New Insights into the Type II Toxins from the Sea Anemone Heteractis crispa


 , ,
, ,  ,
,  , and
, and
Abstract
1. Introduction
2. Results
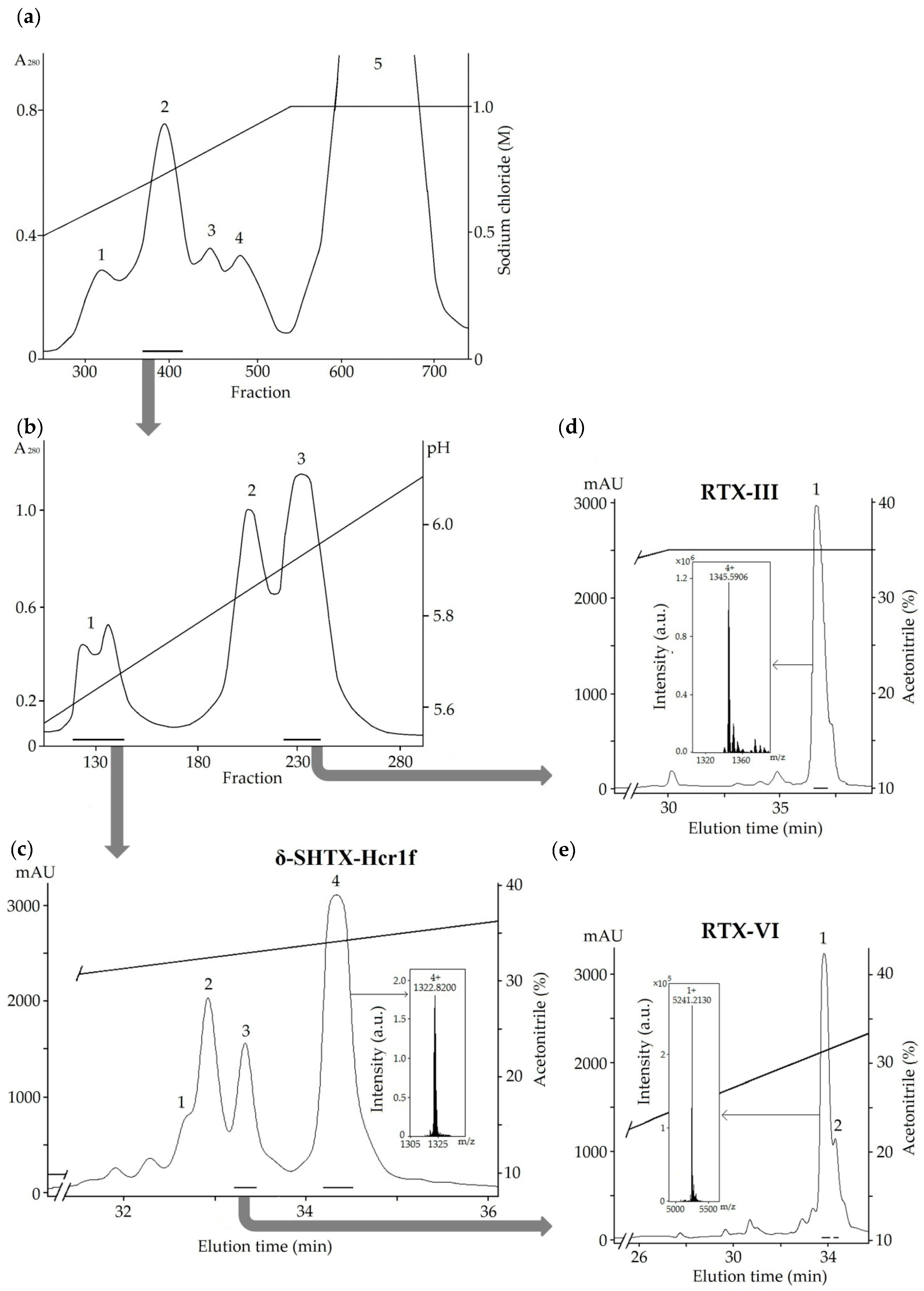
2.1. Isolation of the Toxins
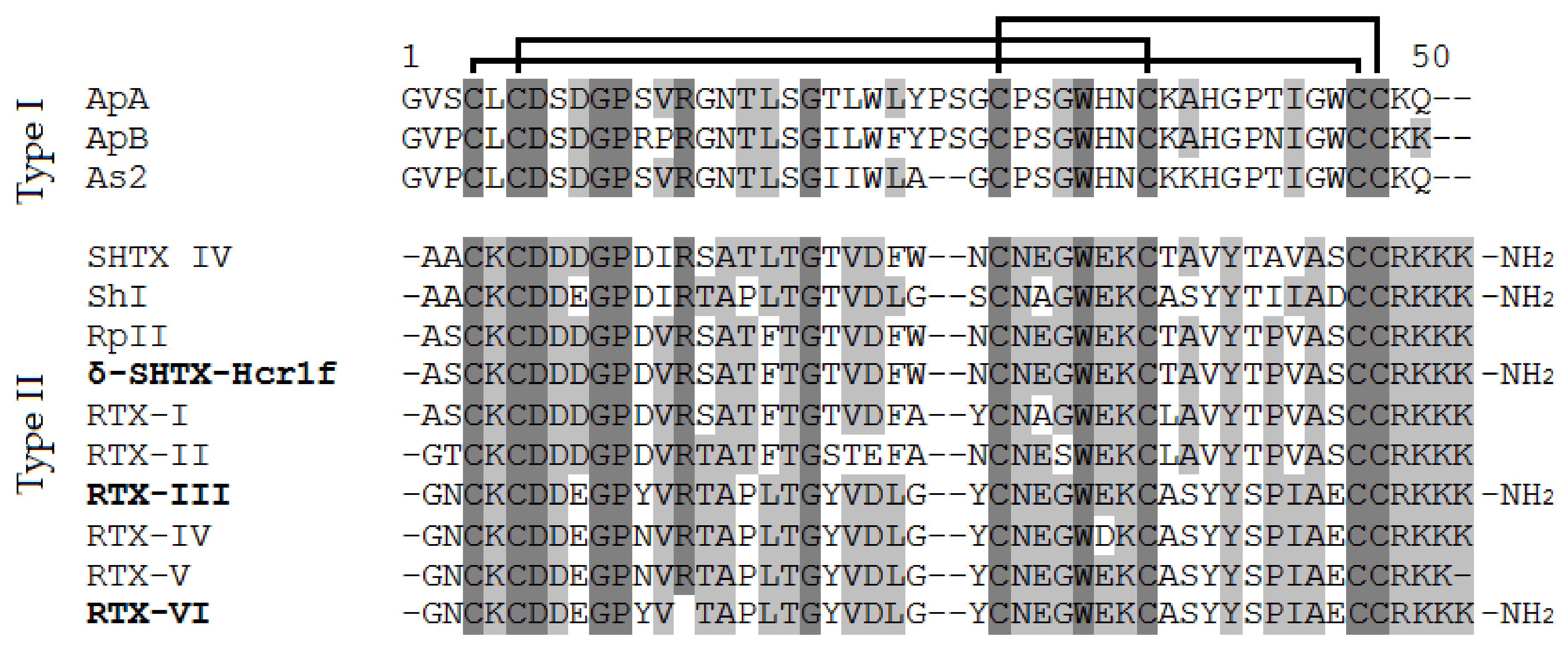
2.2. Structure Determination
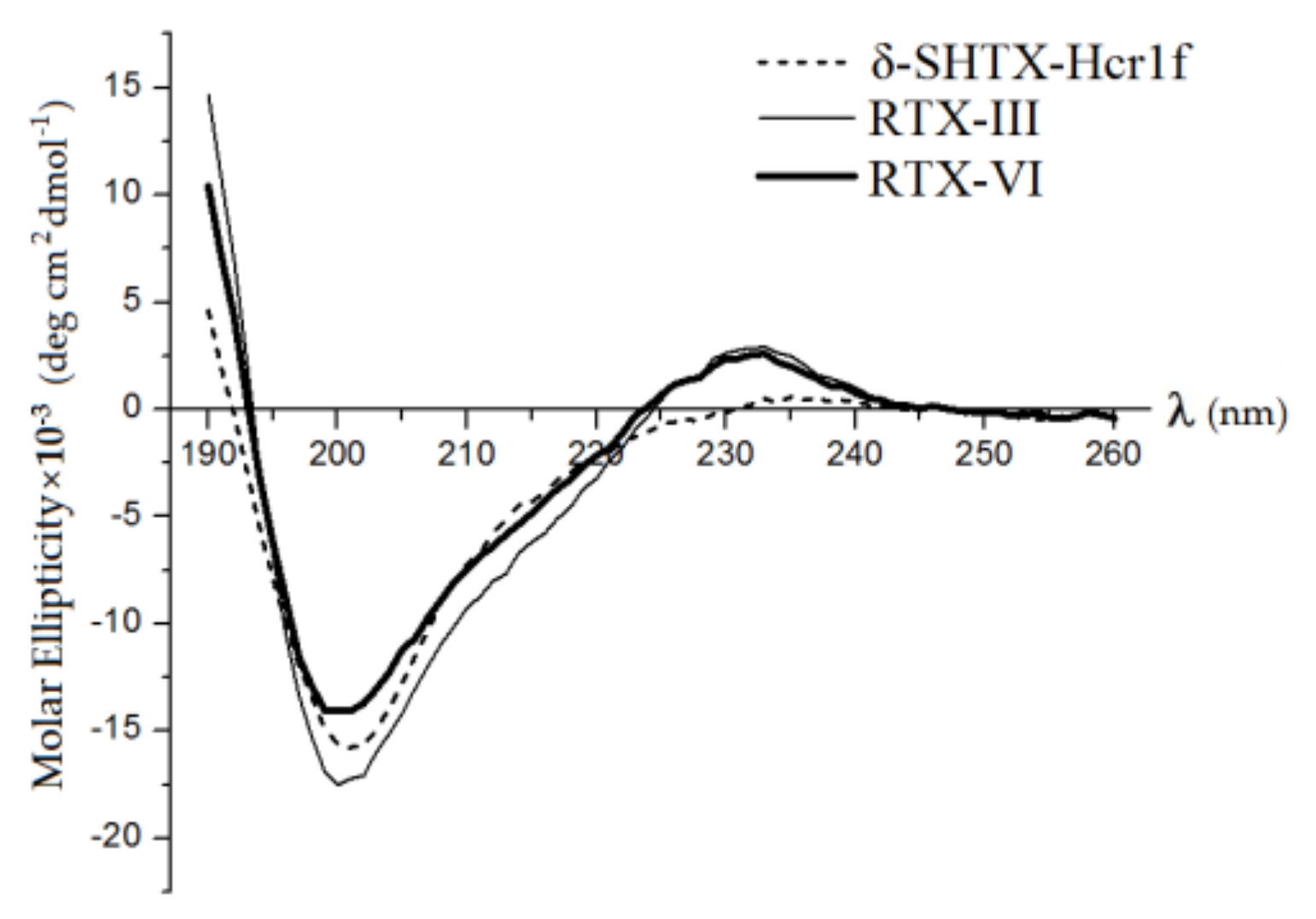
2.3. Secondary Structures of Heteractis Toxins
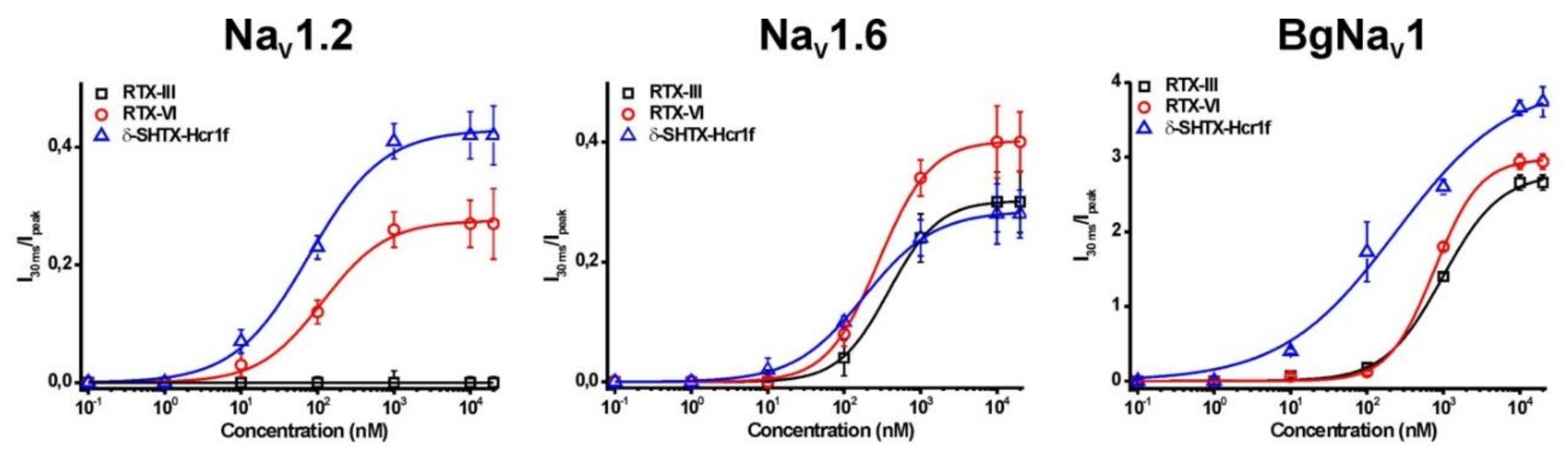
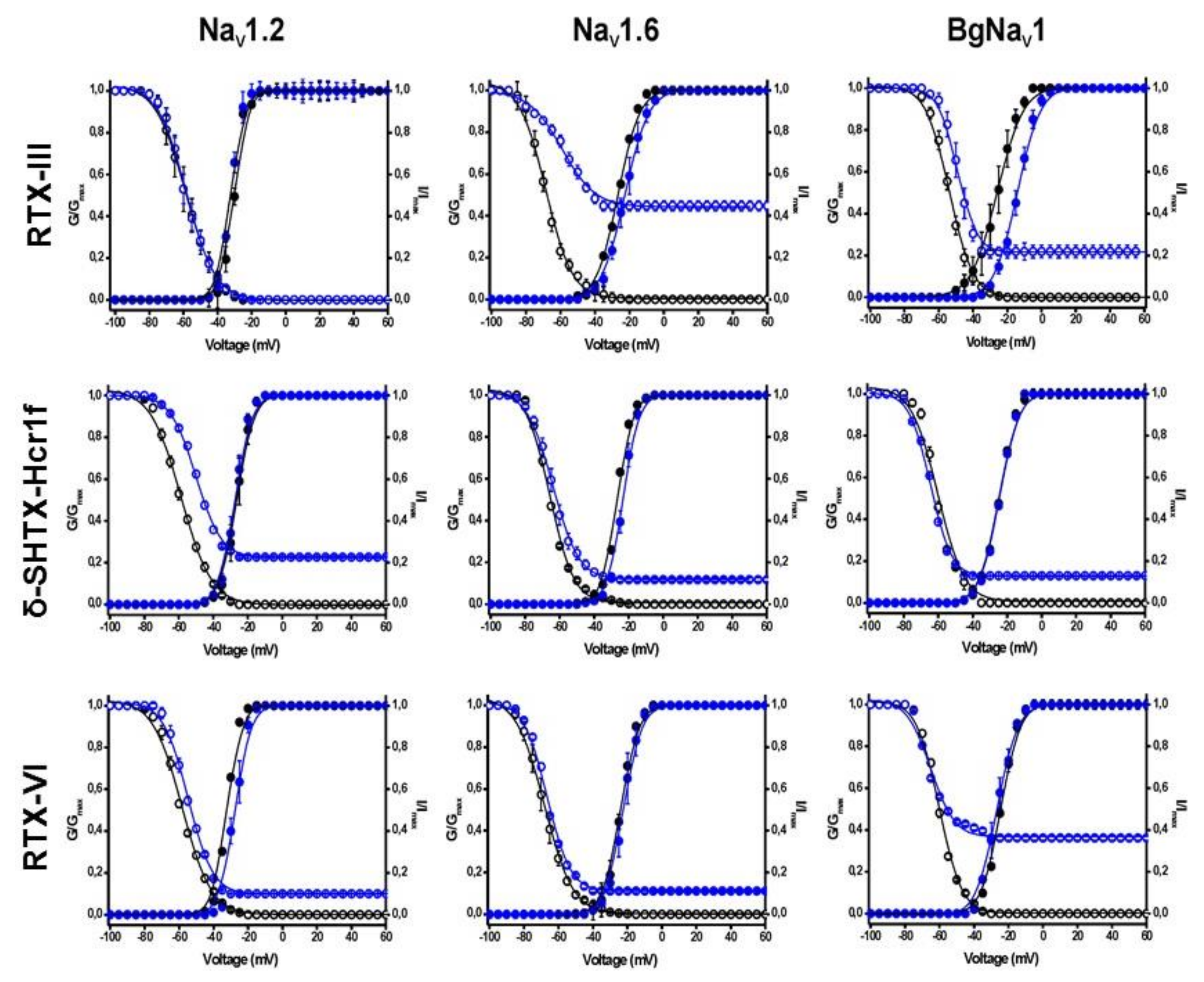
2.4. Electrophysiological Effects on NaV Channels
2.5. Toxicity of Heteractis Toxins
2.6. Homology Models of Heteractis Toxins
2.7. Molecular Modeling of δ-SHTX-Hcr1f Interaction with rNaV1.2
3. Discussion
4. Materials and Methods
4.1. Extraction and Chromatographic Procedure
4.2. Mass Spectrometric Analysis
4.3. Tandem Mass Spectrometry (MS/MS)
4.4. Reduction and Alkylation of Disulfide Bridges
4.5. Sequence Determination
4.6. Circular Dichroism Spectra
4.7. Toxicity
4.8. Expression of Voltage-Gated Ion Channels in Xenopus laevis Oocytes
4.9. Electrophysiological Recordings
4.10. Molecular Modelling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Ou, S.; Wang, Y. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Gillet, L.; Le Guennec, J.Y.; Besson, P. Voltage-gated sodium channels and cancer: Is excitability their primary role? Front. Pharmacol. 2015, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Gamal El-Din, T.M.; Lenaeus, M.J.; Catterall, W.A. Structural and functional analysis of sodium channels viewed from an evolutionary perspective. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 246, pp. 53–72. [Google Scholar] [CrossRef]
