Impact of Bacterial Toxins in the Lungs

 and
and {kind=link}
Abstract
:1. Introduction
2. Staphylococcus aureus (S. aureus)
2.1. Alpha-Hemolysin (Hla)
2.2. Beta-Hemolysin (Hlb)
2.3. Panton-Valentine Leukocidin (PVL)
3. Pseudomonas aeruginosa
3.1. Exotoxin A (P-ExA)
3.2. Exoenzyme S (Exo S) and Exoenzyme T (Exo T)
3.3. Exotoxin U (Exo U)
3.4. Exotoxin Y (Exo Y)
3.5. Exolysin A (Exl A)
3.6. Alkaline Protease (AP)
3.7. Lipopolysaccharide (LPS) from P. aeruginosa
4. Salmonella enterica
LPS of S. enterica (LPSSE)
5. Escherichia coli
5.1. LPS of E. coli (LPSEC)
5.2. Exotoxins of E. coli
6. Bordetella pertussis
6.1. Pertussis Toxin (PTX)
6.2. Adenylate Cyclase Toxin (ACT)
7. Bacillus anthracis
Anthrax Toxins
8. Listeria monocytogenes
Listeriolysin O (LLO)
9. Streptococcus pneumoniae
Pneumolysin (PLY)
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, R.; Feltrup, T.; Kukreja, R.; Patel, K.; Cai, S.; Singh, B.R. Evolutionary features in the structure and function of bacterial toxins. Toxins 2019, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reboud, E.; Basso, P.; Maillard, A.P.; Huber, P.; Attrée, I. Exolysin shapes the virulence of pseudomonas aeruginosa clonal outliers. Toxins 2017, 9, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, J.A. Claudins and alveolar epithelial barrier function in the lung. Ann. New York Acad. Sci. 2012, 1257, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, R.; Verin, A.D.; Black, S.M.; Catravas, J.D. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem. Pharmacol. 2009, 77, 1763–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, J.N.; Lucas, R.; Verin, A.D. The acute respiratory distress syndrome: Mechanisms and perspective therapeutic approaches. Austin J. Vasc. Med. 2015, 2, 1009. [Google Scholar] [PubMed]
- Hamacher, J.; Hadizamani, Y.; Borgmann, M.; Mohaupt, M.; Männel, D.N.; Moehrlen, U.; Lucas, R.F.; Stammberger, U. Cytokine–ion channel interactions in pulmonary inflammation. Front. Immunol. 2017, 8, 1644. [Google Scholar] [CrossRef] [Green Version]
- Sastalla, I.; Monack, D.M.; Kubatzky, K.F. Editorial: Bacterial exotoxins: How bacteria fight the immune system. Front. Immunol. 2016, 7, 300. [Google Scholar] [CrossRef] [Green Version]
- Janga, H.; Cassidy, L.; Wang, F.; Spengler, D.; Oestern-Fitschen, S.; Krause, M.F.; Seekamp, A.; Tholey, A.; Fuchs, S. Site-specific and endothelial-mediated dysfunction of the alveolar-capillary barrier in response to lipopolysaccharides. J. Cell Mol. Med. 2018, 22, 982–998. [Google Scholar] [CrossRef] [Green Version]
- Lucas, R.; Sridhar, S.; Rick, F.G.; Gorshkov, B.; Umapathy, N.S.; Yang, G.; Oseghale, A.; Verin, A.D.; Chakraborty, T.; Matthay, M.A.; et al. Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc. Natl. Acad Sci. USA 2012, 109, 2084–2089. [Google Scholar] [CrossRef] [Green Version]
- Ala’Aldeen, D.A.A.; Wooldridge, K.G. 13-bacterial pathogenicity. In Medical Microbiology, 18th ed.; Greenwood, D., Barer, M., Slack, R., Irving, W., Eds.; Churchill Livingstone: Edinburgh, UK, 2012; pp. 156–167. [Google Scholar] [CrossRef]
- Lucas, R.; Yang, G.; Gorshkov, B.A.; Zemskov, E.A.; Sridhar, S.; Umapathy, N.S.; Jezierska-Drutel, A.; Alieva, I.B.; Leustik, M.; Hossain, H.; et al. Protein kinase C-α and arginase i mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am. J. Respir. Cell Mol. Biol. 2012, 47, 445–453. [Google Scholar] [CrossRef]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [CrossRef]
- Gilbert, R.J.; Jimenez, J.L.; Chen, S.; Tickle, I.J.; Rossjohn, J.; Parker, M.; Andrew, P.W.; Saibil, H.R. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell 1999, 97, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, R.; Malik, A.B.; Minshall, R.D.; Kouklis, P.; Ellis, C.A.; Tiruppathi, C. Ca(2+) signaling and PKC-alpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J. Physiol. 2001, 533, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Stringaris, A.K.; Geisenhainer, J.; Bergmann, F.; Balshusemann, C.; Lee, U.; Zysk, G.; Mitchell, T.J.; Keller, B.U.; Kuhnt, U.; Gerber, J.; et al. Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol. Dis. 2002, 11, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repp, H.; Pamukci, Z.; Koschinski, A.; Domann, E.; Darji, A.; Birringer, J.; Brockmeier, D.; Chakraborty, T.; Dreyer, F. Listeriolysin of Listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cell. Microbiol. 2002, 4, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Birukova, A.; Ramirez, S.I.; Garcia, J.G.; Verin, A.D. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 2003, 28, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Kayal, S.; Charbit, A. Listeriolysin o: A key protein of Listeria monocytogenes with multiple functions. FEMS Microbiol. Rev. 2006, 30, 514–529. [Google Scholar] [CrossRef] [Green Version]
- Sonnen, A.F.; Plitzko, J.M.; Gilbert, R.J. Incomplete pneumolysin oligomers form membrane pores. Open Biol. 2014, 4, 140044. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, J.P. Pore-forming virulence factors of Staphylococcus aureus destabilize epithelial barriers-effects of alpha-toxin in the early phases of airway infection. AIMS Microbiol. 2015, 1, 11–36. [Google Scholar] [CrossRef]
- Yang, G.; Pillich, H.; White, R.; Czikora, I.; Pochic, I.; Yue, Q.; Hudel, M.; Gorshkov, B.; Verin, A.; Sridhar, S.; et al. Listeriolysin O causes ENaC dysfunction in human airway epithelial cells. Toxins 2018, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Vogele, M.; Bhaskara, R.M.; Mulvihill, E.; van Pee, K.; Yildiz, O.; Kuhlbrandt, W.; Muller, D.J.; Hummer, G. Membrane perforation by the pore-forming toxin pneumolysin. Proc. Natl. Acad. Sci. USA 2019, 116, 13352–13357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinfort, C.; Wilson, R.; Mitchell, T.; Feldman, C.; Rutman, A.; Todd, H.; Sykes, D.; Walker, J.; Saunders, K.; Andrew, P.W. Effect of Streptococcus pneumoniae on human respiratory epithelium in vitro. Infect. Immun. 1989, 57, 2006–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, C.; Mitchell, T.J.; Andrew, P.W.; Boulnois, G.J.; Read, R.C.; Todd, H.C.; Cole, P.J.; Wilson, R. The effect of Streptococcus pneumoniae pneumolysin on human respiratory epithelium in vitro. Microb. Pathog. 1990, 9, 275–284. [Google Scholar] [CrossRef]
- Rayner, C.F.; Jackson, A.D.; Rutman, A.; Dewar, A.; Mitchell, T.J.; Andrew, P.W.; Cole, P.J.; Wilson, R. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect. Immun. 1995, 63, 442–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azghani, A.O. Pseudomonas aeruginosa and epithelial permeability: Role of virulence factors elastase and exotoxin a. Am. J. Respir. Cell Mol. Biol. 1996, 15, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Jeon, S.Y.; Min, Y.G.; Rhyoo, C.; Kim, J.W.; Yun, J.B.; Park, S.W.; Kwon, T.Y. Effects of beta-toxin of Staphylococcus aureus on ciliary activity of nasal epithelial cells. Laryngoscope 2000, 110, 2085–2088. [Google Scholar] [CrossRef]
- Mairpady Shambat, S.; Chen, P.; Nguyen Hoang, A.T.; Bergsten, H.; Vandenesch, F.; Siemens, N.; Lina, G.; Monk, I.R.; Foster, T.J.; Arakere, G.; et al. Modelling Staphylococcal pneumonia in a human 3d lung tissue model system delineates toxin-mediated pathology. Dis. Model. Mech. 2015, 8, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Rangel, S.M.; Diaz, M.H.; Knoten, C.A.; Zhang, A.; Hauser, A.R. The role of exoS in dissemination of Pseudomonas aeruginosa during pneumonia. PLoS Pathog. 2015, 11, e1004945. [Google Scholar] [CrossRef] [Green Version]
