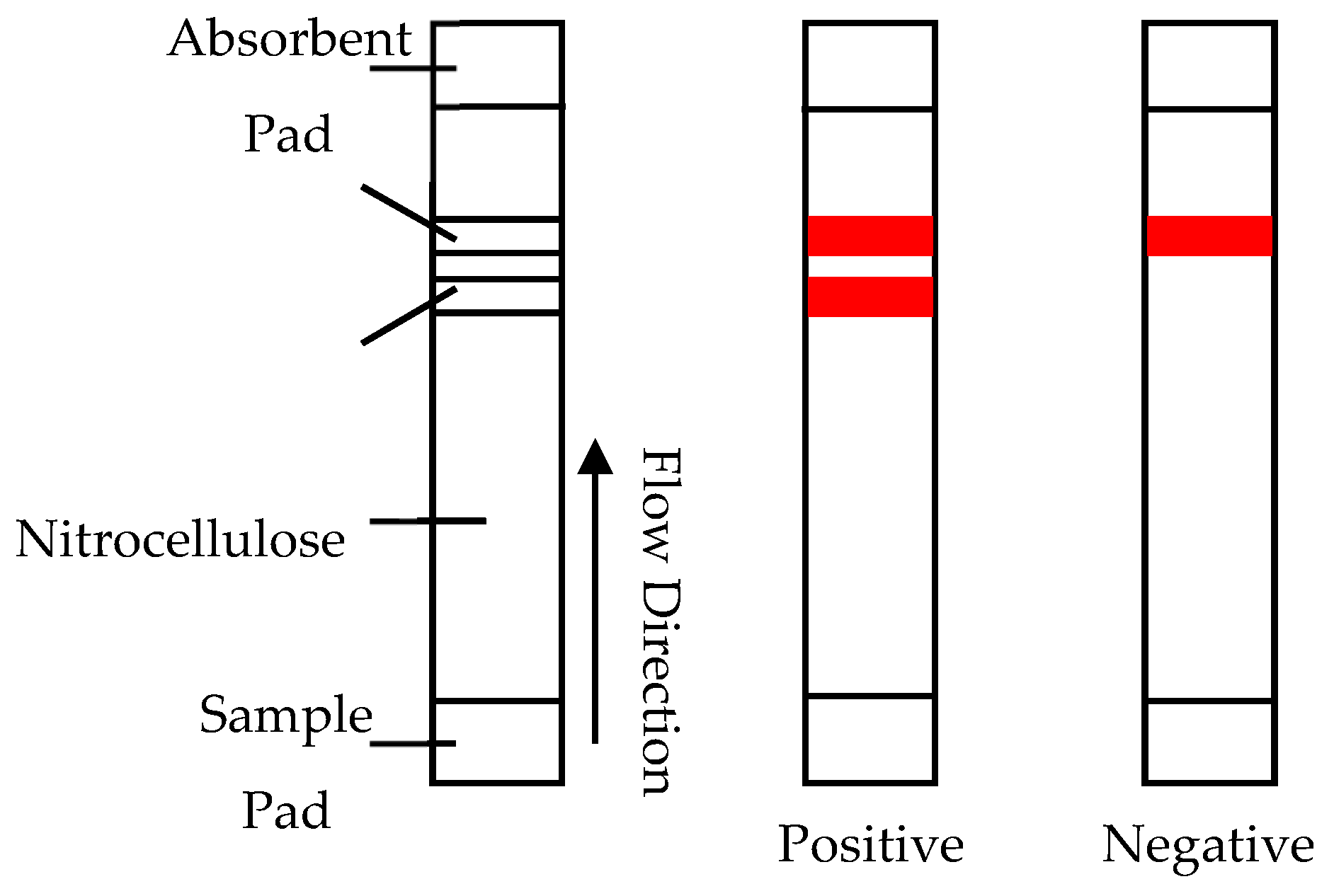
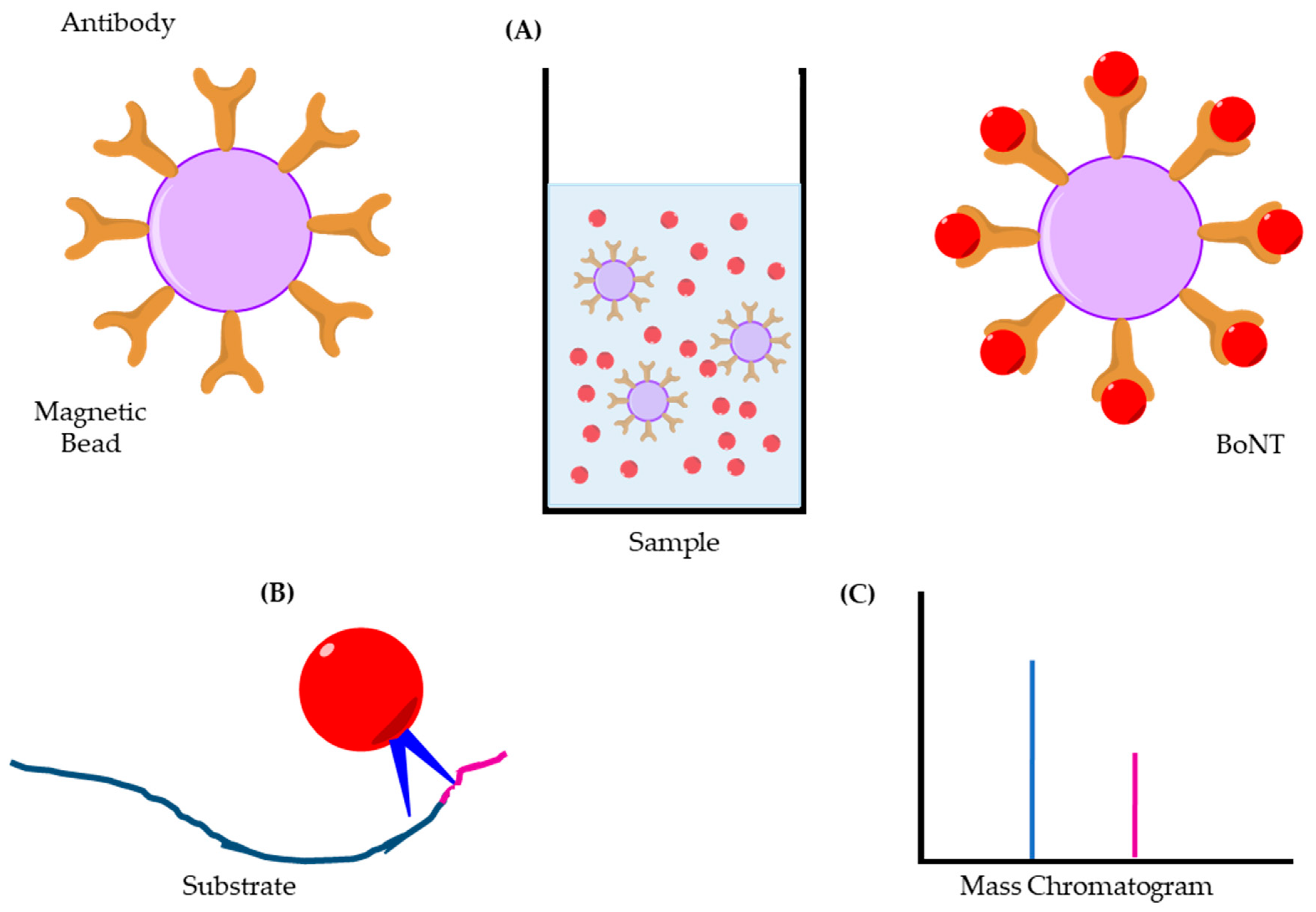
3.3. Immuno Chromatography Assays—Lateral Flow/Column Flow
3.4. Immuno-PCR/Liposome-PCR
3.5. Enzyme-Linked Immunosorbent Assay on a Chip (EOC)
3.6. Biosensors
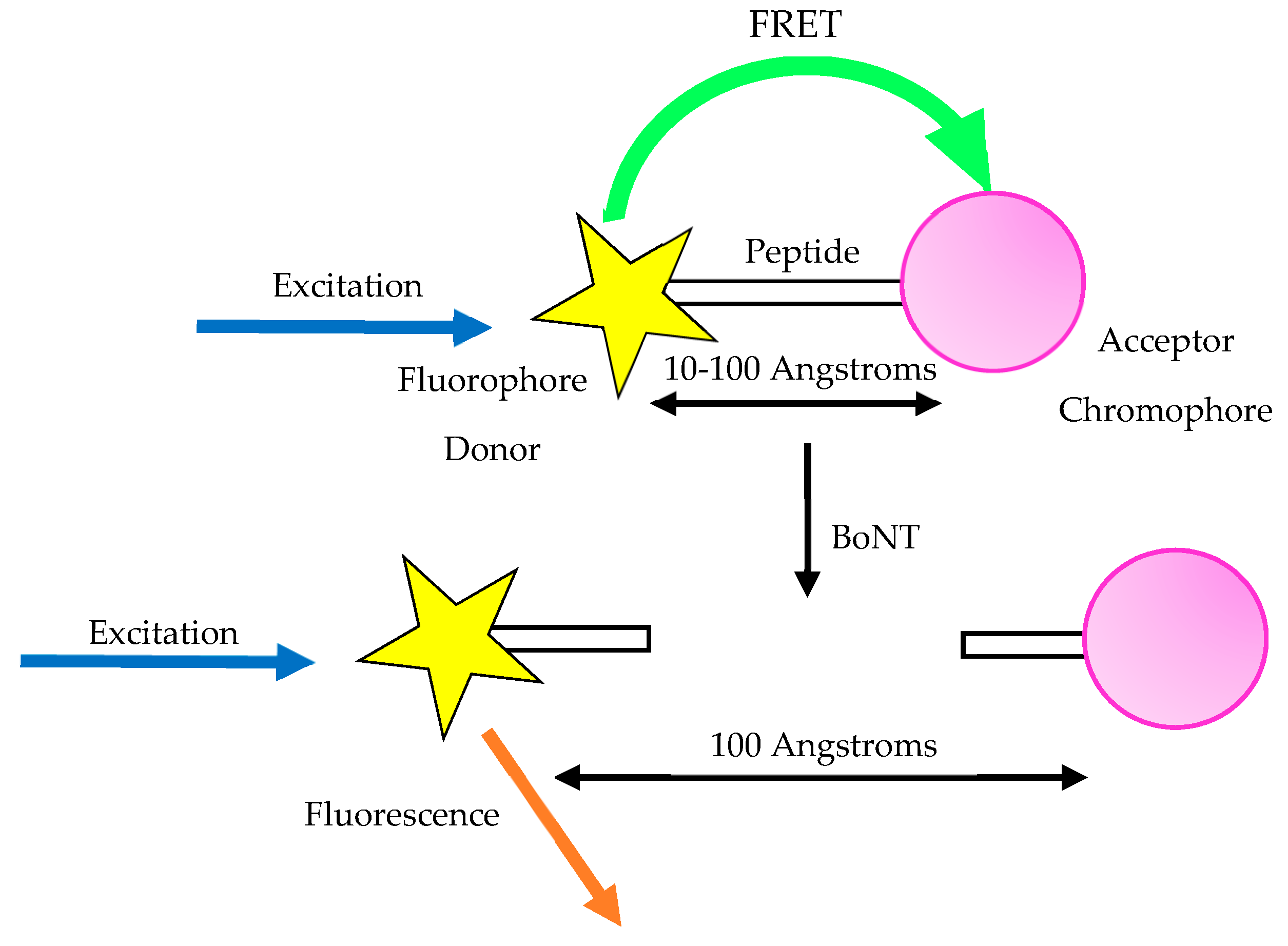
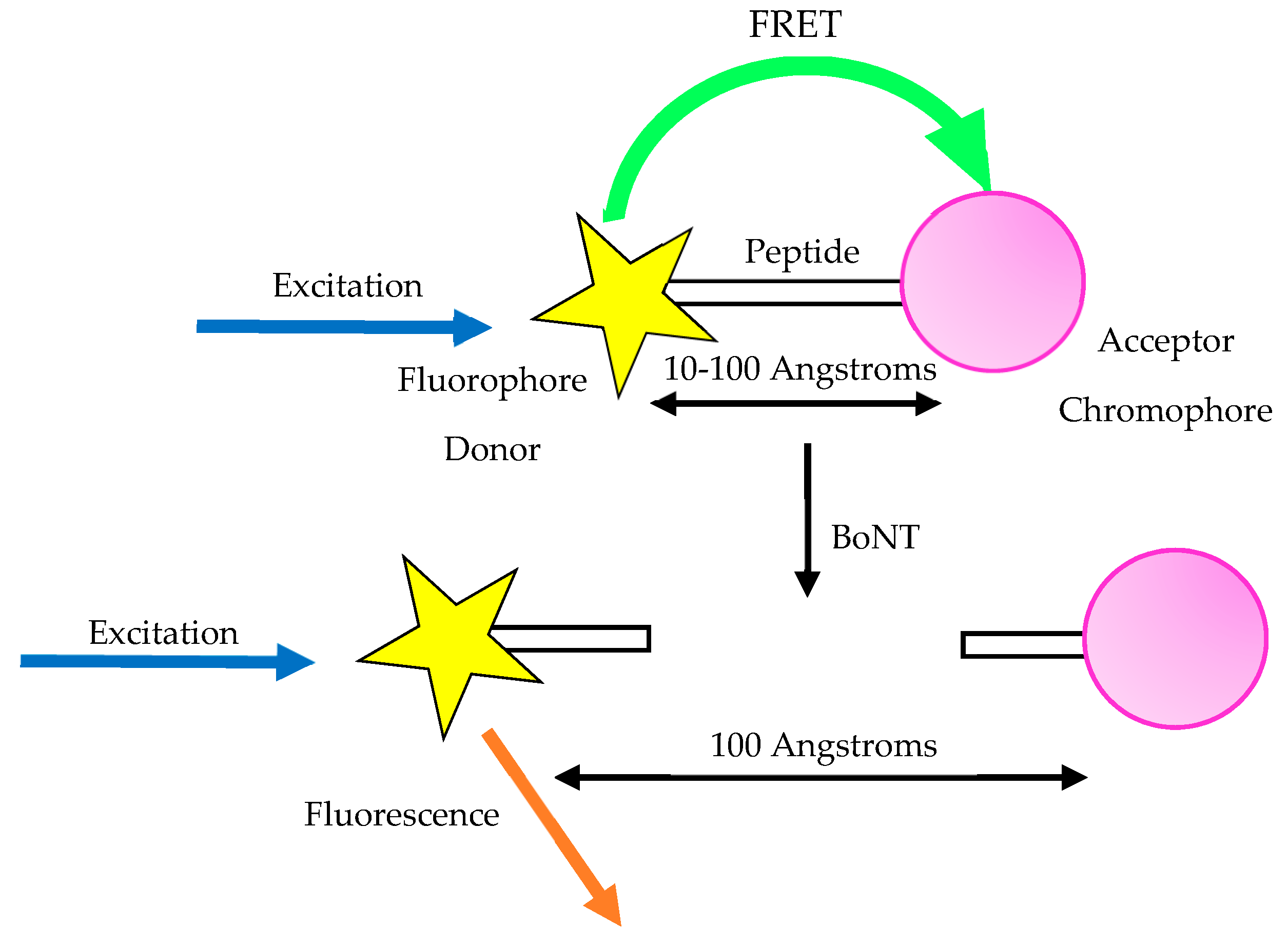
3.7. Fluorescent Resonance Energy Transfer Assay (FRET)
3.8. Flow Cytometry
3.9. Fluorescence Endopeptidase Assay
3.10. Centrifugal Microfluidic Technology
3.11. Electrochemiluminescence Immunoassay
3.12. Cyclic Voltammetry and Electrochemical Impedance Spectroscopy
3.13. Immuno-Detection of Cleavage Product
3.14. Endopeptidase Mass Spectrometry
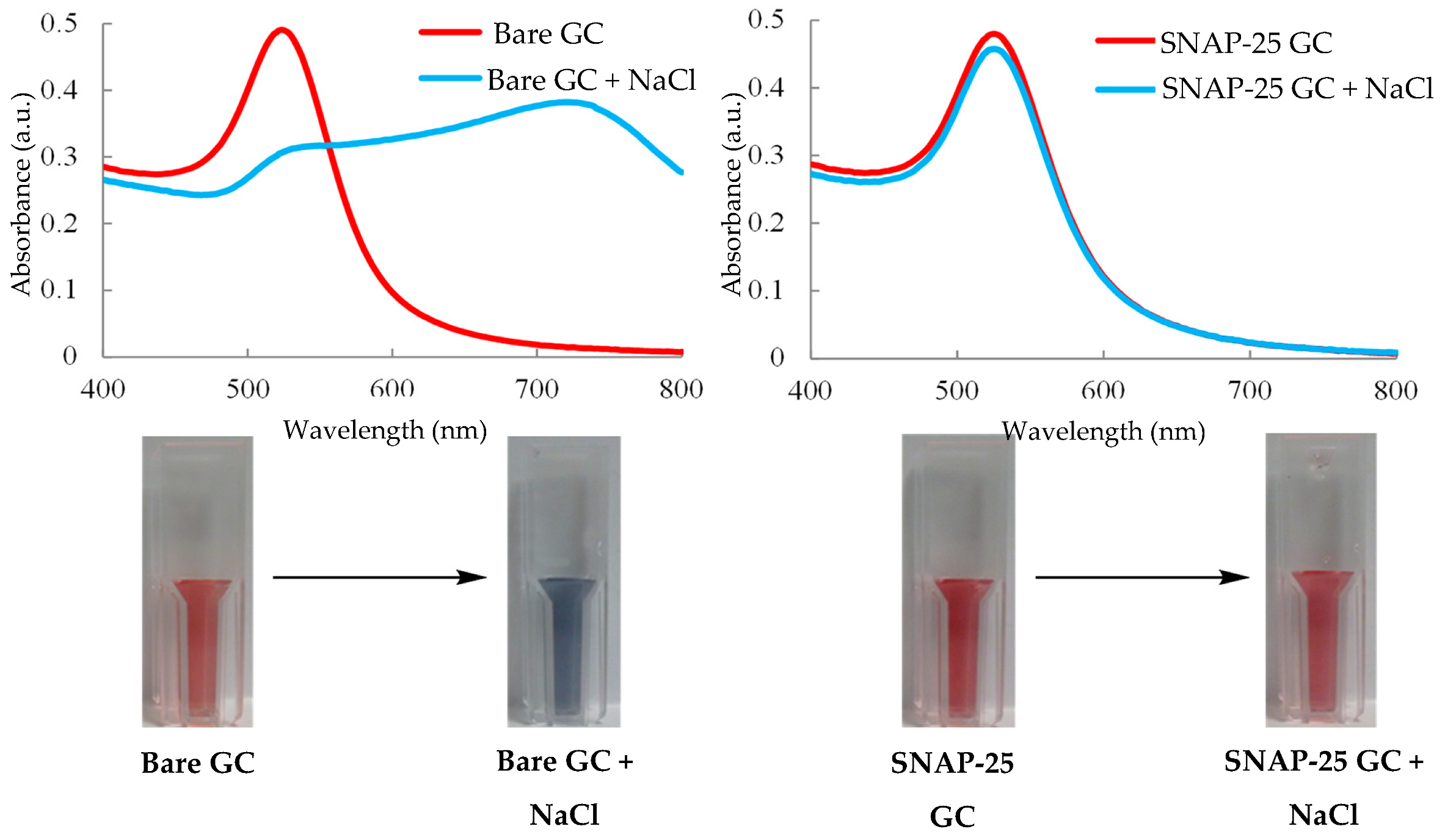
3.15. UV/Visible Spectroscopy—Colorimetric Assay
3.16. SERS Detection
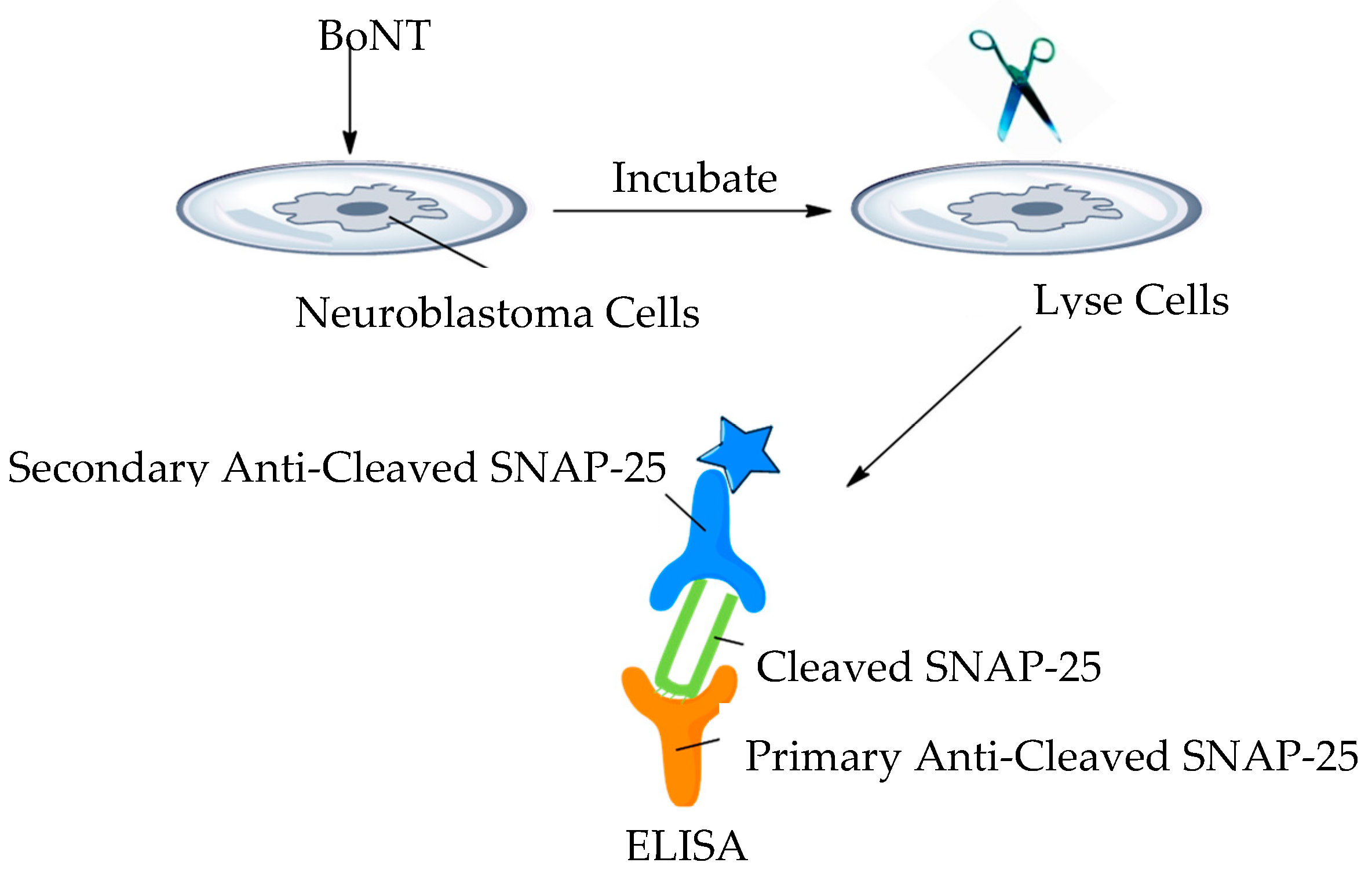
3.17. Cell-Based Assays
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Montecucco, C.; Molgó, J. Botulinal neurotoxins: revival of an old killer. Curr. Opin. Pharmacol. 2005, 5, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.-M.; Hulet, S.W.; McDonough, J.H. The effects of repeated low-dose sarin exposure. Toxicol. Appl. Pharmacol. 2006, 215, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Prigent, J.; Panigai, L.; Lamourette, P.; Sauvaire, D.; Devilliers, K.; Plaisance, M.; Volland, H.; Créminon, C.; Simon, S. Neutralising Antibodies against Ricin Toxin. PLoS ONE 2011, 6, e20166. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.R.; Hayes, B.D.; Erickson, T.B.; Boyer, E.W. Novichok agents: a historical, current, and toxicological perspective. Toxicol. Commun. 2018, 2, 45–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binz, T.; Rummel, A. Cell entry strategy of clostridial neurotoxins. J. Neurochem. 2009, 109, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Čapek, P.; Dickerson, T.J. Sensing the deadliest toxin: Technologies for botulinum neurotoxin detection. Toxins (Basel) 2010, 2, 24–53. [Google Scholar]
- Janik, E.; Ceremuga, M.; Saluk-Bijak, J.; Bijak, M. Biological Toxins as the Potential Tools for Bioterrorism. Int. J. Mol. Sci. 2019, 20, 1181. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D. Clinical applications of botulinum toxin. Curr. Opin. Microbiol. 2012, 15, 325–336. [Google Scholar] [CrossRef]
- Montal, M. Botulinum Neurotoxin: A Marvel of Protein Design. Annu. Rev. Biochem. 2010, 79, 591–617. [Google Scholar] [CrossRef] [Green Version]