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 2017, 127, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Pavasovic, A.; Norton, R.S. Sea anemones: Quiet achievers in the field of peptide toxins. Toxins (Basel) 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Daly, M. Functional and genetic diversity of toxins in sea anemones. In Evolution of Venomous Animals and Their Toxins; Gopalakrishnakone, P., Malhotra, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 87–104. [Google Scholar] [CrossRef]
- Moran, Y.; Weinberger, H.; Sullivan, J.C.; Reitzel, A.M.; Finnerty, J.R.; Gurevitz, M. Concerted evolution of sea anemone neurotoxin genes is revealed through analysis of the Nematostella vectensis genome. Mol. Biol. Evol. 2008, 25, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Schweitz, H.; Bidard, J.N.; Frelin, C.; Pauron, D.; Vijverberg, H.P.M.; Mahasneh, D.M.; Lazdunski, M.; Vilbois, F.; Tsugita, A. Purification, sequence, and pharmacological properties of sea anemone toxins from Radianthus paumotensis. A new class of sea anemone toxins acting on the sodium channel. Biochemistry 1985, 24, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Shiomi, K. Peptide toxins in sea anemones: Structural and functional aspects. Mar. Biotechnol. 2006, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.A.; Vinokurov, L.M.; Kozlovskaya, E.P.; Elyakov, G.B. Amino-acid sequence of neurotoxin III from the sea anemone Radianthus macrodactylus. Bioorg. Khim. 1985, 11, 302–310. [Google Scholar] [CrossRef]
- Groome, J.; Lehmann-Horn, F.; Holzherr, B. Open- and closed-state fast inactivation in sodium channels. Channels 2011, 5, 65–78. [Google Scholar] [CrossRef]
- Benzinger, R.G.; Tonkovich, G.S.; Hanck, D.A. Augmentation of recovery from inactivation by site-3 Na channel toxins. J. Gen. Physiol. 1999, 113, 333–346. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaller, C.; Schulze, C.; Sanchez-Rodriguez, J.; Dannmeier, C.; Ravens, U.; Heubach, J.F.; Eckhardt, K.; Schmidtmayer, J.; Schmidt, H.; et al. Isolation and characterisation of five neurotoxic and cardiotoxic polypeptides from the sea anemone Anthopleura elegantissima. Toxicon 2001, 39, 693–702. [Google Scholar] [CrossRef]