- Galanos, C.; Freudenberg, M.A. Bacterial endotoxins: Biological properties and mechanisms of action. Mediat. Inflamm 1993, 2, S11–S16. [Google Scholar] [CrossRef]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- Steimle, A.; Autenrieth, I.B.; Frick, J.-S. Structure and function: Lipid a modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, S.M.; Ernst, R.K. The role of Pseudomonas lipopolysaccharide in cystic fibrosis airway infection. Subcell. Biochem. 2010, 53, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Li, W.; Jono, H.; Li, Q.; Zhang, S.; Li, J.D.; Shen, H. Reactive oxygen species regulate Pseudomonas aeruginosa lipopolysaccharide-induced muc5ac mucin expression via PKC-NADPH oxidase-ROS-TGF-alpha signaling pathways in human airway epithelial cells. Biochem. Biophys. Res. Commun. 2008, 366, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Mutharia, L.M.; Chan, L.; Darveau, R.P.; Speert, D.P.; Pier, G.B. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: A class of serum-sensitive, non-typeable strains deficient in lipopolysaccharide o side chains. Infect. Immun. 1983, 42, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Vargaftig, B.B. Modifications of experimental bronchopulmonary hyperresponsiveness. Am. J. Respir. Crit. Care Med. 1997, 156, (4 Pt 2). S97–S102. [Google Scholar] [CrossRef]
- Lapa e Silva, J.R.; Possebon da Silva, M.D.; Lefort, J.; Vargaftig, B.B. Endotoxins, asthma, and allergic immune responses. Toxicology 2000, 152, 31–35. [Google Scholar] [CrossRef]
- Curran, C.S.; Bolig, T.; Torabi-Parizi, P. Mechanisms and targeted therapies for Pseudomonas aeruginosa lung infection. Am. J. Respir. Crit. Care Med. 2018, 197, 708–727. [Google Scholar] [CrossRef]
- Lee, S.A.; Lee, S.H.; Kim, J.Y.; Lee, W.S. Effects of glycyrrhizin on lipopolysaccharide-induced acute lung injury in a mouse model. J. Thorac. Dis. 2019, 11, 1287–1302. [Google Scholar] [CrossRef]
- Huszczynski, S.M.; Lam, J.S.; Khursigara, C.M. The role of Pseudomonas aeruginosa lipopolysaccharide in bacterial pathogenesis and physiology. Pathogens 2019, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Panariti, A.; Martin, J.G. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front. Immunol. 2018, 9, 1777. [Google Scholar] [CrossRef] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.R.; Kench, J.A.; Vondracek, A.; Kruk, E.; Daleke, D.L.; Jordan, M.; Marrack, P.; Henson, P.M.; Fadok, V.A. Interaction between phosphatidylserine and the phosphatidylserine receptor inhibits immune responses in vivo. J. Immunol. 2005, 174, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Hristovska, A.M.; Rasmussen, L.E.; Hansen, P.B.; Nielsen, S.S.; Nüsing, R.M.; Narumiya, S.; Vanhoutte, P.; Skøtt, O.; Jensen, B.L. Prostaglandin E2 induces vascular relaxation by E-prostanoid 4 receptor-mediated activation of endothelial nitric oxide synthase. Hypertension 2007, 50, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Madaio, M.P.; Czikora, I.; Kvirkvelia, N.; McMenamin, M.; Yue, Q.; Liu, T.; Flores-Toque, H.; Sridhar, S.; Covington, K.; Alaisami, R.; et al. The TNF-derived TIP peptide activates the epithelial sodium channel and ameliorates experimental nephrotoxic serum nephritis. Kidney Int. 2019, 95, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Dolan, J.M.; Weinberg, J.B.; O’Brien, E.; Abashian, A.; Procario, M.C.; Aronoff, D.M.; Crofford, L.J.; Peters-Golden, M.; Ward, L.; Mancuso, P. Increased lethality and defective pulmonary clearance of Streptococcus pneumoniae in microsomal prostaglandin E synthase-1-knockout mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1111–L1120. [Google Scholar] [CrossRef] [Green Version]
- Harvill, E.T.; Cotter, P.A.; Yuk, M.H.; Miller, J.F. Probing the function of Bordetella bronchiseptica adenylate cyclase toxin by manipulating host immunity. Infect. Immun. 1999, 67, 1493–1500. [Google Scholar] [CrossRef] [Green Version]
- Do Vale, A.; Cabanes, D.; Sousa, S. Bacterial toxins as pathogen weapons against phagocytes. Front. Microbiol. 2016, 7, 42. [Google Scholar] [CrossRef]
- Mehraj, J.; Witte, W.; Akmatov, M.K.; Layer, F.; Werner, G.; Krause, G. Epidemiology of Staphylococcus aureus nasal carriage patterns in the community. Curr. Top. Microbiol. Immunol. 2016, 398, 55–87. [Google Scholar] [CrossRef]
- Foster, T. Staphylococcus. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. Available online: https://www.ncbi.nlm.nih.gov/books/NBK8448/ (accessed on 31 March 2020).
- Gnanamani, A.; Periasamy, H.; Paul Satyaseela, M. Staphylococcus aureus: Overview of bacteriology, clinical diseases, epidemiology, antibiotic resistance and therapeutic approach. In Frontiers in Staphylococcus aureus; Enany, S., Crotty Alexander, L., Eds.; IntechOpen Limited: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Rische, H.; Meyer, W.; Tschape; Voigt, W.; Ziomek, D.; Hummel, R. The taxonomy of staphylococcus aureus. Contrib. Microbiol. Immunol. 1973, 1, 24–29. [Google Scholar] [PubMed]
- Pulverer, G. Taxonomy of staphylococcus aureus. Zentralblatt fuer Bakteriologie, Mikrobiologie, und Hygiene. Ser. A Med Microbiol. Infect. Dis. Virol. Parasitol. 1986, 262, 425–437. [Google Scholar]
- Lowy, F.D. Staphylococcus aureus infections. New Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Corey, G.R. Epidemiology of methicillin-resistant staphylococcus aureus. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46 (Suppl. 5), S344–S349. [Google Scholar] [CrossRef] [Green Version]
- Rasigade, J.P.; Vandenesch, F. Staphylococcus aureus: A pathogen with still unresolved issues. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 21, 510–514. [Google Scholar] [CrossRef]
- Mishra, A.K.; Yadav, P.; Mishra, A. A systemic review on staphylococcal scalded skin syndrome (ssss): A rare and critical disease of neonates. Open Microbiol. J. 2016, 10, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus toxins and their molecular activity in infectious diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J.; Geoghegan, J. Staphylococcus aureus. In Molecular Medical Microbiology; Tang, Y.-W., Sails, A., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 655–674. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of Staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [Green Version]
- Stulik, L.; Malafa, S.; Hudcova, J.; Rouha, H.; Henics, B.Z.; Craven, D.E.; Sonnevend, A.M.; Nagy, E. Alpha-hemolysin activity of methicillin-susceptible Staphylococcus aureus predicts ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 2014, 190, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- McGee, M.P.; Kreger, A.; Leake, E.S.; Harshman, S. Toxicity of staphylococcal alpha toxin for rabbit alveolar macrophages. Infect. Immun. 1983, 39, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, M.E.; Kim, H.K.; Wang, Y.; Bubeck Wardenburg, J. ADAM10 mediates vascular injury induced by staphylococcus aureus alpha-hemolysin. J. Infect. Dis. 2012, 206, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Sheng, S.; Shao, Z. Staphylococcal alpha-hemolysin can form hexamers in phospholipid bilayers. J. Mol. Biol. 1998, 276, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Seilie, E.S.; Bubeck Wardenburg, J. Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol. 2017, 72, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.A.; Fahsel, B.; Kemper, H.; Mayeres, J.; Li, C.; Wilker, B.; Keitsch, S.; Soddemann, M.; Sehl, C.; Kohnen, M.; et al. Staphylococcus aureus alpha-toxin disrupts endothelial-cell tight junctions via acid sphingomyelinase and ceramide. Infect. Immun. 2018, 86, e00606-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walev, I.; Martin, E.; Jonas, D.; Mohamadzadeh, M.; Muller-Klieser, W.; Kunz, L.; Bhakdi, S. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect. Immun 1993, 61, 4972–4979. [Google Scholar] [CrossRef] [Green Version]
- Aksimentiev, A.; Schulten, K. Imaging alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005, 88, 3745–3761. [Google Scholar] [CrossRef] [Green Version]
- Goggel, R.; Winoto-Morbach, S.; Vielhaber, G.; Imai, Y.; Lindner, K.; Brade, L.; Brade, H.; Ehlers, S.; Slutsky, A.S.; Schutze, S.; et al. PAF-mediated pulmonary edema: A new role for acid sphingomyelinase and ceramide. Nat. Med. 2004, 10, 155–160. [Google Scholar] [CrossRef]