- Mahrhold, S.; Rummel, A.; Bigalke, H.; Davletov, B.; Binz, T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006, 580, 2011–2014. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.; Eubanks, L.M.; Dickerson, T.J.; Janda, K.D. The Strange Case of the Botulinum Neurotoxin: Using Chemistry and Biology to Modulate the Most Deadly Poison. Angew. Chemie Int. Ed. 2008, 47, 8360–8379. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tepp, W.H.; Bradshaw, M.; Kalb, S.R.; Dykes, J.K.; Lin, G.; Nawrocki, E.M.; Pier, C.L.; Barr, J.R.; Maslanka, S.E.; et al. Purification and Characterization of Botulinum Neurotoxin FA from a Genetically Modified Clostridium botulinum Strain. mSphere 2016, 1, e00100-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslanka, S.E.; Lúquez, C.; Dykes, J.K.; Tepp, W.H.; Pier, C.L.; Pellett, S.; Raphael, B.H.; Kalb, S.R.; Barr, J.R.; Rao, A.; et al. A Novel Botulinum Neurotoxin, Previously Reported as Serotype H, Has a Hybrid-Like Structure With Regions of Similarity to the Structures of Serotypes A and F and Is Neutralized With Serotype A Antitoxin. J. Infect. Dis. 2016, 213, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Masuyer, G.; Zhang, J.; Shen, Y.; Lundin, D.; Henriksson, L.; Miyashita, S.-I.; Martínez-Carranza, M.; Dong, M.; Stenmark, P. Identification and characterization of a novel botulinum neurotoxin. Nat. Commun. 2017, 8, 14130. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Baudys, J.; Raphael, B.H.; Dykes, J.K.; Lúquez, C.; Maslanka, S.E.; Barr, J.R. Functional Characterization of Botulinum Neurotoxin Serotype H as a Hybrid of Known Serotypes F and A (BoNT F/A). Anal. Chem. 2015, 87, 3911–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Sun, S.; Wang, X.; Fan, F.; Zhou, Q.; Lu, S.; Cao, Y.; Wang, Q.-W.; Dong, M.-Q.; Yao, J.; et al. Mechanistic insights into the SNARE complex disassembly. Sci. Adv. 2019, 5, eaau8164. [Google Scholar] [CrossRef] [Green Version]
- Kukreja, R.V.; Sharma, S.; Cai, S.; Singh, B.R. Role of two active site Glu residues in the molecular action of botulinum neurotoxin endopeptidase. Biochim. Biophys. Acta - Proteins Proteomics 2007, 1774, 213–222. [Google Scholar] [CrossRef]
- Cai, S.; Singh, B.R.; Sharma, S. Botulism Diagnostics: From Clinical Symptoms to in vitro Assays. Crit. Rev. Microbiol. 2007, 33, 109–125. [Google Scholar] [CrossRef]
- Lindström, M.; Keto, R.; Markkula, A.; Nevas, M.; Hielm, S.; Korkeala, H. Multiplex PCR assay for detection and identification of Clostridium botulinum types A, B, E, and F in food and fecal material. Appl. Environ. Microbiol. 2001, 67, 5694–5699. [Google Scholar] [CrossRef]