- Sachkova, M.Y.; Singer, S.A.; Macrander, J.; Reitzel, A.M.; Peigneur, S.; Tytgat, J.; Moran, Y. The birth and death of toxins with distinct functions: A case study in the sea anemone Nematostella. Mol. Biol. Evol. 2019, 36, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.L.; Shafee, T.; Papenfuss, A.T.; Norton, R.S. Evolution of cnidarian trans-defensins: Sequence, structure and exploration of chemical space. Proteins 2019, 87, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.A.; Kozlovskaia, E.P. Amino acid sequence of a neurotoxin from the anemone Radianthus macrodactylus. Bioorg. Khim. 1989, 15, 1301–1306. [Google Scholar] [PubMed]
- Zykova, T.A.; Kozlovskaia, E.P.; Eliakov, G.B. Amino acid sequence of neurotoxin II from the sea anemone Radianthus macrodactylus. Bioorg. Khim. 1988, 14, 878–882. [Google Scholar] [PubMed]
- Zykova, T.A.; Kozlovskaia, E.P.; Eliakov, G.B. Amino acid sequence of neurotoxins IV and V from the sea anemone Radianthus macrodactylus. Bioorg. Khim. 1988, 14, 1489–1494. [Google Scholar] [PubMed]
- Renaud, J.F.; Fosset, M.; Schweitz, H.; Lazdunski, M. The interaction of polypeptide neurotoxins with tetrodotoxin-resistant Na+ channels in mammalian cardiac cells. Correlation with inotropic and arrhythmic effects. Eur. J. Pharmacol. 1986, 120, 161–170. [Google Scholar] [CrossRef]
- Kalina, R.; Gladkikh, I.; Dmitrenok, P.; Chernikov, O.; Koshelev, S.; Kvetkina, A.; Kozlov, S.; Kozlovskaya, E.; Monastyrnaya, M. New APETx-like peptides from sea anemone Heteractis crispa modulate ASIC1a channels. Peptides 2018, 104, 41–49. [Google Scholar] [CrossRef]