- Rose, F.; Dahlem, G.; Guthmann, B.; Grimminger, F.; Maus, U.; Hanze, J.; Duemmer, N.; Grandel, U.; Seeger, W.; Ghofrani, H.A. Mediator generation and signaling events in alveolar epithelial cells attacked by S aureus alpha-toxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L207–L214. [Google Scholar] [CrossRef] [Green Version]
- McElroy, M.C.; Harty, H.R.; Hosford, G.E.; Boylan, G.M.; Pittet, J.-F.; Foster, T.J. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect. Immun. 1999, 67, 5541. [Google Scholar] [CrossRef] [Green Version]
- Hermann, I.; Rath, S.; Ziesemer, S.; Volksdorf, T.; Dress, R.J.; Gutjahr, M.; Muller, C.; Beule, A.G.; Hildebrandt, J.P. Staphylococcus aureus hemolysin a disrupts cell-matrix adhesions in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Baaske, R.; Richter, M.; Moller, N.; Ziesemer, S.; Eiffler, I.; Muller, C.; Hildebrandt, J.P. ATP release from human airway epithelial cells exposed to Staphylococcus aureus alpha-toxin. Toxins 2016, 8, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebert, E.M.; Zsembery, A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim. Et Biophys. Acta 2003, 1615, 7–32. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Kawakami, M.; Sasaki, S.; Katsumata, T.; Mori, H.; Yoshida, H.; Nakahari, T. ATP regulation of ciliary beat frequency in rat tracheal and distal airway epithelium. Exp. Physiol. 2005, 90, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.A.; Sugar, R.A.; Jackson, A.D. Nucleotide-mediated mucin secretion from differentiated human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Danahay, H.; Atherton, H.C.; Jackson, A.D.; Kreindler, J.L.; Poll, C.T.; Bridges, R.J. Membrane capacitance and conductance changes parallel mucin secretion in the human airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L558–L569. [Google Scholar] [CrossRef]
- Douillet, C.D.; Robinson, W.P., 3rd; Milano, P.M.; Boucher, R.C.; Rich, P.B. Nucleotides induce IL-6 release from human airway epithelia via p2y2 and p38 MAPK-dependent pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L734–L746. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.E.; Becker, R.E.N.; Sailer, A.; Turner, J.R.; Bubeck Wardenburg, J. Synergistic action of staphylococcus aureus α-toxin on platelets and myeloid lineage cells contributes to lethal sepsis. Cell Host Microbe 2015, 17, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Ji, Y. Involvement of alpha5beta1-integrin and TNF-alpha in Staphylococcus aureus alpha-toxin-induced death of epithelial cells. Cell Microbiol. 2007, 9, 1809–1821. [Google Scholar] [CrossRef]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009, 4, e7446. [Google Scholar] [CrossRef]
- Bartlett, A.H.; Foster, T.J.; Hayashida, A.; Park, P.W. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in staphylococcus aureus pneumonia. J. Infect. Dis. 2008, 198, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the nlrp3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Bergstralh, D.T.; O’Connor, W.; Morrison, A.C.; Taxman, D.J.; Duncan, J.A.; Barnoy, S.; Venkatesan, M.M.; Flavell, R.A.; Deshmukh, M.; et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components cias1/cryopyrin/nlrp3 and asc. Cell Host Microbe 2007, 2, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Huseby, M.; Shi, K.; Brown, C.K.; Digre, J.; Mengistu, F.; Seo, K.S.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H.; Earhart, C.A. Structure and biological activities of beta toxin from Staphylococcus aureus. J. Bacteriol. 2007, 189, 8719. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, A.; Bartlett, A.H.; Foster, T.J.; Park, P.W. Staphylococcus aureus beta-toxin induces lung injury through syndecan-1. Am. J. Pathol. 2009, 174, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribier, B.; Prevost, G.; Couppie, P.; Finck-Barbancon, V.; Grosshans, E.; Piemont, Y. Staphylococcus aureus leukocidin: A new virulence factor in cutaneous infections? An epidemiological and experimental study. Dermatology 1992, 185, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Panton, P.N.; Valentine, F.C.O. Staphylococcal toxin. Lancet 1932, 219, 506–508. [Google Scholar] [CrossRef]
- Labandeira-Rey, M.; Couzon, F.; Boisset, S.; Brown, E.L.; Bes, M.; Benito, Y.; Barbu, E.M.; Vazquez, V.; Hook, M.; Etienne, J.; et al. Staphylococcus aureus panton-valentine leukocidin causes necrotizing pneumonia. Science 2007, 315, 1130–1133. [Google Scholar] [CrossRef] [Green Version]
- Finck-Barbançon, V.; Duportail, G.; Meunier, O.; Colin, D.A. Pore formation by a two-component leukocidin from Staphyloccocus aureus within the membrane of human polymorphonuclear leukocytes. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 1993, 1182, 275–282. [Google Scholar] [CrossRef]
- Prevost, G.; Cribier, B.; Couppie, P.; Petiau, P.; Supersac, G.; Finck-Barbancon, V.; Monteil, H.; Piemont, Y. Panton-valentine leucocidin and gamma-hemolysin from Staphylococcus aureus atcc 49775 are encoded by distinct genetic loci and have different biological activities. Infect. Immun 1995, 63, 4121–4129. [Google Scholar] [CrossRef] [Green Version]
- Genestier, A.-L.; Michallet, M.-C.; Prévost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.M.; et al. Staphylococcus aureus panton-valentine leukocidin directly targets mitochondria and induces bax-independent apoptosis of human neutrophils. J. Clin. Investig. 2005, 115, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Chan, L.; Tattevin, P.; Kajikawa, O.; Martin, T.R.; Basuino, L.; Mai, T.T.; Marbach, H.; Braughton, K.R.; Whitney, A.R.; et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus panton-valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. USA 2010, 107, 5587–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.C.; Kurdowska, A. Interleukin 8 and acute lung injury. Arch. Pathol. Lab. Med. 2014, 138, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Hensler, T.; Konig, B.; Prevost, G.; Piemont, Y.; Koller, M.; Konig, W. Leukotriene b4 generation and DNA fragmentation induced by leukocidin from Staphylococcus aureus: Protective role of granulocyte-macrophage colony-stimulating factor (GM-CSF and G-CSF for human neutrophils. Infect. Immun. 1994, 62, 2529–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konig, B.; Prevost, G.; Piemont, Y.; Konig, W. Effects of Staphylococcus aureus leukocidins on inflammatory mediator release from human granulocytes. J. Infect. Dis. 1995, 171, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Hensler, T.; Koller, M.; Prevost, G.; Piemont, Y.; Konig, W. GTP-binding proteins are involved in the modulated activity of human neutrophils treated with the panton-valentine leukocidin from Staphylococcus aureus. Infect. Immun. 1994, 62, 5281–5289. [Google Scholar] [CrossRef] [Green Version]
- Iglewski, B. Pseudomonas. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. Available online: https://www.ncbi.nlm.nih.gov/books/NBK8326/ (accessed on 31 March 2020).
- Wu, W.; Jin, Y.; Bai, F.; Jin, S.W. Pseudomonas aeruginosa. In Molecular Medical Microbiology; Tang, Y.-W., Sails, A., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 753–767. [Google Scholar] [CrossRef]
- Wolfgang, M.C.; Jyot, J.; Goodman, A.L.; Ramphal, R.; Lory, S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2004, 101, 6664–6668. [Google Scholar] [CrossRef] [Green Version]
- Kanj, S.S.; Sexton, D.J. Pseudomonas Aeruginosa Infections of The Eye, Ear, Urinary Tract, Gastrointestinal Tract, and Central Nervous System. 2020. Available online: https://www.uptodate.com/contents/pseudomonas-aeruginosa-infections-of-the-eye-ear-urinary-tract-gastrointestinal-tract-and-central-nervous-system (accessed on 31 March 2020).
