Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate



Abstract
:1. Introduction
2. Molecular Mechanisms by Which IS and PCS Induce Vascular Calcification
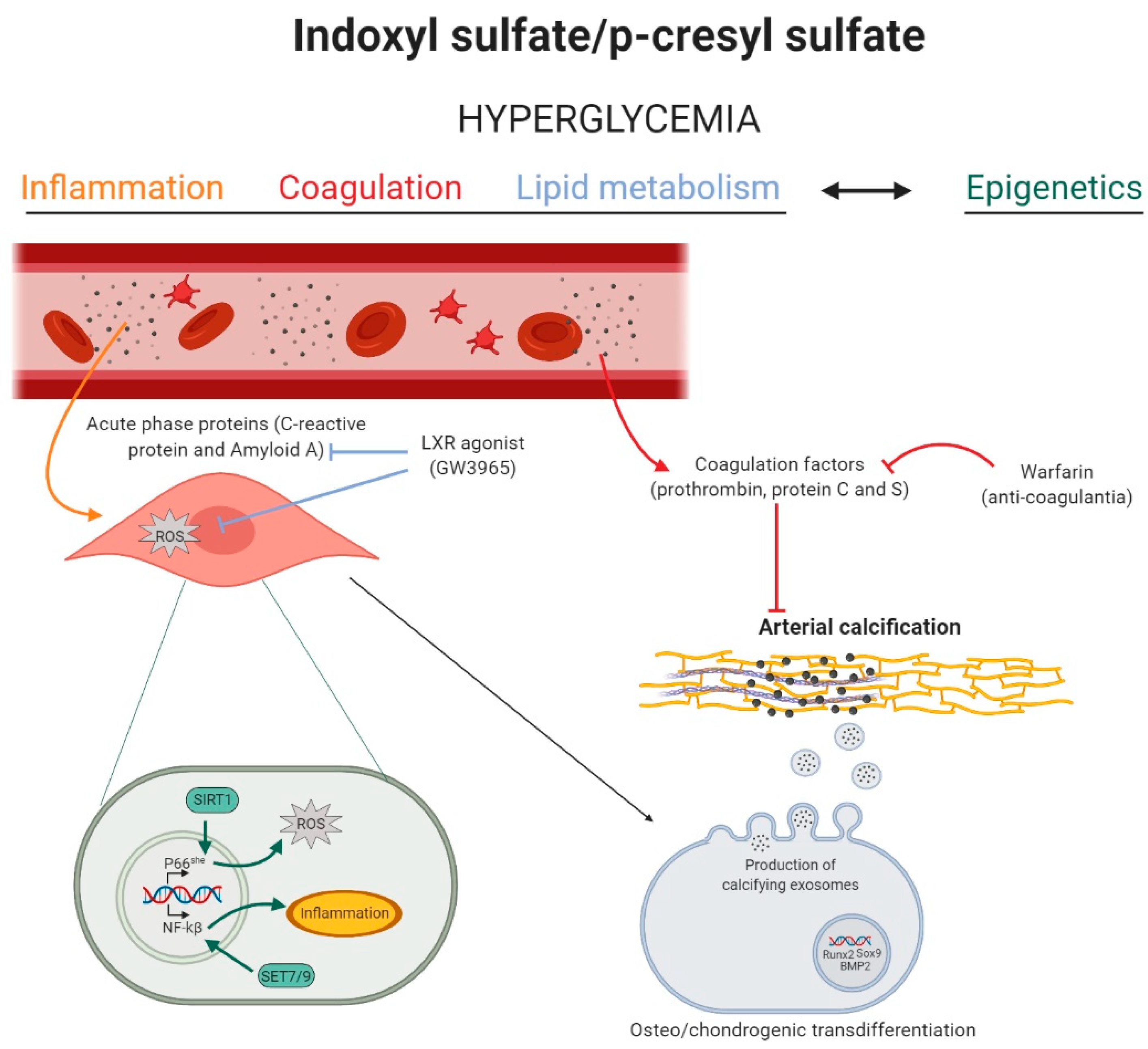
2.1. Inflammation and Coagulation Signaling Pathways
2.2. MicroRNAs, Upcoming Important Epigenetic Regulators in Uremic Toxins-Induced Vascular Calcification
2.3. Novel Research Fields to be Explored
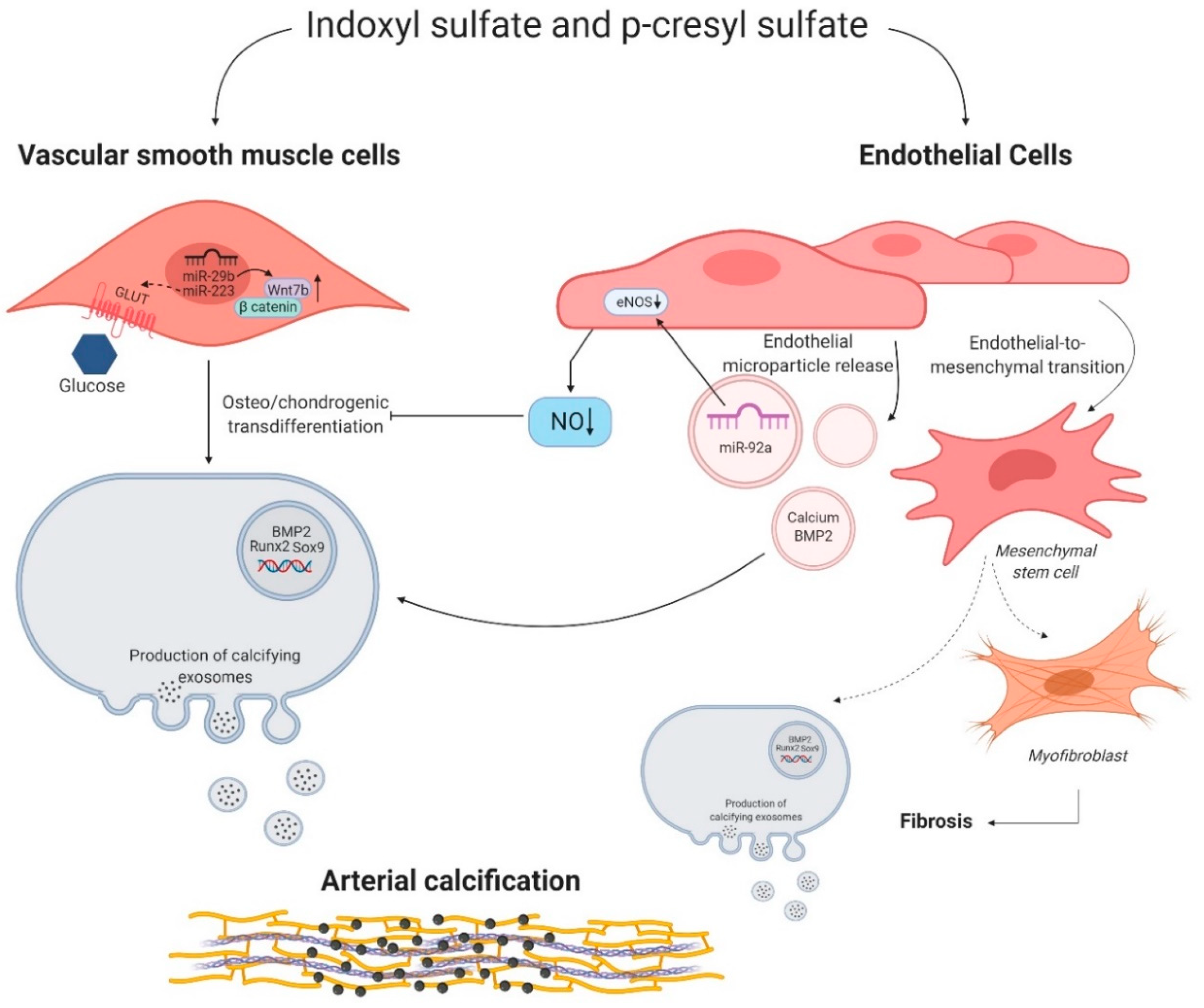
2.3.1. Protein-Bound Uremic Toxins Influence Phenotypic Switch of Multiple Cell Types
2.3.2. The Endothelium, an Overlooked Structure in the Process of Uremic Toxin Induced Vascular Calcification
3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, H.A.; van den Heuvel, L.P.; Ringens, L.H.; Dankers, A.C.; Russel, F.G.; Wetzels, J.F.; Hoenderop, J.G.; Masereeuw, R. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS ONE 2011, 6, e18438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.N.; Wu, I.W.; Huang, Y.F.; Peng, S.Y.; Huang, Y.C.; Ning, H.C. Measuring serum total and free indoxyl sulfate and p-cresyl sulfate in chronic kidney disease using UPLC-MS/MS. J. Food Drug Anal. 2019, 27, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirich, T.L.; Funk, B.A.; Plummer, N.S.; Hostetter, T.H.; Meyer, T.W. Prominent accumulation in hemodialysis patients of solutes normally cleared by tubular secretion. J. Am. Soc. Nephrol. 2014, 25, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephro. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Eloot, S.; Van Biesen, W.; Roels, S.; Delrue, W.; Schepers, E.; Dhondt, A.; Vanholder, R.; Glorieux, G. Spontaneous variability of pre-dialysis concentrations of uremic toxins over time in stable hemodialysis patients. PLoS ONE 2017, 12, e0186010. [Google Scholar] [CrossRef]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transpl. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B.; et al. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef]
- Hsueh, C.H.; Yoshida, K.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.M.; Giacomini, K.M. Identification and Quantitative Assessment of Uremic Solutes as Inhibitors of Renal Organic Anion Transporters, OAT1 and OAT3. Mol. Pharm. 2016, 13, 3130–3140. [Google Scholar] [CrossRef]