- Wemmer, D.E.; Kumar, N.V.; Metrione, R.M.; Lazdunski, M.; Drobny, G.; Kallenbachs, N.R. NMR analysis and sequence of toxin II from the sea anemone Radianthus paumotensis. Biochemistry 1986, 25, 6842–6849. [Google Scholar] [CrossRef]
- Monks, S.A.; Gould, A.R.; Lumley, P.E.; Alewood, P.F.; Kem, W.R.; Goss, N.H.; Norton, R.S. Limited proteolysis study of structure-function relationships in Sh I, a polypeptide neurotoxin from a sea anemone. Biochim. Biophys. Acta 1994, 1207, 93–101. [Google Scholar] [CrossRef]
- Honma, T.; Kawahata, S.; Ishida, M.; Nagai, H.; Nagashima, Y.; Shiomi, K. Novel peptide toxins from the sea anemone Stichodactyla haddoni. Peptides 2008, 29, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Mahnir, V.M.; Kozlovskaya, E.P. Structure-toxicity relationships of neurotoxin RTX-III from the sea anemone Radianthus macrodactylus: Modification of amino groups. Toxicon 1991, 29, 819–826. [Google Scholar] [CrossRef]
- Seibert, A.L.; Liu, J.; Hanck, D.A.; Blumenthal, K.M. Role of Asn-16 and Ser-19 in Anthopleurin B binding. implications for the electrostatic nature of VaV site 3. Biochemistry 2004, 43, 7082–7089. [Google Scholar] [CrossRef] [PubMed]
- Moran, Y.; Cohen, L.; Kahn, R.; Karbat, I.; Gordon, D.; Gurevitz, M. Expression and mutagenesis of the sea anemone toxin Av2 reveals key amino acid residues important for activity on voltage-gated sodium channels. Biochemistry 2006, 45, 8864–8873. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Kem, W.R.; Norton, R.S.; Dunn, B.M. Chemical synthesis of a neurotoxic polypeptide from the sea anemone Stichodactyla helianthus. Int. J. Pept. Protein Res. 2009, 36, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Pallaghy, P.K.; Scanlon, M.J.; Monks, S.A.; Norton, R.S. Three-dimensional structure in solution of the polypeptide cardiac stimulant anthopleurin-A. Biochemistry 1995, 34, 3782–3794. [Google Scholar] [CrossRef] [PubMed]