- Matsui, H.; Grubb, B.R.; Tarran, R.; Randell, S.H.; Gatzy, J.T.; Davis, C.W.; Boucher, R.C. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998, 95, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Pier, G.B.; Grout, M.; Zaidi, T.S.; Olsen, J.C.; Johnson, L.G.; Yankaskas, J.R.; Goldberg, J.B. Role of mutant cftr in hypersusceptibility of cystic fibrosis patients to lung infections. Science 1996, 271, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.J.; Travis, S.M.; Greenberg, E.P.; Welsh, M.J. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996, 85, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Goldman, M.J.; Wilson, J.M. Mouse beta-defensin 1 is a salt-sensitive antimicrobial peptide present in epithelia of the lung and urogenital tract. Infect. Immun. 1998, 66, 1225–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bals, R.; Wang, X.; Wu, Z.; Freeman, T.; Bafna, V.; Zasloff, M.; Wilson, J.M. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J. Clin. Investig. 1998, 102, 874–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tager, A.M.; Wu, J.; Vermeulen, M.W. The effect of chloride concentration on human neutrophil functions: Potential relevance to cystic fibrosis. Am. J. Respir Cell Mol. Biol. 1998, 19, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M. Pseudomonas aeruginosa in cystic fibrosis patients with g551d-cftr treated with ivacaftor. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 60, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Iglewski, B.H.; Kabat, D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc. Natl. Acad. Sci. USA 1975, 72, 2284–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianciotto, N.P. Type II secretion: A protein secretion system for all seasons. Trends Microbiol. 2005, 13, 581–588. [Google Scholar] [CrossRef]
- Basso, P.; Ragno, M.; Elsen, S.; Reboud, E.; Golovkine, G.; Bouillot, S.; Huber, P.; Lory, S.; Faudry, E.; Attree, I. Pseudomonas aeruginosa pore-forming exolysin and type iv pili cooperate to induce host cell lysis. mBio 2017, 8, e02250-16. [Google Scholar] [CrossRef] [Green Version]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin a of Pseudomonas aeruginosa at 3.0-angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef] [Green Version]
- Rasper, D.M.; Merrill, A.R. Evidence for the modulation of Pseudomonas aeruginosa exotoxin a-induced pore formation by membrane surface charge density. Biochemistry 1994, 33, 12981–12989. [Google Scholar] [CrossRef]
- Zalman, L.S.; Wisnieski, B.J. Characterization of the insertion of Pseudomonas exotoxin a into membranes. Infect. Immun. 1985, 50, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.J.; Rijneveld, A.W.; Florquin, S.; Speelman, P.; Van Deventer, S.J.; van der Poll, T. Impairment of host defense by exotoxin a in Pseudomonas aeruginosa pneumonia in mice. J. Med. Microbiol. 2001, 50, 822–827. [Google Scholar] [CrossRef] [Green Version]
- Wieland, C.W.; Siegmund, B.; Senaldi, G.; Vasil, M.L.; Dinarello, C.A.; Fantuzzi, G. Pulmonary inflammation induced by Pseudomonas aeruginosa lipopolysaccharide, phospholipase c, and exotoxin a: Role of interferon regulatory factor 1. Infect. Immun. 2002, 70, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Bourke, W.J.; O’Connor, C.M.; FitzGerald, M.X.; McDonnell, T.J. Pseudomonas aeruginosa exotoxin a induces pulmonary endothelial cytotoxicity: Protection by dibutyryl-cAMP. Eur. Respir. J. 1994, 7, 1754–1758. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, J.T.; Sun, J. Pseudomonas aeruginosa exo S and exo T. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 79–92. [Google Scholar] [CrossRef]
- Kulich, S.M.; Yahr, T.L.; Mende-Mueller, L.M.; Barbieri, J.T.; Frank, D.W. Cloning the structural gene for the 49-kda form of exoenzyme s (exo S) from pseudomonas aeruginosa strain 388. J. Biol. Chem. 1994, 269, 10431–10437. [Google Scholar]
- Iglewski, B.H.; Sadoff, J.; Bjorn, M.J.; Maxwell, E.S. Pseudomonas aeruginosa exoenzyme s: An adenosine diphosphate ribosyltransferase distinct from toxin a. Proc. Natl. Acad. Sci. USA 1978, 75, 3211–3215. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.; Ehrmantraut, M.E.; Banwart, B.D.; Frank, D.W.; Barbieri, J.T. Sera from adult patients with cystic fibrosis contain antibodies to Pseudomonas aeruginosa type III apparatus. Infect. Immun. 2001, 69, 1185–1188. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Shaver, C.M.; Hauser, A.R. Relative contributions of Pseudomonas aeruginosa exo U, exo S, and exo T to virulence in the lung. Infect. Immun. 2004, 72, 6969–6977. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.E.; Hwang, W.S.; Shahrabadi, M.S.; Que, J.U. Alteration of pulmonary structure by Pseudomonas aeruginosa exoenzyme s. J. Med. Microbiol. 1988, 26, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Ganter, M.; Roux, J.; Su, G.; Lynch, S.; Deutschman, C.; Weiss, Y.G.; Christiaans, S.C.; Miyazawa, B.; Kipnis, E.; Wiener-Kronish, J.P.; et al. Role of small GTPases and αvβ5 integrin in P. aeruginosa-induced increase in lung permeability. Am. J. Respir. Cell Mol. Biol. 2009, 40, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Huber, P.; Bouillot, S.; Elsen, S.; Attree, I. Sequential inactivation of rho GTPases and lim kinase by Pseudomonas aeruginosa toxins exos and exot leads to endothelial monolayer breakdown. Cell. Mol. Life Sci. CMLS 2014, 71, 1927–1941. [Google Scholar] [CrossRef]
- Rangel, S.M.; Logan, L.K.; Hauser, A.R. The ADP-ribosyltransferase domain of the effector protein Exo S inhibits phagocytosis of Pseudomonas aeruginosa during pneumonia. mBio 2014, 5, e01080-14. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Stack, D.; Bell, C.; Wong, E.; Neely, G.G.; Krutzik, S.; Miyake, K.; Kubes, P.; Zbytnuik, L.D.; Ma, L.L.; et al. Different domains of Pseudomonas aeruginosa exoenzyme s activate distinct TLRs. J. Immunol. 2004, 173, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Yahr, T.L.; Mende-Mueller, L.M.; Friese, M.B.; Frank, D.W. Identification of type III secreted products of the Pseudomonas aeruginosa exoenzyme s regulon. J. Bacteriol. 1997, 179, 7165–7168. [Google Scholar] [CrossRef] [Green Version]
- Geiser, T.K.; Kazmierczak, B.I.; Garrity-Ryan, L.K.; Matthay, M.A.; Engel, J.N. Pseudomonas aeruginosa exo T inhibits in vitro lung epithelial wound repair. Cell. Microbiol. 2001, 3, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Garrity-Ryan, L.; Kazmierczak, B.; Kowal, R.; Comolli, J.; Hauser, A.; Engel, J.N. The arginine finger domain of exo T contributes to actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect. Immun. 2000, 68, 7100–7113. [Google Scholar] [CrossRef] [Green Version]
- Shafikhani, S.H.; Morales, C.; Engel, J. The Pseudomonas aeruginosa type III secreted toxin Exo T is necessary and sufficient to induce apoptosis in epithelial cells. Cell. Microbiol. 2008, 10, 994–1007. [Google Scholar] [CrossRef]
- Sawa, T. The molecular mechanism of acute lung injury caused by Pseudomonas aeruginosa: From bacterial pathogenesis to host response. J. Intensive Care 2014, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Vourc’h, M.; Roquilly, A.; Broquet, A.; David, G.; Hulin, P.; Jacqueline, C.; Caillon, J.; Retiere, C.; Asehnoune, K. Exoenzyme T plays a pivotal role in the IFN-gamma production after Pseudomonas challenge in IL-12 primed natural killer cells. Front. Immunol. 2017, 8, 1283. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Frank, D.W.; Hillard, C.J.; Feix, J.B.; Pankhaniya, R.R.; Moriyama, K.; Finck-Barbançon, V.; Buchaklian, A.; Lei, M.; Long, R.M.; et al. The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, exou. EMBO J. 2003, 22, 2959–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, J.; Balachandran, P. Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microbiol. 2009, 12, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Sitkiewicz, I.; Stockbauer, K.E.; Musser, J.M. Secreted bacterial phospholipase A2 enzymes: Better living through phospholipolysis. Trends Microbiol. 2007, 15, 63–69. [Google Scholar] [CrossRef]
- Galle, M.; Jin, S.; Bogaert, P.; Haegman, M.; Vandenabeele, P.; Beyaert, R. The Pseudomonas aeruginosa type III secretion system has an exotoxin s/t/y independent pathogenic role during acute lung infection. PLoS ONE 2012, 7, e41547. [Google Scholar] [CrossRef]
- Finck-Barbancon, V.; Goranson, J.; Zhu, L.; Sawa, T.; Wiener-Kronish, J.P.; Fleiszig, S.M.; Wu, C.; Mende-Mueller, L.; Frank, D.W. Exo U expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 1997, 25, 547–557. [Google Scholar] [CrossRef]
- Machado, G.B.; de Assis, M.C.; Leao, R.; Saliba, A.M.; Silva, M.C.; Suassuna, J.H.; de Oliveira, A.V.; Plotkowski, M.C. Exo U-induced vascular hyperpermeability and platelet activation in the course of experimental Pseudomonas aeruginosa pneumosepsis. Shock 2010, 33, 315–321. [Google Scholar] [CrossRef]
- Diaz, M.H.; Hauser, A.R. Pseudomonas aeruginosa cytotoxin exo U is injected into phagocytic cells during acute pneumonia. Infect. Immun. 2010, 78, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- De Lima, C.D.; Calegari-Silva, T.C.; Pereira, R.M.; Santos, S.A.; Lopes, U.G.; Plotkowski, M.C.; Saliba, A.M. Exo U activates NF-kappaB and increases IL-8/KC secretion during Pseudomonas aeruginosa infection. PLoS ONE 2012, 7, e41772. [Google Scholar] [CrossRef]