- Toth-Manikowski, S.M.; Sirich, T.L.; Meyer, T.W.; Hostetter, T.H.; Hwang, S.; Plummer, N.S.; Hai, X.; Coresh, J.; Powe, N.R.; Shafi, T.; et al. Contribution of ‘clinically negligible’ residual kidney function to clearance of uremic solutes. Nephrol. Dial. Transpl. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Pawlak, D. Indoxyl sulfate - the uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Wu, V.; Wu, P.C.; Wu, C.J. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Sirich, T.L.; Meyer, T.W.; Hostetter, T.H.; Plummer, N.S.; Hwang, S.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R.; et al. Results of the HEMO Study suggest that p-cresol sulfate and indoxyl sulfate are not associated with cardiovascular outcomes. Kidney Int. 2017, 92, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transpl. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney. Dis. 2004, 44, 1024–1030. [Google Scholar] [CrossRef]
- Sigrist, M.K.; Taal, M.W.; Bungay, P.; McIntyre, C.W. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 1241–1248. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.C.; Mitsnefes, M.M. Cardiovascular disease in CKD in children: update on risk factors, risk assessment, and management. Am. J. Kidney. Dis. 2009, 54, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Ishimura, E.; Okuno, S.; Kitatani, K.; Kim, M.; Shoji, T.; Nakatani, T.; Inaba, M.; Nishizawa, Y. Different risk factors for peripheral vascular calcification between diabetic and non-diabetic haemodialysis patients--importance of glycaemic control. Diabetologia 2002, 45, 1446–1448. [Google Scholar] [CrossRef] [Green Version]
- Katz, R.; Budoff, M.J.; O’Brien, K.D.; Wong, N.D.; Nasir, K. The metabolic syndrome and diabetes mellitus as predictors of thoracic aortic calcification as detected by non-contrast computed tomography in the Multi-Ethnic Study of Atherosclerosis. Diabetic Med. 2016, 33, 912–919. [Google Scholar] [CrossRef] [Green Version]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Mare, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E.; et al. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Gruys, E.; Toussaint, M.J.; Niewold, T.A.; Koopmans, S.J. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. Sci. B 2005, 6, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Venteclef, N.; Jakobsson, T.; Steffensen, K.R.; Treuter, E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrin. Met. 2011, 22, 333–343. [Google Scholar] [CrossRef]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Saldanha, J.F.; Yi, D.; Mafra, D.; Fouque, D.; Soulage, C.O. The uremic toxin indoxyl sulfate exacerbates reactive oxygen species production and inflammation in 3T3-L1 adipose cells. Free Radical Res. 2016, 50, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Bessueille, L.; Magne, D. Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell. Mol. Life Sci. 2015, 72, 2475–2489. [Google Scholar] [CrossRef]