- Monks, S.A.; Pallaghy, P.K.; Scanlon, M.J.; Norton, R.S. Solution structure of the cardiostimulant polypeptide anthopleurin-B and comparison with anthopleurin-A. Structure 1995, 3, 791–803. [Google Scholar] [CrossRef]
- Widmer, H.; Billeter, M.; Wüthrich, K. Three-dimensional structure of the neurotoxin ATX Ia from Anemonia sulcata in aqueous solution determined by nuclear magnetic resonance spectroscopy. Proteins Struct. Funct. Genet. 1989, 6, 357–371. [Google Scholar] [CrossRef]
- Salceda, E.; Pérez-Castells, J.; López-Méndez, B.; Garateix, A.; Salazar, H.; López, O.; Aneiros, A.; Ständker, L.; Béress, L.; Forssmann, W.-G.; et al. CgNa, a type I toxin from the giant Caribbean sea anemone Condylactis gigantea shows structural similarities to both type I and II toxins, as well as distinctive structural and functional properties 1. Biochem. J. 2007, 406, 67–76. [Google Scholar] [CrossRef]
- Fogh, R.H.; Kem, W.R.; Norton, R.S. Solution structure of neurotoxin I from the sea anemone Stichodactyla helianthus. A nuclear magnetic resonance, distance geometry, and restrained molecular dynamics study. J. Biol. Chem. 1990, 265, 13016–13028. [Google Scholar]
- Billen, B.; Debaveye, S.; Béress, L.; Garateix, A.; Tytgat, J. Phyla- and subtype-selectivity of CgNa, a Na+ channel toxin from the venom of the Giant Caribbean sea anemone Condylactis gigantea. Front. Pharmacol. 2010, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Aneiros, A.; Tytgat, J. The sea anemone Bunodosoma granulifera contains surprisingly efficacious and potent insect-selective toxins. FEBS Lett. 2002, 532, 131–134. [Google Scholar] [CrossRef]