- Yahr, T.L.; Goranson, J.; Frank, D.W. Exoenzyme s of Pseudomonas aeruginosa is secreted by a type iii pathway. Mol. Microbiol. 1996, 22, 991–1003. [Google Scholar] [CrossRef]
- Yahr, T.L.; Vallis, A.J.; Hancock, M.K.; Barbieri, J.T.; Frank, D.W. Exo Y, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad Sci. USA 1998, 95, 13899–13904. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Kumar, P.; Bhatnagar, R. The adenylate cyclase toxins. Crit. Rev. Microbiol. 2004, 30, 187–196. [Google Scholar] [CrossRef]
- Kloth, C.; Schirmer, B.; Munder, A.; Stelzer, T.; Rothschuh, J.; Seifert, R. The role of Pseudomonas aeruginosa exo Y in an acute mouse lung infection model. Toxins 2018, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- Cowell, B.A.; Evans, D.J.; Fleiszig, S.M. Actin cytoskeleton disruption by exo Y and its effects on Pseudomonas aeruginosa invasion. FEMS Microbiol. Lett. 2005, 250, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Stevens, T.C.; Ochoa, C.D.; Morrow, K.A.; Robson, M.J.; Prasain, N.; Zhou, C.; Alvarez, D.F.; Frank, D.W.; Balczon, R.; Stevens, T. The Pseudomonas aeruginosa exoenzyme Y impairs endothelial cell proliferation and vascular repair following lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L915–L924. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, C.D.; Alexeyev, M.; Pastukh, V.; Balczon, R.; Stevens, T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J. Biol. Chem. 2012, 287, 25407–25418. [Google Scholar] [CrossRef] [Green Version]
- Balczon, R.; Prasain, N.; Ochoa, C.; Prater, J.; Zhu, B.; Alexeyev, M.; Sayner, S.; Frank, D.W.; Stevens, T. Pseudomonas aeruginosa exotoxin Y-mediated tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS ONE 2013, 8, e74343. [Google Scholar] [CrossRef]
- Sayner, S.L.; Frank, D.W.; King, J.; Chen, H.; VandeWaa, J.; Stevens, T. Paradoxical camp-induced lung endothelial hyperpermeability revealed by pseudomonas aeruginosa exo Y. Circ. Res. 2004, 95, 196–203. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Zhou, Y.; Liu, F.; Liu, H.; Tan, H.; Jin, S.; Wu, W.; Ge, B. Bacterial nucleotidyl cyclase inhibits the host innate immune response by suppressing tak1 activation. Infect. Immun. 2017, 85, e00239-17. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Kim, Y.J.; Shin, H.; Ha, U.H. T3ss effector exo Y reduces inflammasome-related responses by suppressing bacterial motility and delaying activation of NF-kappab and caspase-1. FEBS J. 2017, 284, 3392–3403. [Google Scholar] [CrossRef] [Green Version]
- Elsen, S.; Huber, P.; Bouillot, S.; Coute, Y.; Fournier, P.; Dubois, Y.; Timsit, J.F.; Maurin, M.; Attree, I. A type III secretion negative clinical strain of Pseudomonas aeruginosa employs a two-partner secreted exolysin to induce hemorrhagic pneumonia. Cell Host Microbe 2014, 15, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Reboud, E.; Elsen, S.; Bouillot, S.; Golovkine, G.; Basso, P.; Jeannot, K.; Attree, I.; Huber, P. Phenotype and toxicity of the recently discovered exla-positive Pseudomonas aeruginosa strains collected worldwide. Environ. Microbiol. 2016, 18, 3425–3439. [Google Scholar] [CrossRef] [PubMed]
- Bouillot, S.; Munro, P.; Gallet, B.; Reboud, E.; Cretin, F.; Golovkine, G.; Schoehn, G.; Attree, I.; Lemichez, E.; Huber, P. Pseudomonas aeruginosa exolysin promotes bacterial growth in lungs, alveolar damage and bacterial dissemination. Sci. Rep. 2017, 7, 2120. [Google Scholar] [CrossRef]
- Reboud, E.; Bouillot, S.; Patot, S.; Beganton, B.; Attree, I.; Huber, P. Pseudomonas aeruginosa exla and Serratia marcescens shla trigger cadherin cleavage by promoting calcium influx and ADAM10 activation. PLoS Pathog. 2017, 13, e1006579. [Google Scholar] [CrossRef]
- Klockgether, J.; Tummler, B. Recent advances in understanding Pseudomonas aeruginosa as a pathogen. F1000Res 2017, 6, 1261. [Google Scholar] [CrossRef]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Duong, F.; Bonnet, E.; Geli, V.; Lazdunski, A.; Murgier, M.; Filloux, A. The aprx protein of Pseudomonas aeruginosa: A new substrate for the apr type i secretion system. Gene 2001, 262, 147–153. [Google Scholar] [CrossRef]
- Kida, Y.; Higashimoto, Y.; Inoue, H.; Shimizu, T.; Kuwano, K. A novel secreted protease from Pseudomonas aeruginosa activates NF-kappaB through protease-activated receptors. Cell. Microbiol. 2008, 10, 1491–1504. [Google Scholar] [CrossRef]
- Zhang, L.; Conway, J.F.; Thibodeau, P.H. Calcium-induced folding and stabilization of the pseudomonas aeruginosa alkaline protease. J. Biol. Chem. 2012, 287, 4311–4322. [Google Scholar] [CrossRef] [Green Version]
- Caballero, A.R.; Moreau, J.M.; Engel, L.S.; Marquart, M.E.; Hill, J.M.; O’Callaghan, R.J. Pseudomonas aeruginosa protease iv enzyme assays and comparison to other Pseudomonas proteases. Anal. Biochem. 2001, 290, 330–337. [Google Scholar] [CrossRef]
- Leidal, K.G.; Munson, K.L.; Johnson, M.C.; Denning, G.M. Metalloproteases from Pseudomonas aeruginosa degrade human rantes, mcp-1, and ena-78. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2003, 23, 307–318. [Google Scholar] [CrossRef]
- Guyot, N.; Bergsson, G.; Butler, M.W.; Greene, C.M.; Weldon, S.; Kessler, E.; Levine, R.L.; O’Neill, S.J.; Taggart, C.C.; McElvaney, N.G. Functional study of elafin cleaved by Pseudomonas aeruginosa metalloproteinases. Biol. Chem. 2010, 391, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterworth, M.B.; Zhang, L.; Heidrich, E.M.; Myerburg, M.M.; Thibodeau, P.H. Activation of the epithelial sodium channel (enac) by the alkaline protease from Pseudomonas aeruginosa. J. Biol. Chem. 2012, 287, 32556–32565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, S. The role of bacterial proteases in the pathogenesis of cystic fibrosis. Am. J. Respir. Crit. Care Med. 1994, 150, S118–S122. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57Bl/10sccr mice: Mutations in TLR4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C. Immunology. Sensing endotoxins from within. Science 2013, 341, 1184–1185. [Google Scholar] [CrossRef] [Green Version]
- Hachim, M.Y.; Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Pyroptosis: The missing puzzle among innate and adaptive immunity crosstalk. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Nova, Z.; Skovierova, H.; Calkovska, A. Alveolar-capillary membrane-related pulmonary cells as a target in endotoxin-induced acute lung injury. Int. J. Mol. Sci. 2019, 20, 831. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Huber, M.; Kalis, C.; Keck, S.; Galanos, C.; et al. CD14 is required for MYD88-independent LPS signaling. Nat. Immunol. 2005, 6, 565–570. [Google Scholar] [CrossRef]
- Wieland, C.W.; Florquin, S.; Maris, N.A.; Hoebe, K.; Beutler, B.; Takeda, K.; Akira, S.; van der Poll, T. The myd88-dependent, but not the MYD88-independent, pathway of TLR4 signaling is important in clearing nontypeable Hemophilus influenzae from the mouse lung. J. Immunol. 2005, 175, 6042–6049. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Su, X.; Pan, P.; Zhang, L.; Hu, Y.; Tan, H.; Wu, D.; Liu, B.; Li, H.; Li, H.; et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 2016, 6, 37252. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Non-canonical inflammasome activation by intracellular lps independent of tlr4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. Gsdmd p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, J.B.; Pier, G.B. Pseudomonas aeruginosa lipopolysaccharides and pathogenesis. Trends Microbiol. 1996, 4, 490–494. [Google Scholar] [CrossRef]
- Boncoeur, E.; Tardif, V.; Tessier, M.C.; Morneau, F.; Lavoie, J.; Gendreau-Berthiaume, E.; Grygorczyk, R.; Dagenais, A.; Berthiaume, Y. Modulation of epithelial sodium channel activity by lipopolysaccharide in alveolar type ii cells: Involvement of purinergic signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L417–L426. [Google Scholar] [CrossRef]
- Migneault, F.; Boncoeur, E.; Morneau, F.; Pascariu, M.; Dagenais, A.; Berthiaume, Y. Cycloheximide and lipopolysaccharide downregulate alpha-ENaC via different mechanisms in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L747–L755. [Google Scholar] [CrossRef] [Green Version]
- Buyck, J.M.; Verriere, V.; Benmahdi, R.; Higgins, G.; Guery, B.; Matran, R.; Harvey, B.J.; Faure, K.; Urbach, V.P. aeruginosa LPS stimulates calcium signaling and chloride secretion via cftr in human bronchial epithelial cells. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2013, 12, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Baines, D.L.; Baker, E.H. Chapter 3—Glucose transport and homeostasis in lung epithelia. In Lung epithelial Biology in the Pathogenesis of Pulmonary Disease; Sidhaye, V.K., Koval, M., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 33–57. [Google Scholar] [CrossRef]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef]
- Li, W.; Yan, F.; Zhou, H.; Lin, X.; Wu, Y.; Chen, C.; Zhou, N.; Chen, Z.; Li, J.D.; Shen, H.P. aeruginosa lipopolysaccharide-induced muc5ac and clca3 expression is partly through duox1 in vitro and in vivo. PLoS ONE 2013, 8, e63945. [Google Scholar] [CrossRef] [Green Version]