- Sobel, J. Botulism. Clin. Infect. Dis. 2005, 41, 1167–1173. [Google Scholar] [CrossRef]
- Smith, L.A. Botulism and vaccines for its prevention. Vaccine 2009, 27, D33–D39. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.; Korkeala, H. Laboratory diagnostics of botulism. Clin. Microbiol. Rev. 2006, 19, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, N.; Johnson, E.; Pillai, S.; Hodge, D.; Stanker, L.; Wentz, T.; Singh, B.; Venkateswaran, K.; McNutt, P.; Adler, M.; et al. Botulinum Neurotoxin Detection Methods for Public Health Response and Surveillance. Front. Bioeng. Biotechnol. 2018, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.W. Clostridium botulinum and the safety of minimally heated, chilled foods: An emerging issue? J. Appl. Microbiol. 2006, 101, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Rosow, L.K.; Strober, J.B. Infant Botulism: Review and Clinical Update. Pediatr. Neurol. 2015, 52, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, D.; Grant, K.A.; McLauchlin, J. Improvement in laboratory diagnosis of wound botulism and tetanus among injecting illicit-drug users by use of real-time PCR assays for neurotoxin gene fragments. J. Clin. Microbiol. 2005, 43, 4342–4348. [Google Scholar] [CrossRef] [PubMed]
- Holzer, E. Botulism caused by inhalation. Med. Klin. 1962, 57, 1735–1738. [Google Scholar]
- Dembek, Z.F.; Smith, L.A.; Rusnak, J.M. Botulism: Cause, Effects, Diagnosis, Clinical and Laboratory Identification, and Treatment Modalities. Disaster Med. Public Health Prep. 2007, 1, 122–134. [Google Scholar] [CrossRef]
- Oie, S.; Obayashi, A.; Yamasaki, H.; Furukawa, H.; Kenri, T.; Takahashi, M.; Kawamoto, K.; Makino, S. Disinfection methods for spores of Bacillus atrophaeus, B. anthracis, Clostridium tetani, C. botulinum and C. difficile. Biol. Pharm. Bull. 2011, 34, 1325–1329. [Google Scholar] [CrossRef]
- Bakheit, A.M.; Ward, C.D.; McLellan, D.L. Generalised botulism-like syndrome after intramuscular injections of botulinum toxin type A: a report of two cases. J. Neurol. Neurosurg. Psychiatry 1997, 62, 198. [Google Scholar] [CrossRef]
- Mezaki, T.; Kaji, R.; Kohara, N.; Kimura, J. Development of general weakness in a patient with amyotrophic lateral sclerosis after focal botulinum toxin injection. Neurology 1996, 46, 845–846. [Google Scholar] [PubMed]