- Avramovski, P.; Avramovska, M.; Sotiroski, K.; Sikole, A. Acute-phase proteins as promoters of abdominal aortic calcification in chronic dialysis patients. Saudi J. Kidney Dis. Transpl. 2019, 30, 376–386. [Google Scholar] [CrossRef]
- Henze, L.A.; Luong, T.T.D.; Boehme, B.; Masyout, J.; Schneider, M.P.; Brachs, S.; Lang, F.; Pieske, B.; Pasch, A.; Eckardt, K.U.; et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging 2019, 11, 5445–5462. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Wang, S. Serum Amyloid a Induces a Vascular Smooth Muscle Cell Phenotype Switch through the p38 MAPK Signaling Pathway. Biomed Res. Int. 2017, 2017, 4941379. [Google Scholar] [CrossRef]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Hsieh, M.Y.; Hung, S.C.; Kuo, K.L.; Tsai, T.H.; Lai, C.L.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C.; et al. Serum Indoxyl Sulfate Associates with Postangioplasty Thrombosis of Dialysis Grafts. J. Am. Soc. Nephrol. 2016, 27, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Spronk, H.M.; ten Cate, H.; Hofstra, L. Accelerated in vivo thrombin formation independently predicts the presence and severity of CT angiographic coronary atherosclerosis. JACC Cardiovasc. Imag. 2012, 5, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Horn, P.; Erkilet, G.; Veulemans, V.; Kropil, P.; Schurgers, L.; Zeus, T.; Heiss, C.; Kelm, M.; Westenfeld, R. Microparticle-Induced Coagulation Relates to Coronary Artery Atherosclerosis in Severe Aortic Valve Stenosis. PLoS ONE 2016, 11, e0151499. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Schoppet, M.; Schurgers, L.J.; Reynolds, J.L.; McNair, R.; Heiss, A.; Jahnen-Dechent, W.; Hackeng, T.M.; Schlieper, G.; Harrison, P.; et al. Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arter. Thromb. Vasc. Biol. 2017, 37, e22–e32. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, J.; Dhondt, A. The role of vitamin K in vascular calcification of patients with chronic kidney disease. Acta Clin. Belg. 2016, 71, 462–467. [Google Scholar] [CrossRef]
- De Mare, A.; Maudsley, S.; Azmi, A.; Hendrickx, J.O.; Opdebeeck, B.; Neven, E.; D’Haese, P.C.; Verhulst, A. Sclerostin as Regulatory Molecule in Vascular Media Calcification and the Bone-Vascular Axis. Toxins 2019, 11, 428. [Google Scholar] [CrossRef] [Green Version]
- De Vriese, A.S.; Caluwe, R.; Pyfferoen, L.; De Bacquer, D.; De Boeck, K.; Delanote, J.; De Surgeloose, D.; Van Hoenacker, P.; Van Vlem, B.; Verbeke, F.; et al. Multicenter Randomized Controlled Trial of Vitamin K Antagonist Replacement by Rivaroxaban with or without Vitamin K2 in Hemodialysis Patients with Atrial Fibrillation: the Valkyrie Study. J. Am. Soc. Nephrol. 2020, 31, 186–196. [Google Scholar] [CrossRef]