- Eutamene, H.; Theodorou, V.; Schmidlin, F.; Tondereau, V.; Garcia-Villar, R.; Salvador-Cartier, C.; Chovet, M.; Bertrand, C.; Bueno, L. LPS-induced lung inflammation is linked to increased epithelial permeability: Role of mlck. Eur. Respir. J. 2005, 25, 789–796. [Google Scholar] [CrossRef]
- Le, B.V.; Khorsi-Cauet, H.; Bach, V.; Gay-Queheillard, J. Mast cells mediate Pseudomonas aeruginosa lipopolysaccharide-induced lung inflammation in rat. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2012, 31, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Thorley, A.J.; Ford, P.A.; Giembycz, M.A.; Goldstraw, P.; Young, A.; Tetley, T.D. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type ii epithelial cells and macrophages. J. Immunol. 2007, 178, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Su, L.H.; Chiu, C.H. Salmonella: Clinical importance and evolution of nomenclature. Chang. Gung Med. J. 2007, 30, 210–219. [Google Scholar] [PubMed]
- Ibarra, J.A.; Steele-Mortimer, O. Salmonella—The ultimate insider. Salmonella virulence factors that modulate intracellular survival. Cell. Microbiol. 2009, 11, 1579–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, L.; Moffatt, C.R.M.; Fearnley, E.; Miller, M.; Gregory, J.; Sloan-Gardner, T.S.; Polkinghorne, B.G.; Bell, R.; Franklin, N.; Williamson, D.A.; et al. The epidemiology of Salmonella enterica outbreaks in Australia, 2001–2016. Front. Sustain. Food Syst. 2018, 2, 86. [Google Scholar] [CrossRef]
- Dodrill, M.W.; Fedan, J.S. Lipopolysaccharide hyperpolarizes guinea pig airway epithelium by increasing the activities of the epithelial Na(+) channel and the Na(+)-K(+) pump. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L550–L558. [Google Scholar] [CrossRef] [Green Version]
- Dodrill, M.W.; Beezhold, D.H.; Meighan, T.; Kashon, M.L.; Fedan, J.S. Lipopolysaccharide increases Na+,K+-pump, but not ENaC, expression in guinea-pig airway epithelium. Eur. J. Pharmacol. 2011, 651, 176–186. [Google Scholar] [CrossRef]
- Wheeldon, E.B.; Walker, M.E.; Murphy, D.J.; Turner, C.R. Intratracheal aerosolization of endotoxin in the rat: A model of the adult respiratory distress syndrome (ards). Lab. Anim. 1992, 26, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.; Yoon, J.; Hovde, C.J. A brief overview of Escherichia coli o157:H7 and its plasmid o157. J. Microbiol Biotechnol 2010, 20, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Poolman, J.T. Escherichia coli. In International Encyclopedia of Public Health, 2nd ed.; Quah, S.R., Ed.; Academic Press: Oxford, UK, 2017; pp. 585–593. [Google Scholar] [CrossRef]
- Wu, H.; Santoni-Rugiu, E.; Ralfkiaer, E.; Porse, B.T.; Moser, C.; Hoiby, N.; Borregaard, N.; Cowland, J.B. Lipocalin 2 is protective against E coli pneumonia. Respir. Res. 2010, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Yayan, J.; Ghebremedhin, B.; Rasche, K. No development of imipenem resistance in pneumonia caused by Escherichia coli. Medicine 2015, 94, e1020. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kuwano, K.; Kunitake, R.; Hagimoto, N.; Miyazaki, H.; Kaneko, Y.; Kawasaki, M.; Maeyama, T.; Hara, N. Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int. Arch. Allergy Immunol. 1998, 117, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Li, C.Y.; Tong, J.; Zhang, W.; Wang, D.X. Regulation of ENaC-mediated alveolar fluid clearance by insulin via pi3k/akt pathway in lps-induced acute lung injury. Respir. Res. 2012, 13, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.; Zhang, L.; Nguyen, C.; Vogel, S.N.; Goldblum, S.E.; Blackwelder, W.C.; Cross, A.S. Neuraminidase reprograms lung tissue and potentiates lipopolysaccharide-induced acute lung injury in mice. J. Immunol. 2013, 191, 4828–4837. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Wang, Y.; Li, W.; Mu, Q.; Li, H.; Yao, H.; Zhang, H. Protective effects of isofraxidin against lipopolysaccharide-induced acute lung injury in mice. Int. Immunopharmacol. 2015, 24, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Wang, Z.; Tian, J.; Liu, T.; Zhou, H. Glycyrrhizin inactivates toll-like receptor (TLR) signaling pathway to reduce lipopolysaccharide-induced acute lung injury by inhibiting TLR2. J. Cell Physiol. 2019, 234, 4597–4607. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Sun, X.; Hou, Y.; Yang, X.; Chen, H.; Zhang, P.; Wu, J. Protective effect of oxytocin on LPS-induced acute lung injury in mice. Sci. Rep. 2019, 9, 2836. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Kajikawa, O.; Martin, T.R.; Sharar, S.R.; Harlan, J.M.; Winn, R.K. The role of leukocyte emigration and IL-8 on the development of lipopolysaccharide-induced lung injury in rabbits. J. Immunol. 1998, 161, 5704–5709. [Google Scholar]
- Bosmann, M.; Grailer, J.J.; Russkamp, N.F.; Ruemmler, R.; Zetoune, F.S.; Sarma, J.V.; Ward, P.A. Cd11c+ alveolar macrophages are a source of IL-23 during lipopolysaccharide-induced acute lung injury. Shock 2013, 39, 447–452. [Google Scholar] [CrossRef] [Green Version]
- O’Brien-Ladner, A.R.; Nelson, M.E.; Cowley, B.D., Jr.; Bailey, K.; Wesselius, L.J. Hyperoxia amplifies TNF-alpha production in LPS-stimulated human alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 1995, 12, 275–279. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to irf-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.Y.; Shin, M.H.; Chung, K.S.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; Kim, Y.S.; Kim, S.K.; Chang, J.; Park, M.S. Epha2 receptor signaling mediates inflammatory responses in lipopolysaccharide-induced lung injury. Tuberc. Respir. Dis. 2015, 78, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.J.; Nie, Y.C.; Lin, F.; Li, P.B.; Liu, M.H.; Xie, C.S.; Long, C.F.; Su, W.W. Biphasic modulation of alpha-ENaC expression by lipopolysaccharide in vitro and in vivo. Mol. Med. Rep. 2014, 10, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ferrari, J.D.; Cao, Y.; Ramirez, M.I.; Jones, M.R.; Quinton, L.J.; Mizgerd, J.P. Type i alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 2012, 189, 2450–2459. [Google Scholar] [CrossRef]
- Hauber, H.P.; Goldmann, T.; Vollmer, E.; Wollenberg, B.; Hung, H.L.; Levitt, R.C.; Zabel, P. LPS-induced mucin expression in human sinus mucosa can be attenuated by hclca inhibitors. J. Endotoxin Res. 2007, 13, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Essler, M.; Staddon, J.M.; Weber, P.C.; Aepfelbacher, M. Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of rho/rho kinase signaling. J. Immunol. 2000, 164, 6543–6549. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Xing, J.; Fu, P.; Yakubov, B.; Dubrovskyi, O.; Fortune, J.A.; Klibanov, A.M.; Birukov, K.G. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: Role of pak1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L652–L663. [Google Scholar] [CrossRef]
- Hoyt, D.G.; Mannix, R.J.; Rusnak, J.M.; Pitt, B.R.; Lazo, J.S. Collagen is a survival factor against LPS-induced apoptosis in cultured sheep pulmonary artery endothelial cells. Am. J. Physiol. 1995, 269, L171–L177. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80, 2012–2020. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.M.; Kellner, M.; Wang, T.; Lu, Q.; Sun, X.; Zemskov, E.A.; Noonepalle, S.; Kangath, A.; Kumar, S.; Gonzalez-Garay, M.; et al. LPS-induced acute lung injury involves nf-κb-mediated downregulation of sox18. Am. J. Respir. Cell Mol. Biol. 2018, 58, 614–624. [Google Scholar] [CrossRef]
- Welch, R.A. Uropathogenic Escherichia coli-Associated Exotoxins. Microbiol Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, F.; Kiss, L.; Grimminger, F.; Mayer, K.; Grandel, U.; Seeger, W.; Bieniek, E.; Sibelius, U. E. coli hemolysin-induced lipid mediator metabolism in alveolar macrophages: Impact of eicosapentaenoic acid. Am. J. Physiol Lung Cell Mol. Physiol. 2000, 279, L100–L109. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A. RTX toxin structure and function: A story of numerous anomalies and few analogies in toxin biology. Curr. Top. Microbiol. Immunol. 2001, 257, 85–111. [Google Scholar] [PubMed]
- Munro, P.; Flatau, G.; Doye, A.; Boyer, L.; Oregioni, O.; Mege, J.L.; Landraud, L.; Lemichez, E. Activation and proteasomal degradation of rho GTPases by cytotoxic necrotizing factor-1 elicit a controlled inflammatory response. J. Biol. Chem. 2004, 279, 35849–35857. [Google Scholar] [CrossRef] [Green Version]
- Hofman, P.; Le Negrate, G.; Mograbi, B.; Hofman, V.; Brest, P.; Alliana-Schmid, A.; Flatau, G.; Bouquet, P.; Rossi, B. Escherichia coli cytotoxic necrotizing factor-1 (CNF-1) increases the adherence to epithelia and the oxidative burst of human polymorphonuclear leukocytes but decreases bacteria phagocytosis. J. Leukoc. Biol. 2000, 68, 522–528. [Google Scholar]
- Parreira, V.R.; Gyles, C.L. A novel pathogenicity island integrated adjacent to the thrW tRNA gene of avian pathogenic Escherichia coli encodes a vacuolating autotransporter toxin. Infect. Immun. 2003, 71, 5087–5096. [Google Scholar] [CrossRef] [Green Version]
- Finger, H.; Von Koenig, C. Bordetella. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. Available online: https://www.ncbi.nlm.nih.gov/books/NBK7813/ (accessed on 31 March 2020).