- Oliveira, J.S.; Redaelli, E.; Zaharenko, A.J.; Cassulini, R.R.; Konno, K.; Pimenta, D.C.; Freitas, J.C.; Clare, J.J.; Wanke, E. Binding specificity of sea anemone toxins to NaV1.1-1.6 sodium channels. Unexpected contributions from differences in the IV/S3-S4 outer loop. J. Biol. Chem. 2004, 279, 33323–33335. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Beress, L.; Moller, C.; Mari, F.; Forssmann, W.-G.; Tytgat, J. A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. FASEB J. 2012, 26, 5141–5151. [Google Scholar] [CrossRef]
- Maier, S.K.G.; Westenbroek, R.E.; Schenkman, K.A.; Feigl, E.O.; Scheuer, T.; Catterall, W.A. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. USA 2002, 99, 4073–4078. [Google Scholar] [CrossRef]
- Poffers, M.; Bühne, N.; Herzog, C.; Thorenz, A.; Chen, R.; Güler, F.; Hage, A.; Leffler, A.; Echtermeyer, F. Sodium channel Nav1.3 Is expressed by polymorphonuclear neutrophils during mouse heart and kidney ischemia in vivo and regulates adhesion, transmigration, and chemotaxis of human and mouse neutrophils In Vitro. Anesthesiology 2018, 128, 1151–1166. [Google Scholar] [CrossRef]
- Noda, M.; Ikeda, T.; Kayano, T.; Suzuki, H.; Takeshima, H.; Kurasaki, M.; Takahashi, H.; Numa, S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature 1986, 320, 188–192. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel NaV1.4 in complex with β1. Science 2018, 362. [Google Scholar] [CrossRef]
- Madio, B.; King, G.F.; Undheim, E.A.B. Sea anemone toxins: A structural overview. Mar. Drugs 2019, 17, 325. [Google Scholar] [CrossRef]
- Langenegger, N.; Koua, D.; Schürch, S.; Heller, M.; Nentwig, W.; Kuhn-Nentwig, L. Identification of a precursor processing protease from the spider Cupiennius salei essential for venom neurotoxin maturation. J. Biol. Chem. 2018, 293, 2079–2090. [Google Scholar] [CrossRef]
- Khera, P.K.; Benzinger, G.R.; Lipkind, G.; Drum, C.L.; Hanck, D.A.; Blumenthal, K.M. Multiple cationic residues of Anthopleurin B that determine high affinity and channel isoform discrimination. Biochemistry 1995, 34, 8533–8541. [Google Scholar] [CrossRef] [PubMed]