- Bi, X.; Song, J.; Gao, J.; Zhao, J.; Wang, M.; Scipione, C.A.; Koschinsky, M.L.; Wang, Z.V.; Xu, S.; Fu, G.; et al. Activation of liver X receptor attenuates lysophosphatidylcholine-induced IL-8 expression in endothelial cells via the NF-kappaB pathway and SUMOylation. J. Cell. Mol. Med. 2016, 20, 2249–2258. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Even-Or, O.; Xu, X.; van Rosmalen, M.; Lim, L.; Gadde, S.; Farokhzad, O.C.; Fisher, E.A. Nanoparticles containing a liver X receptor agonist inhibit inflammation and atherosclerosis. Adv. Healthc. Mater. 2015, 4, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhang, D.; Zhu, M.; Wang, Y.; Guo, S.; Xu, B.; Hou, G.; Feng, Y.; Liu, G. Liver X receptor alpha is targeted by microRNA-1 to inhibit cardiomyocyte apoptosis through a ROS-mediated mitochondrial pathway. Biochem. Cell. Biol. 2018, 96, 11–18. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.; Zhang, H.; Ning, Y.; Song, N.; Hu, J.; Cai, J.; Shi, Y.; Ding, X.; Zhang, X.; et al. Amelioration of Uremic Toxin Indoxyl Sulfate-Induced Osteoblastic Calcification by SET Domain Containing Lysine Methyltransferase 7/9 Protein. Nephron 2019, 141, 287–294. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D.; et al. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef] [Green Version]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallee, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [Green Version]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X.; et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012, 61, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xiao, J.; Li, R.; Qin, X.; Wang, F.; Mao, Y.; Liang, W.; Sheng, X.; Guo, M.; Song, Y.; et al. Metformin alleviates vascular calcification induced by vitamin D3 plus nicotine in rats via the AMPK pathway. Vasc. Pharmacol. 2016, 81, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138.e3113. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol.-Renal. 2014, 307, F891–F900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [Green Version]
- Kanno, Y.; Into, T.; Lowenstein, C.J.; Matsushita, K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc. Res. 2008, 77, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Sambe, T.; Mason, R.P.; Dawoud, H.; Bhatt, D.L.; Malinski, T. Metformin treatment decreases nitroxidative stress, restores nitric oxide bioavailability and endothelial function beyond glucose control. Biomed. Pharmacother. 2018, 98, 149–156. [Google Scholar] [CrossRef]