- Pittman, M. Pertussis toxin: The cause of the harmful effects and prolonged immunity of whooping cough. A hypothesis. Rev. Infect. Dis. 1979, 1, 401–412. [Google Scholar] [CrossRef]
- Monack, D.; Munoz, J.J.; Peacock, M.G.; Black, W.J.; Falkow, S. Expression of pertussis toxin correlates with pathogenesis in bordetella species. J. Infect. Dis 1989, 159, 205–210. [Google Scholar] [CrossRef]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Scanlon, K.; Skerry, C.; Carbonetti, N. Association of pertussis toxin with severe pertussis disease. Toxins 2019, 11, 373. [Google Scholar] [CrossRef] [Green Version]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heininger, U. 32—Pertussis and other Bordetella infections of the respiratory tract. In Kendig’s Disorders of the Respiratory Tract in Children, 9th ed.; Wilmott, R.W., Deterding, R., Li, A., Ratjen, F., Sly, P., Zar, H.J., Bush, A., Eds.; Elsevier: Philadelphia, PA, USA, 2019; pp. 528–534.e522. [Google Scholar]
- Teter, K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins 2019, 11, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkel, A.R.; Pauza, C.D. Pertussis toxin treatment in vivo reduces surface expression of the adhesion integrin leukocyte function antigen-1 (LFA-1). Cell Adhes. Commun. 1999, 7, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Connelly, C.E.; Sun, Y.; Carbonetti, N.H. Pertussis toxin exacerbates and prolongs airway inflammatory responses during Bordetella pertussis infection. Infect. Immun. 2012, 80, 4317–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonetti, N.H.; Artamonova, G.V.; Mays, R.M.; Worthington, Z.E. Pertussis toxin plays an early role in respiratory tract colonization by bordetella pertussis. Infect. Immun 2003, 71, 6358–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.G.; Wang, P.; Schaphorst, K.L.; Becker, P.M.; Borbiev, T.; Liu, F.; Birukova, A.; Jacobs, K.; Bogatcheva, N.; Verin, A.D. Critical involvement of p38 MAP kinase in pertussis toxin-induced cytoskeletal reorganization and lung permeability. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1064–1076. [Google Scholar] [CrossRef]
- Dudek, S.M.; Camp, S.M.; Chiang, E.T.; Singleton, P.A.; Usatyuk, P.V.; Zhao, Y.; Natarajan, V.; Garcia, J.G. Pulmonary endothelial cell barrier enhancement by fty720 does not require the S1P1 receptor. Cell Signal. 2007, 19, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Patterson, C.E.; Stasek, J.E.; Schaphorst, K.L.; Davis, H.W.; Garcia, J.G. Mechanisms of pertussis toxin-induced barrier dysfunction in bovine pulmonary artery endothelial cell monolayers. Am. J. Physiol. 1995, 268, L926–L934. [Google Scholar] [CrossRef]
- Tsan, M.F.; Cao, X.; White, J.E.; Sacco, J.; Lee, C.Y. Pertussis toxin-induced lung edema. Role of manganese superoxide dismutase and protein kinase c. Am. J. Respir. Cell Mol. Biol. 1999, 20, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Clerch, L.B.; Neithardt, G.; Spencer, U.; Melendez, J.A.; Massaro, G.D.; Massaro, D. Pertussis toxin treatment alters manganese superoxide dismutase activity in lung. Evidence for lung oxygen toxicity in air-breathing rats. J. Clin. Investig. 1994, 93, 2482–2489. [Google Scholar] [CrossRef]
- Saha, C.; Nigam, S.K.; Denker, B.M. Involvement of Galphai2 in the maintenance and biogenesis of epithelial cell tight junctions. J. Biol. Chem. 1998, 273, 21629–21633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirimanjeswara, G.S.; Agosto, L.M.; Kennett, M.J.; Bjornstad, O.N.; Harvill, E.T. Pertussis toxin inhibits neutrophil recruitment to delay antibody-mediated clearance of bordetella pertussis. J. Clin. Investig. 2005, 115, 3594–3601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasen, C.; Carbonetti, N.H. Pertussis toxin inhibits early chemokine production to delay neutrophil recruitment in response to Bordetella pertussis respiratory tract infection in mice. Infect. Immun. 2008, 76, 5139–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasen, C.; Powell, D.A.; Carbonetti, N.H. Pertussis toxin stimulates IL-17 production in response to bordetella pertussis infection in mice. PLoS ONE 2009, 4, e7079. [Google Scholar] [CrossRef]
- Carbonetti, N.H.; Artamonova, G.V.; Van Rooijen, N.; Ayala, V.I. Pertussis toxin targets airway macrophages to promote Bordetella pertussis infection of the respiratory tract. Infect. Immun. 2007, 75, 1713–1720. [Google Scholar] [CrossRef] [Green Version]
- Murray, E.L.; Nieves, D.; Bradley, J.S.; Gargas, J.; Mason, W.H.; Lehman, D.; Harriman, K.; Cherry, J.D. Characteristics of severe Bordetella pertussis infection among infants ≤90 days of age admitted to pediatric intensive care units—Southern California, september 2009–june 2011. J. Pediatric Infect. Dis. Soc. 2013, 2, 1–6. [Google Scholar] [CrossRef]
- Vojtova-Vodolanova, J.; Basler, M.; Osicka, R.; Knapp, O.; Maier, E.; Cerny, J.; Benada, O.; Benz, R.; Sebo, P. Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 2831–2843. [Google Scholar] [CrossRef]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.; Kulkarni, N.N.; Asbjarnarson, A.; Linhartova, I.; Osicka, R.; Sebo, P.; Gudmundsson, G.H. Bordetella pertussis adenylate cyclase toxin disrupts functional integrity of bronchial epithelial layers. Infect. Immun. 2018, 86, e00445-17. [Google Scholar] [CrossRef] [Green Version]
- Hanski, E. Invasive adenylate cyclase toxin of bordetella pertussis. Trends Biochem. Sci. 1989, 14, 459–463. [Google Scholar] [CrossRef]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic activity of adenylate cyclase toxin from bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finley, N.L. Revealing how an adenylate cyclase toxin uses bait and switch tactics in its activation. PLoS Biol. 2018, 16, e2005356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonetti, N.H.; Artamonova, G.V.; Andreasen, C.; Bushar, N. Pertussis toxin and adenylate cyclase toxin provide a one-two punch for establishment of bordetella pertussis infection of the respiratory tract. Infect. Immun. 2005, 73, 2698–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, G.; Bianco, M.; Ausiello, C.M. The virulence factors of Bordetella pertussis: Talented modulators of host immune response. Arch. Immunol. Et Ther. Exp. 2013, 61, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Donato, G.M.; Goldsmith, C.S.; Paddock, C.D.; Eby, J.C.; Gray, M.C.; Hewlett, E.L. Delivery of Bordetella pertussis adenylate cyclase toxin to target cells via outer membrane vesicles. FEBS Lett. 2012, 586, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimova, G.; Fayolle, C.; Gmira, S.; Ullmann, A.; Leclerc, C.; Ladant, D. Charge-dependent translocation of Bordetella pertussis adenylate cyclase toxin into eukaryotic cells: Implication for the in vivo delivery of CD8(+) t cell epitopes into antigen-presenting cells. Proc. Natl. Acad. Sci. USA 1998, 95, 12532–12537. [Google Scholar] [CrossRef] [Green Version]
- Ladant, D.; Ullmann, A. Bordetella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Hewlett, E.L.; Donato, G.M.; Gray, M.C. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: More than just making cyclic AMP! Mol. Microbiol. 2006, 59, 447–459. [Google Scholar] [CrossRef]
- Khelef, N.; Zychlinsky, A.; Guiso, N. Bordetella pertussis induces apoptosis in macrophages: Role of adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 4064–4071. [Google Scholar] [CrossRef] [Green Version]
- Khelef, N.; Gounon, P.; Guiso, N. Internalization of Bordetella pertussis adenylate cyclase-haemolysin into endocytic vesicles contributes to macrophage cytotoxicity. Cell. Microbiol. 2001, 3, 721–730. [Google Scholar] [CrossRef]
- Boyd, A.P.; Ross, P.J.; Conroy, H.; Mahon, N.; Lavelle, E.C.; Mills, K.H. Bordetella pertussis adenylate cyclase toxin modulates innate and adaptive immune responses: Distinct roles for acylation and enzymatic activity in immunomodulation and cell death. J. Immunol. 2005, 175, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueirard, P.; Druilhe, A.; Pretolani, M.; Guiso, N. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect. Immun. 1998, 66, 1718–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, C.; Gómez-Bilbao, G.; Ostolaza, H. Bordetella adenylate cyclase toxin promotes calcium entry into both cd11b+ and cd11b- cells through cAMP-dependent l-type-like calcium channels. J. Biol. Chem. 2010, 285, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Bullon, D.; Uribe, K.B.; Largo, E.; Guembelzu, G.; Garcia-Arribas, A.B.; Martin, C.; Ostolaza, H. Membrane permeabilization by Bordetella adenylate cyclase toxin involves pores of tunable size. Biomolecules 2019, 9, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, K.A.; Chatterjee, K. Anthrax. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507773/ (accessed on 31 March 2020).