- Sato, Y.; Wigle, W.L.; Gallagher, S.; Johnson, A.L.; Sweeney, R.W.; Wakenell, P.S. Outbreak of Type C Botulism in Commercial Layer Chickens. Avian Dis. 2016, 60, 90–94. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Júnior, C.A.; Silva, R.O.S.; Olinda, R.G.; Lobato, F.C.F.; de Oliveira Júnior, C.A.; Silva, R.O.S.; Olinda, R.G.; Lobato, F.C.F. Botulism in non-ruminants in Brazil. Ciência Rural 2016, 46, 2158–2165. [Google Scholar] [CrossRef] [Green Version]
- Lamoureux, A.; Pouzot-Nevoret, C.; Escriou, C. A case of type B botulism in a pregnant bitch. J. Small Anim. Pract. 2015, 56, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.L.; McAdams-Gallagher, S.C.; Aceto, H. Accuracy of a Mouse Bioassay for the Diagnosis of Botulism in Horses. J. Vet. Intern. Med. 2016, 30, 1293–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef]
- Jankovic, J. Botulinum toxin: State of the art. Mov. Disord. 2017, 32, 1131–1138. [Google Scholar] [CrossRef]
- Dressler, D.; Hallett, M. Immunological aspects of Botox, Dysport and Myobloc/NeuroBloc. Eur. J. Neurol. 2006, 13, 11–15. [Google Scholar] [CrossRef]
- Rust, A.; Doran, C.; Hart, R.; Binz, T.; Stickings, P.; Sesardic, D.; Peden, A.A.; Davletov, B. A Cell Line for Detection of Botulinum Neurotoxin Type B. Front. Pharmacol. 2017, 8, 796. [Google Scholar] [CrossRef]
- Xavier, B.; Perobelli, R.; Walter, M.; da Silva, F.; Dalmora, S.; Xavier, B.; Perobelli, R.F.; Walter, M.E.; da Silva, F.S.; Dalmora, S.L. Content/Potency Assessment of Botulinum Neurotoxin Type-A by Validated Liquid Chromatography Methods and Bioassays. Toxins (Basel) 2019, 11, 35. [Google Scholar] [CrossRef]
- Singh, A.K.; Stanker, L.H.; Sharma, S.K. Botulinum neurotoxin: Where are we with detection technologies? Crit. Rev. Microbiol. 2013, 39, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Sesardic, D. Pioneering Better Science: 2013 Research Review; The National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs): London, UK, 2013; pp. 84–87. [Google Scholar]
- Stern, D.; von Berg, L.; Skiba, M.; Dorner, M.B.; Dorner, B.G. Replacing the mouse bioassay for diagnostics and potential testing of botulinum neurotoxins - progress and challenges - Open Access - Vetline. Berl. Munch. Tierarztl. Wochenschr. 2018, 1, 1–21. [Google Scholar]
- Fernández-Salas, E.; Wang, J.; Molina, Y.; Nelson, J.B.; Jacky, B.P.S.; Aoki, K.R. Botulinum Neurotoxin Serotype a Specific Cell-Based Potency Assay to Replace the Mouse Bioassay. PLoS ONE 2012, 7, e49516. [Google Scholar] [CrossRef] [PubMed]
- Merz Pharma GmbH, Landmark Change for Botulinum Neurotoxin: Alternative Test Method Approved in the U.S. Available online: https://www.merz.com/Ablog/news/botulinum-neurotoxin/ (accessed on 27 June 2018).