- Du, Y.; Gao, C.; Liu, Z.; Wang, L.; Liu, B.; He, F.; Zhang, T.; Wang, Y.; Wang, X.; Xu, M.; et al. Upregulation of a disintegrin and metalloproteinase with thrombospondin motifs-7 by miR-29 repression mediates vascular smooth muscle calcification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2580–2588. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Yu, Y.; Guan, X.; Nie, L.; Liu, Y.; He, T.; Xiong, J.; Xu, X.; Li, Y.; Yang, K.; Wang, Y.; et al. DNA hypermethylation of sFRP5 contributes to indoxyl sulfate-induced renal fibrosis. J. Mol. Med. 2017, 95, 601–613. [Google Scholar] [CrossRef]
- Massy, Z.A.; Metzinger-Le Meuth, V.; Metzinger, L. MicroRNAs Are Associated with Uremic Toxicity, Cardiovascular Calcification, and Disease. Contrib. Nephrol. 2017, 189, 160–168. [Google Scholar] [CrossRef]
- Lu, H.; Buchan, R.J.; Cook, S.A. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 2010, 86, 410–420. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Metzinger-Le Meuth, V.; Henaut, L.; Djelouat, M.S.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: evidence for the involvement of miR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; De Schutter, T.M.; De Broe, M.E.; D’Haese, P.C. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011, 79, 1166–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transpl. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transpl. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Yu, M.A.; Ryu, E.S.; Jang, Y.H.; Kang, D.H. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab. Invest. 2012, 92, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [Green Version]
- Bolati, D.; Shimizu, H.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces epithelial-to-mesenchymal transition in rat kidneys and human proximal tubular cells. Am. J. Nephrol. 2011, 34, 318–323. [Google Scholar] [CrossRef]
- Bolati, D.; Shimizu, H.; Niwa, T. AST-120 ameliorates epithelial-to-mesenchymal transition and interstitial fibrosis in the kidneys of chronic kidney disease rats. J. Renal. Nutr. 2012, 22, 176–180. [Google Scholar] [CrossRef]
- Chang, L.C.; Sun, H.L.; Tsai, C.H.; Kuo, C.W.; Liu, K.L.; Lii, C.K.; Huang, C.S.; Li, C.C. 1,25(OH)2 D3 attenuates indoxyl sulfate-induced epithelial-to-mesenchymal cell transition via inactivation of PI3K/Akt/beta-catenin signaling in renal tubular epithelial cells. Nutrition 2019, 69, 110554. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, D.; Olsen, B.R. The role of endothelial-mesenchymal transition in heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Guo, Y.; Moon, J.H.; Jumabay, M.; Bostrom, K.I.; Yao, Y. Serine Protease Activation Essential for Endothelial-Mesenchymal Transition in Vascular Calcification. Circ. Res. 2015, 117, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Cubberly, M.R.; Bostrom, K.I. A role for the endothelium in vascular calcification. Circ. Res. 2013, 113, 495–504. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephro. 2011, 6, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Lu, L.; Hua, Y.; Huang, K.; Wang, I.; Huang, L.; Fu, Q.; Chen, A.; Chan, P.; Fan, H.; et al. Vasculopathy in the setting of cardiorenal syndrome: roles of protein-bound uremic toxins. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1–H13. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal. Bioanal. Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol.-Cell Physiol. 2012, 303, C126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. Senescence and dysfunction of proximal tubular cells are associated with activated p53 expression by indoxyl sulfate. Am. J. Physiol.-Cell Physiol. 2010, 299, C1110–C1117. [Google Scholar] [CrossRef] [PubMed]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Drozdov, I.; Shroff, R.; Beltran, L.E.; Shanahan, C.M. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ. Res. 2013, 112, e99–e109. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Buendia, P.; Montes de Oca, A.; Madueno, J.A.; Merino, A.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Rodriguez, M.; Carracedo, J. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. 2015, 29, 173–181. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| CKD | Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 |
|---|---|---|---|---|---|
| N | 29 | 49 | 64 | 40 | 22 |
| Total IS (mg/L) | 1.03 ± 0.85 | 1.54 ± 1.11 | 2.22 ± 1.79 | 4.74 ± 4.34 | 18.21 ± 15.06 |
| Total PCS (mg/L) | 2.69 ± 4.34 | 4.42 ± 4.47 | 6.45 ± 7.12 | 16.10 ± 13.98 | 27.00 ± 17.66 |
| Free IS (mg/L) | 0.08 ± 0.06 | 0.11 ± 0.09 | 0.17 ± 0.13 | 0.49 ± 0.72 | 2.36 ± 2.64 |
| Free PCS (mg/L) | 0.15 ± 0.20 | 0.24 ± 0.29 | 0.36 ± 0.37 | 1.36 ± 2.58 | 2.38 ± 2.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58. https://doi.org/10.3390/toxins12010058
Opdebeeck B, D’Haese PC, Verhulst A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins. 2020; 12(1):58. https://doi.org/10.3390/toxins12010058
Chicago/Turabian StyleOpdebeeck, Britt, Patrick C. D’Haese, and Anja Verhulst. 2020. "Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate" Toxins 12, no. 1: 58. https://doi.org/10.3390/toxins12010058
APA StyleOpdebeeck, B., D’Haese, P. C., & Verhulst, A. (2020). Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins, 12(1), 58. https://doi.org/10.3390/toxins12010058