- Clairfeuille, T.; Cloake, A.; Infield, D.T.; Llongueras, J.P.; Arthur, C.P.; Li, Z.R.; Jian, Y.; Martin-Eauclaire, M.-F.; Bougis, P.E.; Ciferri, C.; et al. Structural basis of α-scorpion toxin action on NaV channels. Science 2019, 363, eaav8573. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Huang, X.; Huang, G.; Gao, S.; Shen, H.; Liu, L.; Lei, J.; Yan, N. Molecular basis for pore blockade of human Na+ channel NaV1.2 by the m-conotoxin KIIIA. Science 2019, 363, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Sokotun, I.N.; Leĭchenko, E.V.; Vakorina, T.I.; Es’kov, A.A.; Il’ina, A.P.; Monastyrnaia, M.M.; Kozlovskaia, E.P. A serine protease inhibitor from the anemone Radianthus macrodactylus: Isolation and physicochemical characteristics. Bioorg. Khim. 2007, 33, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Moriyama, E.N. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 2004, 5, 378–388. [Google Scholar] [CrossRef]
- Provencher, S.W.; Gloeckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef]
- Yamanaka, S.; Hashimoto, M.; Tobe, M.; Kobayashi, K.; Sekizawa, J.; Nishimura, M. A simple method for screening assessment of acute toxicity of chemicals. Arch. Toxicol. 1990, 64, 262–268. [Google Scholar] [CrossRef]
- Peigneur, S.; Billen, B.; Derua, R.; Waelkens, E.; Debaveye, S.; Béress, L.; Tytgat, J. A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties. Biochem. Pharmacol. 2011, 82, 81–90. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucl. Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins Struct. Funct. Bioinform. 2017, 85, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE). 2019.01; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, H3A 2R7, Montreal, QC, Canada. 2019. Available online: https://www.chemcomp.com/ (accessed on 7 January 2020).