- Taylor, K.; Gericke, C.; Alvarez, L.R. Botulinum toxin testing on animals is still a Europe-wide issue. ALTEX 2019, 36, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.W.; Onisko, B.; Johnson, E.A.; Reader, J.R.; Griffey, S.M.; Larson, A.E.; Tepp, W.H.; Stanker, L.H.; Brandon, D.L.; Carter, J.M. Effects of purification on the bioavailability of botulinum neurotoxin type A. Toxicology 2008, 249, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Soloman, H.M.; Lilly, T., Jr. Bacteriological Analytical Manual: Chapter 17 - Clostridium Botulinum; Jackson, G.J., Merker, R.I., Bandler, R., Eds.; 8th ed.; U.S. Food and Drug Administration, Center for Food Safety and Applied Nutrition: College Park, MD, USA, 2001.
- Rasooly, R.; Do, P.M. Development of an in vitro activity assay as an alternative to the mouse bioassay for Clostridium botulinum neurotoxin type A. Appl. Environ. Microbiol. 2008, 74, 4309–4313. [Google Scholar] [CrossRef]
- Pellett, S.; Tepp, W.H.; Toth, S.I.; Johnson, E.A. Comparison of the primary rat spinal cord cell (RSC) assay and the mouse bioassay for botulinum neurotoxin type A potency determination. J. Pharmacol. Toxicol. Methods 2010, 61, 304–310. [Google Scholar] [CrossRef]
- Asensio, L.; González, I.; García, T.; Martín, R. Determination of food authenticity by enzyme-linked immunosorbent assay (ELISA). Food Control 2008, 19, 1–8. [Google Scholar] [CrossRef]
- Ng, A.H.C.; Uddayasankar, U.; Wheeler, A.R. Immunoassays in microfluidic systems. Anal. Bioanal. Chem. 2010, 397, 991–1007. [Google Scholar] [CrossRef]
- Krska, R.; Becalski, A.; Braekevelt, E.; Koerner, T.; Cao, X.-L.; Dabeka, R.; Godefroy, S.; Lau, B.; Moisey, J.; Rawn, D.F.K.; et al. Challenges and trends in the determination of selected chemical contaminants and allergens in food. Anal. Bioanal. Chem. 2012, 402, 139–162. [Google Scholar] [CrossRef]
- Eteshola, E.; Leckband, D. Development and characterization of an ELISA assay in PDMS microfluidic channels. Sensors Actuators B Chem. 2001, 72, 129–133. [Google Scholar] [CrossRef]
- Zeng, X.; Shen, Z.; Mernaugh, R. Recombinant antibodies and their use in biosensors. Anal. Bioanal. Chem. 2012, 402, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Burry, R.W. Controls for Immunocytochemistry. J. Histochem. Cytochem. 2011, 59, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Meulenberg, E.P. Immunochemical Methods for Ochratoxin A Detection: A Review. Toxins (Basel) 2012, 4, 244–266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Ferreira, J.L.; Eblen, B.S.; Whiting, R.C. Detection of Type A, B, E, and F Clostridium botulinum Neurotoxins in Foods by Using an Amplified Enzyme-Linked Immunosorbent Assay with Digoxigenin-Labeled Antibodies. Appl. Environ. Microbiol. 2006, 72, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Szílagyi, M.; Rivera, V.R.; Neal, D.; Merrill, G.A.; Poli, M.A. Development of sensitive colorimetric capture elisas for Clostridium botulinum neurotoxin serotypes A and B. Toxicon 2000, 38, 381–389. [Google Scholar] [CrossRef]
- Stanker, L.H.; Merrill, P.; Scotcher, M.C.; Cheng, L.W. Development and partial characterization of high-affinity monoclonal antibodies for botulinum toxin type A and their use in analysis of milk by sandwich ELISA. J. Immunol. Methods 2008, 336, 1–8. [Google Scholar] [CrossRef]
- Mazuet, C.; Dano, J.; Popoff, M.R.; Créminon, C.; Volland, H. Characterization of Botulinum Neurotoxin Type A Neutralizing Monoclonal Antibodies and Influence of Their Half-Lives on Therapeutic Activity. PLoS ONE 2010, 5, e12416. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Razai, A.; Geren, I.; Lou, J.; Conrad, F.; Wen, W.-H.; Farr-Jones, S.; Smith, T.; Brown, J.; Skerry, J.; et al. A Three Monoclonal Antibody Combination Potently Neutralizes Multiple Botulinum Neurotoxin Serotype E Subtypes. Toxins (Basel) 2018, 10, 105. [Google Scholar] [CrossRef]
- Naumann, M.; Boo, L.M.; Ackerman, A.H.; Gallagher, C.J. Immunogenicity of botulinum toxins. J. Neural Transm. 2013, 120, 275–290. [Google Scholar] [CrossRef]
- Springer, C.J.; Niculescu-Duvaz, I. Chapter 8 - Gene-directed enzyme prodrug therapy. In Anticancer Drug Development; Kerr, B.C.B.J., Ed.; Academic Press: San Diego, CA, USA, 2002; pp. 137–155. ISBN 9780120726516. [Google Scholar]
- Benecke, R. Clinical relevance of botulinum toxin immunogenicity. BioDrugs 2012, 26, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Ching, K.H.; Lin, A.; McGarvey, J.A.; Stanker, L.H.; Hnasko, R. Rapid and selective detection of botulinum neurotoxin serotype-A and -B with a single immunochromatographic test strip. J. Immunol. Methods 2012, 380, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiao, D.-J.; Shyu, R.-H.; Hu, C.-S.; Chiang, H.-Y.; Tang, S.-S. Colloidal gold-based immunochromatographic assay for detection of botulinum neurotoxin type B. J. Chromatogr. B 2004, 809, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Gessler, F.; Pagel-Wieder, S.; Avondet, M.-A.; Böhnel, H. Evaluation of lateral flow assays for the detection of botulinum neurotoxin type A and their application in laboratory diagnosis of botulism. Diagn. Microbiol. Infect. Dis. 2007, 57, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.B.; Rigsby, P.; Sesardic, D.; Marks, J.D.; Jones, R.G.A. A functional dual-coated (FDC) microtiter plate method to replace the botulinum toxin LD50 test. Anal. Biochem. 2012, 425, 28–35. [Google Scholar] [CrossRef]
- Liu, J.; Gao, S.; Kang, L.; Ji, B.; Xin, W.; Kang, J.; Li, P.; Gao, J.; Wang, H.; Wang, J.; et al. An Ultrasensitive Gold Nanoparticle-based Lateral Flow Test for the Detection of Active Botulinum Neurotoxin Type A. Nanoscale Res. Lett. 2017, 12, 227. [Google Scholar] [CrossRef]
- Wu, H.C.; Huang, Y.L.; Lai, S.C.; Huang, Y.Y.; Shaio, M.F. Shaio Detection of Clostridium botulinum neurotoxin type A using immuno-PCR. Lett. Appl. Microbiol. 2001, 32, 321–325. [Google Scholar] [CrossRef]
- Chao, H.-Y.; Wang, Y.-C.; Tang, S.-S.; Liu, H.-W. A highly sensitive immuno-polymerase chain reaction assay for Clostridium botulinum neurotoxin type A. Toxicon 2004, 43, 27–34. [Google Scholar] [CrossRef]
- Mason, J.T.; Xu, L.; Sheng, Z.M.; O’Leary, T.J. A liposome-PCR assay for the ultrasensitive detection of biological toxins. Nat. Biotechnol. 2006, 24, 555–557. [Google Scholar] [CrossRef]
- Mason, J.T.; Xu, L.; Sheng, Z.; He, J.; O’Leary, T.J. Liposome polymerase chain reaction assay for the sub-attomolar detection of cholera toxin and botulinum neurotoxin type A. Nat. Protoc. 2006, 1, 2003–2011. [Google Scholar] [CrossRef]
- Han, S.-M.; Cho, J.-H.; Cho, I.-H.; Paek, E.-H.; Oh, H.-B.; Kim, B.-S.; Ryu, C.; Lee, K.; Kim, Y.-K.; Paek, S.-H. Plastic enzyme-linked immunosorbent assays (ELISA)-on-a-chip biosensor for botulinum neurotoxin A. Anal. Chim. Acta 2007, 587, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Eivazzadeh-Keihan, R.; Pashazadeh-Panahi, P.; Baradaran, B.; de la Guardia, M.; Hejazi, M.; Sohrabi, H.; Mokhtarzadeh, A.; Maleki, A. Recent progress in optical and electrochemical biosensors for sensing of Clostridium botulinum neurotoxin. TrAC Trends Anal. Chem. 2018, 103, 184–197. [Google Scholar] [CrossRef]
- Singh, B.R.; Silvia, M.A. Detection of botulinum neurotoxins using optical fiber-based biosensor. Adv. Exp. Med. Biol. 1996, 391, 499–508. [Google Scholar] [PubMed]
- Rowe-Taitt, C.A.; Golden, J.P.; Feldstein, M.J.; Cras, J.J.; Hoffman, K.E.; Ligler, F.S. Array biosensor for detection of biohazards. Biosens. Bioelectron. 2000, 14, 785–794. [Google Scholar] [CrossRef]
- Sapsford, K.E.; Taitt, C.R.; Loo, N.; Ligler, F.S. Biosensor detection of botulinum toxoid A and staphylococcal enterotoxin B in food. Appl. Environ. Microbiol. 2005, 71, 5590–5592. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Ho, C.-M. Aptamer-based electrochemical biosensor for Botulinum neurotoxin. Anal. Bioanal. Chem. 2009, 393, 1943–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.; Halevi, S.; Melman, P.; Schwartz, J.; Cai, S.; Singh, B.; Patel, K.; Halevi, S.; Melman, P.; Schwartz, J.; et al. A Novel Surface Plasmon Resonance Biosensor for the Rapid Detection of Botulinum Neurotoxins. Biosensors 2017, 7, 32. [Google Scholar] [CrossRef]
- Guo, J.; Xu, C.; Li, X.; Chen, S. A Simple, Rapid and Sensitive FRET Assay for Botulinum Neurotoxin Serotype B Detection. PLoS ONE 2014, 9, e114124. [Google Scholar] [CrossRef]
- Bagramyan, K.; Barash, J.R.; Arnon, S.S.; Kalkum, M. Attomolar Detection of Botulinum Toxin Type A in Complex Biological Matrices. PLoS ONE 2008, 3, e2041. [Google Scholar] [CrossRef]
- Duracova, M.; Klimentova, J.; Fucikova, A.; Dresler, J.; Duracova, M.; Klimentova, J.; Fucikova, A.; Dresler, J. Proteomic Methods of Detection and Quantification of Protein Toxins. Toxins (Basel) 2018, 10, 99. [Google Scholar] [CrossRef]
- Pauly, D.; Kirchner, S.; Stoermann, B.; Schreiber, T.; Kaulfuss, S.; Schade, R.; Zbinden, R.; Avondet, M.-A.; Dorner, M.B.; Dorner, B.G. Simultaneous quantification of five bacterial and plant toxins from complex matrices using a multiplexed fluorescent magnetic suspension assay. Analyst 2009, 134, 2028. [Google Scholar] [CrossRef] [PubMed]
- Ozanich, R.M.; Bruckner-Lea, C.J.; Warner, M.G.; Miller, K.; Antolick, K.C.; Marks, J.D.; Lou, J.; Grate, J.W. Rapid Multiplexed Flow Cytometric Assay for Botulinum Neurotoxin Detection Using an Automated Fluidic Microbead-Trapping Flow Cell for Enhanced Sensitivity. Anal. Chem. 2009, 81, 5783–5793. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.J.; Stafford, R.G.; Millard, C.B. High-Throughput Assays for Botulinum Neurotoxin Proteolytic Activity: Serotypes A, B, D, and F. Anal. Biochem. 2001, 296, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.L.; Tepp, W.H.; Johnson, E.A.; Beebe, D.J. Self-assembled peptide monolayers as a toxin sensing mechanism within arrayed microchannels. Anal. Chem. 2009, 81, 2760–2767. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.L.; Berthier, E.; Tepp, W.H.; Johnson, E.A.; Beebe, D.J. Bead-based microfluidic toxin sensor integrating evaporative signal amplification. Lab Chip 2008, 8, 1793. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.-Y.; Schaff, U.Y.; Piccini, M.E.; Stanker, L.H.; Cheng, L.W.; Ravichandran, E.; Singh, B.-R.; Sommer, G.J.; Singh, A.K. Centrifugal Microfluidic Platform for Ultrasensitive Detection of Botulinum Toxin. Anal. Chem. 2015, 87, 922–928. [Google Scholar] [CrossRef]
- Gross, E.M.; Durant, H.E.; Hipp, K.N.; Lai, R.Y. Electrochemiluminescence Detection in Paper-Based and Other Inexpensive Microfluidic Devices. ChemElectroChem 2017, 4, 1594–1603. [Google Scholar] [CrossRef] [Green Version]
- Gatto-Menking, D.L.; Yu, H.; Bruno, J.G.; Goode, M.T.; Miller, M.; Zulich, A.W. Sensitive detection of biotoxoids and bacterial spores using an immunomagnetic electrocheminescence sensor. Biosens. Bioelectron. 1995, 10, 501–507. [Google Scholar] [CrossRef]
- Cheng, L.W.; Stanker, L.H. Detection of Botulinum Neurotoxin Serotypes A and B Using a Chemiluminescent versus Electrochemiluminescent Immunoassay in Food and Serum. J. Agric. Food Chem. 2013, 61, 755–760. [Google Scholar] [CrossRef]