- Case, D.A.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; et al. AMBER 2017; University of California: San Francisco, CA, USA, 2017; Available online: http://ambermd.org (accessed on 7 January 2020).
- Newport, T.D.; Sansom, M.S.P.; Stansfeld, P.J. The MemProtMD database: A resource for membrane-embedded protein structures and their lipid interactions. Nucl. Acids Res. 2019, 47, D390–D397. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | α-Helices | β-Turns | β-Turns | Unordered | ||||
|---|---|---|---|---|---|---|---|---|
| αR | αD | αtot | βR | βD | βtot | |||
| δ-SHTX-Hcr1f | 0.4 | 4.7 | 5.1 | 22.6 | 12.1 | 34.7 | 23.5 | 36.7 |
| RTX-III | 0.1 | 0.4 | 0.5 | 27.8 | 13.3 | 41.1 | 18.3 | 40.1 |
| RTX-VI | 0.0 | 0.3 | 0.3 | 26.9 | 13.6 | 40.5 | 20.9 | 38.3 |
| Toxin | Voltage-Gated Sodium Channel Subtype | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| NaV1.1 | NaV1.2 | NaV1.3 | NaV1.4 | NaV1.5 | NaV1.6 | NaV1.8 | BgNaV1 | VdNaV1 | |
| δ-SHTX-Hcr1f | + | + | - | - | - | + | - | + | + |
| RTX-III | - | - | + | - | - | + | - | + | + |
| RTX-VI | - | + | - | - | - | + | - | + | + |
| Toxin | EC50 (nM) | ||
|---|---|---|---|
| NaV1.2 | NaV1.6 | BgNaV1 | |
| δ-SHTX-Hcr1f | 79.5 ± 4.3 | 183.5 ± 8.2 | 226.1 ± 12.3 |
| RTX-III | Not active | 381.8 ± 5.8 | 978.1 ± 62.3 |
| RTX-VI | 120.9 ± 19.3 | 282.3 ± 7.0 | 760.5 ± 25.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalina, R.S.; Peigneur, S.; Zelepuga, E.A.; Dmitrenok, P.S.; Kvetkina, A.N.; Kim, N.Y.; Leychenko, E.V.; Tytgat, J.; Kozlovskaya, E.P.; Monastyrnaya, M.M.; et al. New Insights into the Type II Toxins from the Sea Anemone Heteractis crispa. Toxins 2020, 12, 44. https://doi.org/10.3390/toxins12010044
Kalina RS, Peigneur S, Zelepuga EA, Dmitrenok PS, Kvetkina AN, Kim NY, Leychenko EV, Tytgat J, Kozlovskaya EP, Monastyrnaya MM, et al. New Insights into the Type II Toxins from the Sea Anemone Heteractis crispa. Toxins. 2020; 12(1):44. https://doi.org/10.3390/toxins12010044
Chicago/Turabian StyleKalina, Rimma S., Steve Peigneur, Elena A. Zelepuga, Pavel S. Dmitrenok, Aleksandra N. Kvetkina, Natalia Y. Kim, Elena V. Leychenko, Jan Tytgat, Emma P. Kozlovskaya, Margarita M. Monastyrnaya, and et al. 2020. "New Insights into the Type II Toxins from the Sea Anemone Heteractis crispa" Toxins 12, no. 1: 44. https://doi.org/10.3390/toxins12010044
APA StyleKalina, R. S., Peigneur, S., Zelepuga, E. A., Dmitrenok, P. S., Kvetkina, A. N., Kim, N. Y., Leychenko, E. V., Tytgat, J., Kozlovskaya, E. P., Monastyrnaya, M. M., & Gladkikh, I. N. (2020). New Insights into the Type II Toxins from the Sea Anemone Heteractis crispa. Toxins, 12(1), 44. https://doi.org/10.3390/toxins12010044