- Halliwell, J.; Savage, A.C.; Buckley, N.; Gwenin, C. Electrochemical impedance spectroscopy biosensor for detection of active botulinum neurotoxin. Sens. Bio-Sens. Res. 2014, 2, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Davis, J.J. Electrical biosensors and the label free detection of protein disease biomarkers. Chem. Soc. Rev. 2013, 42, 5944. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.M.; Ryu, M.Y.; Kim, J.H.; Cho, C.H.; Park, T.J.; Park, J.P. An electrochemical biosensor for detection of the sepsis-related biomarker procalcitonin. RSC Adv. 2017, 7, 36562–36565. [Google Scholar] [CrossRef] [Green Version]
- Bertok, T.; Lorencova, L.; Chocholova, E.; Jane, E.; Vikartovska, A.; Kasak, P.; Tkac, J. Electrochemical Impedance Spectroscopy Based Biosensors: Mechanistic Principles, Analytical Examples and Challenges towards Commercialization for Assays of Protein Cancer Biomarkers. ChemElectroChem 2019, 6, 989–1003. [Google Scholar] [CrossRef]
- Ye, W.; Guo, J.; Chen, S.; Yang, M. Nanoporous membrane based impedance sensors to detect the enzymatic activity of botulinum neurotoxin A. J. Mater. Chem. B 2013, 1, 6544. [Google Scholar] [CrossRef]
- Barreiros dos Santos, M.; Agusil, J.P.; Prieto-Simón, B.; Sporer, C.; Teixeira, V.; Samitier, J. Highly sensitive detection of pathogen Escherichia coli O157:H7 by electrochemical impedance spectroscopy. Biosens. Bioelectron. 2013, 45, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Chiriacò, M.; Parlangeli, I.; Sirsi, F.; Poltronieri, P.; Primiceri, E.; Chiriacò, M.S.; Parlangeli, I.; Sirsi, F.; Poltronieri, P.; Primiceri, E. Impedance Sensing Platform for Detection of the Food Pathogen Listeria monocytogenes. Electronics 2018, 7, 347. [Google Scholar] [CrossRef]
- Savage, A.; Buckley, N.; Halliwell, J.; Gwenin, C.; Savage, A.C.; Buckley, N.; Halliwell, J.; Gwenin, C. Botulinum Neurotoxin Serotypes Detected by Electrochemical Impedance Spectroscopy. Toxins (Basel) 2015, 7, 1544–1555. [Google Scholar] [CrossRef] [Green Version]
- Makhotkina, O.; Kilmartin, P.A. The use of cyclic voltammetry for wine analysis: Determination of polyphenols and free sulfur dioxide. Anal. Chim. Acta 2010, 668, 155–165. [Google Scholar] [CrossRef]
- Bishop, G.W.; Ahiadu, B.K.; Smith, J.L.; Patterson, J.D. Use of Redox Probes for Characterization of Layer-by-Layer Gold Nanoparticle-Modified Screen-Printed Carbon Electrodes. J. Electrochem. Soc. 2017, 164, 23–28. [Google Scholar] [CrossRef]
- Lisdat, F.; Schäfer, D. The use of electrochemical impedance spectroscopy for biosensing. Anal. Bioanal. Chem. 2008, 391, 1555–1567. [Google Scholar] [CrossRef]
- Yadirgi, G.; Stickings, P.; Rajagopal, S.; Liu, Y.; Sesardic, D. Immuno-detection of cleaved SNAP-25 from differentiated mouse embryonic stem cells provides a sensitive assay for determination of botulinum A toxin and antitoxin potency. J. Immunol. Methods 2017, 451, 90–99. [Google Scholar] [CrossRef]
- Kalb, S.R.; Smith, T.J.; Moura, H.; Hill, K.; Lou, J.; Geren, I.N.; Garcia-Rodriguez, C.; Marks, J.D.; Smith, L.A.; Pirkle, J.L.; et al. The use of Endopep-MS to detect multiple subtypes of botulinum neurotoxins A, B, E, and F. Int. J. Mass Spectrom. 2008, 278, 101–108. [Google Scholar] [CrossRef]
- Kalb, S.R.; Moura, H.; Boyer, A.E.; McWilliams, L.G.; Pirkle, J.L.; Barr, J.R. The use of Endopep–MS for the detection of botulinum toxins A, B, E, and F in serum and stool samples. Anal. Biochem. 2006, 351, 84–92. [Google Scholar] [CrossRef]
- Rosen, O.; Feldberg, L.; Yamin, T.S.; Dor, E.; Barnea, A.; Weissberg, A.; Zichel, R. Development of a multiplex Endopep-MS assay for simultaneous detection of botulinum toxins A, B and E. Sci. Rep. 2017, 7, 14859. [Google Scholar] [CrossRef]
- Kalb, S.R.; Krilich, J.C.; Dykes, J.K.; Lúquez, C.; Maslanka, S.E.; Barr, J.R. Detection of Botulinum Toxins A, B, E, and F in Foods by Endopep-MS. J. Agric. Food Chem. 2015, 63, 1133–1141. [Google Scholar] [CrossRef]
- Aldewachi, H.; Chalati, T.; Woodroofe, M.N.; Bricklebank, N.; Sharrack, B.; Gardiner, P. Gold nanoparticle-based colorimetric biosensors. Nanoscale 2018, 10, 18–33. [Google Scholar] [CrossRef]
- António, M.; Nogueira, J.; Vitorino, R.; Daniel-da-Silva, A.; António, M.; Nogueira, J.; Vitorino, R.; Daniel-da-Silva, A.L. Functionalized Gold Nanoparticles for the Detection of C-Reactive Protein. Nanomaterials 2018, 8, 200. [Google Scholar] [CrossRef]
- Zuber, A.; Purdey, M.; Schartner, E.; Forbes, C.; van der Hoek, B.; Giles, D.; Abell, A.; Monro, T.; Ebendorff-Heidepriem, H. Detection of gold nanoparticles with different sizes using absorption and fluorescence based method. Sensors Actuators B Chem. 2016, 227, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, B.; Qi, Y.; Jin, Y. Label-free aptamer-based colorimetric detection of mercury ions in aqueous media using unmodified gold nanoparticles as colorimetric probe. Anal. Bioanal. Chem. 2009, 393, 2051–2057. [Google Scholar] [CrossRef]
- Wang, G.; Sun, W. Optical Limiting of Gold Nanoparticle Aggregates Induced by Electrolytes. J. Phys. Chem. B 2006, 110, 20901–20905. [Google Scholar] [CrossRef]
- Dominguez-Medina, S.; Blankenburg, J.; Olson, J.; Landes, C.F.; Link, S. Adsorption of a Protein Monolayer via Hydrophobic Interactions Prevents Nanoparticle Aggregation under Harsh Environmental Conditions. ACS Sustain. Chem. Eng. 2013, 1, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, J.; Gwenin, C.; Halliwell, J.; Gwenin, C. A Label Free Colorimetric Assay for the Detection of Active Botulinum Neurotoxin Type A by SNAP-25 Conjugated Colloidal Gold. Toxins (Basel) 2013, 5, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chu, L.T.; Chen, T.-H. Colorimetric detection of active botulinum neurotoxin using Cu2+ mediated gold nanoparticles agglomeration. Sensors Actuators B Chem. 2016, 235, 563–567. [Google Scholar] [CrossRef]
- Lim, C.Y.; Granger, J.H.; Porter, M.D. SERS detection of Clostridium botulinum neurotoxin serotypes A and B in buffer and serum: Towards the development of a biodefense test platform. Anal. Chim. Acta X 2019, 1, 100002. [Google Scholar] [CrossRef]
- Dong, M.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Using fluorescent sensors to detect botulinum neurotoxin activity in vitro and in living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14701–14706. [Google Scholar] [CrossRef]
- Hakami, R.M.; Ruthel, G.; Stahl, A.M.; Bavari, S. Gaining ground: assays for therapeutics against botulinum neurotoxin. Trends Microbiol. 2010, 18, 164–172. [Google Scholar] [CrossRef]
- Ladd, J.; Taylor, A.D.; Homola, J.; Jiang, S. Detection of botulinum neurotoxins in buffer and honey using a surface plasmon resonance (SPR) sensor. Sens. Actuators B Chem. 2008, 130, 129–134. [Google Scholar] [CrossRef]
- Marconi, S.; Ferracci, G.; Berthomieu, M.; Kozaki, S.; Miquelis, R.; Boucraut, J.; Seagar, M.; Lévêque, C. A protein chip membrane-capture assay for botulinum neurotoxin activity. Toxicol. Appl. Pharmacol. 2008, 233, 439–446. [Google Scholar] [CrossRef]
- Lévêque, C.; Ferracci, G.; Maulet, Y.; Mazuet, C.; Popoff, M.R.; Blanchard, M.-P.; Seagar, M.; El Far, O. An optical biosensor assay for rapid dual detection of Botulinum neurotoxins A and E. Sci. Rep. 2016, 5, 17953. [Google Scholar] [CrossRef]
- Peruski, A.H.; Johnson, L.H.; Peruski, L.F. Rapid and sensitive detection of biological warfare agents using time-resolved fluorescence assays. J. Immunol. Methods 2002, 263, 35–41. [Google Scholar] [CrossRef]
- Klaubert, B.; Vujtovic-Ockenga, N.; Wermter, R.; Schad, K.; von Meyer, L. Determination of botulinum toxins after peptic sample pre-treatment by multidimensional nanoscale liquid chromatography and nano-electrospray ion-trap mass spectrometry. J. Chromatogr. B 2009, 877, 1084–1092. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Zhang, J.; Aili, D.; Liedberg, B. Time-resolved botulinum neurotoxin A activity monitored using peptide-functionalized Au nanoparticle energy transfer sensors. Chem. Sci. 2014, 5, 2651–2656. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Kang, Y.; Zhang, Z.; Cui, Z.; Zhou, Y.; Zhang, X.-E. Micellar electrokinetic chromatography and laser induced fluorescence detection of botulinum neurotoxin type A activity using a dual-labelled substrate. Int. J. Environ. Anal. Chem. 2008, 88, 947–956. [Google Scholar] [CrossRef]
- Liu, W.; Montana, V.; Chapman, E.R.; Mohideen, U.; Parpura, V. Botulinum toxin type B micromechanosensor. Proc. Natl. Acad. Sci. USA 2003, 100, 13621–13625. [Google Scholar] [CrossRef] [Green Version]
- Luka, G.S.; Nowak, E.; Kawchuk, J.; Hoorfar, M.; Najjaran, H. Portable device for the detection of colorimetric assays. R. Soc. Open Sci. 2017, 4, 171025. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Group | Serotype | Human Botulism |
|---|---|---|---|
| C. Botulinum | I | A,B,F | Yes |
| II | E,B,F | Yes | |
| III | C,D | No | |
| C. Argentinense | IV | G | No |
| C. Baratii | V | F | Yes |
| C. Butyricum | VI | E | Yes |
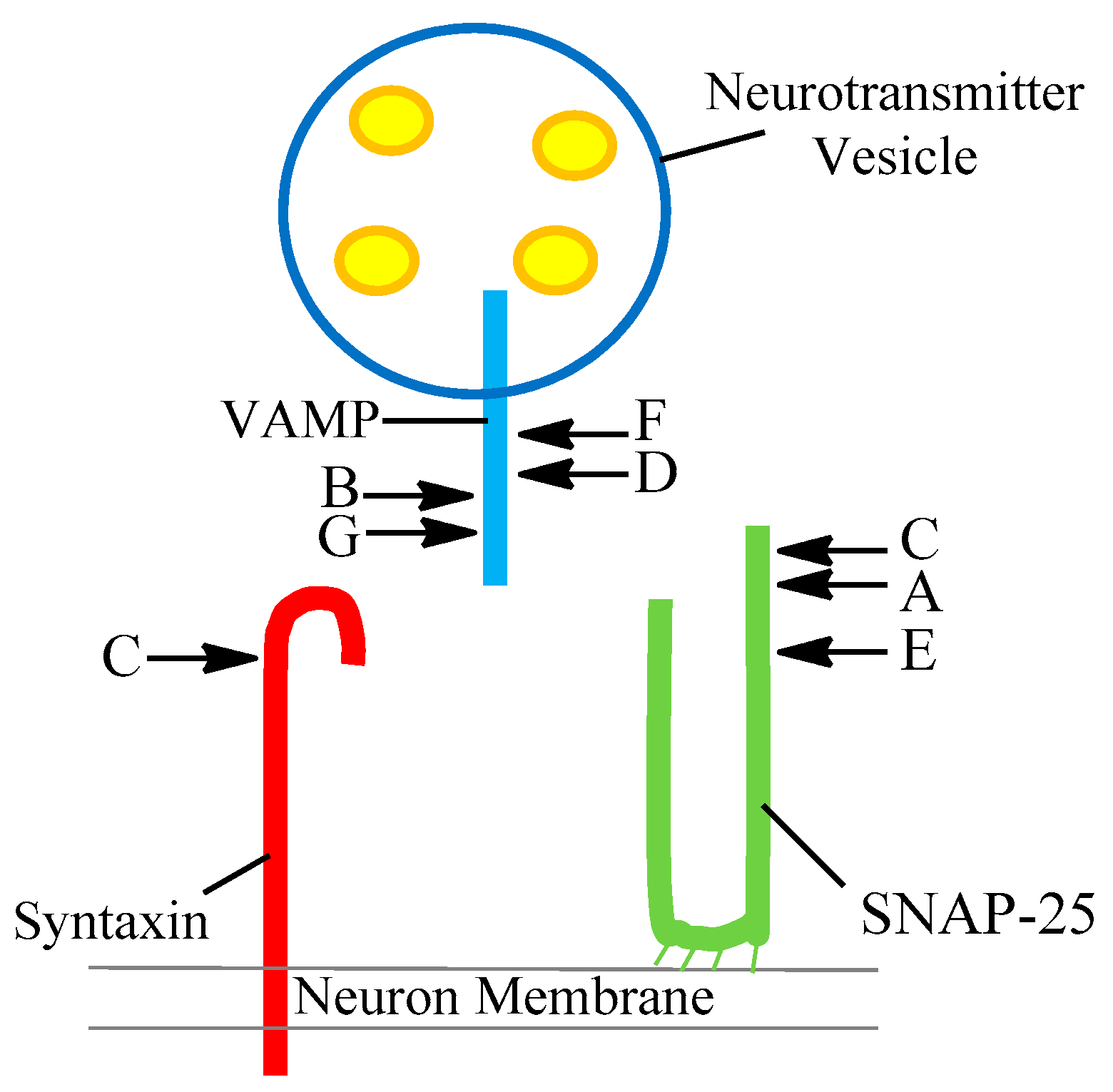
| Serotype | SNARE Protein | Cleavage Site |
|---|---|---|
| A | SNAP-25 | 197–198 |
| B | Synaptobrevin (VAMP) | 76–77 |
| C | SNAP-25 Syntaxin | 198–199 253–254 |
| D | Synaptobrevin (VAMP) | 59–60 |
| E | SNAP-25 | 180–181 |
| F | Synaptobrevin (VAMP) | 58–59 |
| G | Synaptobrevin (VAMP) | 81–82 |
| FA | Synaptobrevin (VAMP) | 54–55 |
| Detection Method | Sensitivity of Analysis (LoD) | Speed of Analysis | Estimated Cost (Per Test) |
|---|---|---|---|
| Mouse Bioassay (MBA) [6,41] | 10 pg/mL | 4–6 days | £320 |
| Enzyme-linked immunosorbent assay (ELISA) [6,41] | 2 pg/mL | 6 h | £25 |
| Lateral Flow Assay (LFA) [67,70] | 50 pg/mL 20 pg/mL● | ~10 min ~12 h | £3 £25 |
| Immuno-PCR [72] | 1 pg/mL | 6–9 h | £45 |
| Liposome-PCR [73] | 20 ag/mL | 7–9 h | £25 |
| Enzyme-Linked Immunosorbent Assay on a Chip (EOC) [75] | 2 ng/mL | ~30 min | £20 |
| Biosensor—Fluorescence-based [77] | 150 pg/mL | ~10 min | £20 |
| Biosensor—Aptamer-based [80] | 40 pg/mL | ~24 h | £12 |
| Biosensor—SPR-based [81] | 6.76 pg/mL | ~20 min | £28 |
| Fluorescent Resonance Energy Transfer Assay (FRET) [6,83] | 60 pg/mL 1 fg/mL▲ | 3 h 2 h | £11 £50 |
| Flow Cytometry [86] | 50 pg/mL | ~4 h | £40 |
| Fluorescence Endopeptidase Assay [6,88] | 3 pg/mL | 3 h | £20 |
| Centrifugal Microfluidic Technology [90] | 90 fg/mL | ~30 min | £2 |
| Electrochemiluminescence Immunoassay (ECL) [92] | 5 pg/mL | ~40 min | £50 |
| Cyclic Voltammetry (CV) [94] | 250 pg/mL | ~15 min | £20 |
| Electrochemical Impedance Spectroscopy (EIS) [101] | 25 fg/mL | ~35 min | £24 |
| Immuno detection of Cleavage Product [105] | 440 fg/mL | 6 h | £40 |
| Endopeptidase Mass Spectrometry [109] | 100 fg/mL | 4–8 h | £40 |
| Colorimetric Assay [116] | 370 fg/mL (cuvette) 600 fg/mL (96-well plate) | ~10 min ~7 min | £2 £1 |
| Surface-enhanced Ramon scattering (SERS) [118] | 84-700 pg/mL | ~23 h | £25 |
| Cell Based Assays [6,39,50,120] | 1–10 ng/mL 3 pg/mL† | 2–3 days | £35 £45 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hobbs, R.J.; Thomas, C.A.; Halliwell, J.; Gwenin, C.D. Rapid Detection of Botulinum Neurotoxins—A Review. Toxins 2019, 11, 418. https://doi.org/10.3390/toxins11070418
Hobbs RJ, Thomas CA, Halliwell J, Gwenin CD. Rapid Detection of Botulinum Neurotoxins—A Review. Toxins. 2019; 11(7):418. https://doi.org/10.3390/toxins11070418
Chicago/Turabian StyleHobbs, Robert J., Carol A. Thomas, Jennifer Halliwell, and Christopher D. Gwenin. 2019. "Rapid Detection of Botulinum Neurotoxins—A Review" Toxins 11, no. 7: 418. https://doi.org/10.3390/toxins11070418
APA StyleHobbs, R. J., Thomas, C. A., Halliwell, J., & Gwenin, C. D. (2019). Rapid Detection of Botulinum Neurotoxins—A Review. Toxins, 11(7), 418. https://doi.org/10.3390/toxins11070418