- Brooks, G.F.; Jawetz, E.; Melnick, J.L.; Adelberg, E.A. Jawetz, Melnick and Adelberg’s Medical Microbiology; McGraw-Hill Medical: New York, NY, USA, 2013. [Google Scholar]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. MMBR 2004, 68, 373–402, table of contents. [Google Scholar] [CrossRef] [Green Version]
- Bann, J.G. Anthrax toxin protective antigen—Insights into molecular switching from prepore to pore. Protein Sci. 2012, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fabre, L.; Santelli, E.; Mountassif, D.; Donoghue, A.; Biswas, A.; Blunck, R.; Hanein, D.; Volkmann, N.; Liddington, R.; Rouiller, I. Structure of anthrax lethal toxin prepore complex suggests a pathway for efficient cell entry. J. Gen. Physiol. 2016, 148, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Friebe, S.; van der Goot, F.G.; Burgi, J. The ins and outs of anthrax toxin. Toxins 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef]
- Leuber, M.; Kronhardt, A.; Tonello, F.; Dal Molin, F.; Benz, R. Binding of N-terminal fragments of anthrax edema factor (ef(n)) and lethal factor (lf(n)) to the protective antigen pore. Biochim. Et Biophys. Acta 2008, 1778, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, R.C. Bacillus anthracis. J. Clin. Pathol. 2003, 56, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, A.K. Anthrax: A disease of biowarfare and public health importance. World J. Clin. Cases 2015, 3, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.N.; Quesnel-Hellmann, A.; Cleret, A.; Vidal, D.R. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell. Microbiol. 2007, 9, 555–565. [Google Scholar] [CrossRef]
- Goossens, P.L.; Tournier, J.N. Crossing of the epithelial barriers by Bacillus anthracis: The known and the unknown. Front. Microbiol 2015, 6, 1122. [Google Scholar] [CrossRef] [Green Version]
- Guidi-Rontani, C. The alveolar macrophage: The trojan horse of bacillus anthracis. Trends Microbiol. 2002, 10, 405–409. [Google Scholar] [CrossRef]
- Cleret, A.; Quesnel-Hellmann, A.; Vallon-Eberhard, A.; Verrier, B.; Jung, S.; Vidal, D.; Mathieu, J.; Tournier, J.N. Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J. Immunol. 2007, 178, 7994–8001. [Google Scholar] [CrossRef] [Green Version]
- Shetron-Rama, L.M.; Herring-Palmer, A.C.; Huffnagle, G.B.; Hanna, P. Transport of bacillus anthracis from the lungs to the draining lymph nodes is a rapid process facilitated by CD11c+ cells. Microb. Pathog. 2010, 49, 38–46. [Google Scholar] [CrossRef]
- Weiner, Z.P.; Glomski, I.J. Updating perspectives on the initiation of Bacillus anthracis growth and dissemination through its host. Infect. Immun. 2012, 80, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Russell, B.H.; Vasan, R.; Keene, D.R.; Koehler, T.M.; Xu, Y. Potential dissemination of Bacillus anthracis utilizing human lung epithelial cells. Cell. Microbiol. 2008, 10, 945–957. [Google Scholar] [CrossRef]
- Golden, H.B.; Watson, L.E.; Lal, H.; Verma, S.K.; Foster, D.M.; Kuo, S.R.; Sharma, A.; Frankel, A.; Dostal, D.E. Anthrax toxin: Pathologic effects on the cardiovascular system. Front. Biosci. 2009, 14, 2335–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guichard, A.; Nizet, V.; Bier, E. New insights into the biological effects of anthrax toxins: Linking cellular to organismal responses. Microbes Infect. 2012, 14, 97–118. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, P.; Michailidis, G.; Zielke, R.; Walker, A.; Patel, N.; Strahler, J.; Driks, A.; Andrews, P.; Maddock, J. Early events of Bacillus anthracis germination identified by time-course quantitative proteomics. Proteomics 2006, 6, 5199–5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, J.; Hutchison, J.; Hess, B.; Straub, T. Bacillus anthracis spores germinate extracellularly at air–liquid interface in an in vitro lung model under serum-free conditions. J. Appl. Microbiol. 2015, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Xu, W.; Neupane, P.; Weiser-Schlesinger, A.; Weng, R.; Pockros, B.; Li, Y.; Moayeri, M.; Leppla, S.H.; Fitz, Y.; et al. Bacillus anthracis lethal toxin, but not edema toxin, increases pulmonary artery pressure and permeability in isolated perfused rat lungs. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1076–H1090. [Google Scholar] [CrossRef]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun 1991, 59, 3472–3477. [Google Scholar] [CrossRef] [Green Version]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Investig. 2003, 112, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Moayeri, M.; Li, Y.; Li, X.; Haley, M.; Fitz, Y.; Correa-Araujo, R.; Banks, S.M.; Leppla, S.H.; Eichacker, P.Q. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R699–R709. [Google Scholar] [CrossRef]
- Culley, N.C.; Pinson, D.M.; Chakrabarty, A.; Mayo, M.S.; LeVine, S.M. Pathophysiological manifestations in mice exposed to anthrax lethal toxin. Infect. Immun. 2005, 73, 7006. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Milia, E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J. Cell. Physiol. 2012, 227, 1438–1445. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Warburton, R.R.; Hill, N.S.; Kayyali, U.S. Anthrax lethal toxin-induced lung injury and treatment by activating mk2. J. Appl. Physiol. 2015, 119, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, M.; Duggan, E.S.; Booth, J.L.; Patel, V.I.; Zander, R.A.; Silasi-Mansat, R.; Ramani, V.; Veres, T.Z.; Prenzler, F.; Sewald, K.; et al. Bacillus anthracis lethal toxin reduces human alveolar epithelial barrier function. Infect. Immun. 2012, 80, 4374–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, M.; Noack, D.; Wood, M.; Perego, M.; Knaus, U.G. Lung epithelial injury by b. Anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS ONE 2009, 4, e4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, B.; Batsche, E.; Boutillon, F.; Wu, Y.Z.; Leduc, D.; Balloy, V.; Raoust, E.; Muchardt, C.; Goossens, P.L.; Touqui, L. Anthrax lethal toxin impairs IL-8 expression in epithelial cells through inhibition of histone H3 modification. PLoS Pathog. 2009, 5, e1000359. [Google Scholar] [CrossRef] [Green Version]
- Tessier, J.; Green, C.; Padgett, D.; Zhao, W.; Schwartz, L.; Hughes, M.; Hewlett, E. Contributions of histamine, prostanoids, and neurokinins to edema elicited by edema toxin from Bacillus anthracis. Infect. Immun 2007, 75, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Mehta, H.; Chakrabarty, K.; Booth, J.L.; Duggan, E.S.; Patel, K.B.; Ballard, J.D.; Coggeshall, K.M.; Metcalf, J.P. Resistance of human alveolar macrophages to bacillus anthracis lethal toxin. J. Immunol. 2009, 183, 5799–5806. [Google Scholar] [CrossRef] [Green Version]
- Lomonaco, S.; Nucera, D.; Filipello, V. The evolution and epidemiology of Listeria monocytogenes in Europe and the united states. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2015, 35, 172–183. [Google Scholar] [CrossRef]
- O’Neil, H.S.; Marquis, H. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect. Immun. 2006, 74, 6675–6681. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, G.A. Listeria monocytogenes. In Pediatric Clinical Advisor, 2nd ed.; Garfunkel, L.C., Kaczorowski, J.M., Christy, C., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 339–340. [Google Scholar] [CrossRef]
- Hamon, M.A.; Ribet, D.; Stavru, F.; Cossart, P. Listeriolysin O: The swiss army knife of Listeria. Trends Microbiol. 2012, 20, 360–368. [Google Scholar] [CrossRef]
- Ananthraman, A.; Israel, R.H.; Magnussen, C.R. Pleural-pulmonary aspects of Listeria monocytogenes infection. Respiration 1983, 44, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Lerolle, N.; Zahar, J.R.; Duboc, V.; Tissier, F.; Rabbat, A. Pneumonia involving Legionella pneumophila and listeria monocytogenes in an immunocompromised patient: An unusual coinfection. Respiration 2002, 69, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.F.; Sobel, J.; Mazaki-Tovi, S.; Kusanovic, J.P.; Vaisbuch, E.; Kim, S.K.; Uldbjerg, N.; Romero, R. Listeriosis in human pregnancy: A systematic review. J. Perinat. Med. 2011, 39, 227–236. [Google Scholar] [CrossRef]
- Cossart, P. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc. Natl. Acad. Sci USA 2011, 108, 19484–19491. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, K.; J Czuprynski, C. Pathogenesis of listeriosis during pregnancy. Anim. Health Res. Rev. 2013, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Morgand, M.; Leclercq, A.; Maury, M.M.; Bracq-Dieye, H.; Thouvenot, P.; Vales, G.; Lecuit, M.; Charlier, C. Listeria monocytogenes-associated respiratory infections: A study of 38 consecutive cases. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2018, 24, 1339.e1–1339.e5. [Google Scholar] [CrossRef] [Green Version]
- Palmer, M. The family of thiol-activated, cholesterol-binding cytolysins. Toxicon 2001, 39, 1681–1689. [Google Scholar] [CrossRef]
- Koster, S.; van Pee, K.; Hudel, M.; Leustik, M.; Rhinow, D.; Kuhlbrandt, W.; Chakraborty, T.; Yildiz, O. Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 2014, 5, 3690. [Google Scholar] [CrossRef] [Green Version]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signaling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell. Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef]
- Gekara, N.O.; Groebe, L.; Viegas, N.; Weiss, S. Listeria monocytogenes desensitizes immune cells to subsequent ca2+ signaling via listeriolysin o-induced depletion of intracellular Ca2+ stores. Infect. Immun. 2008, 76, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Hummler, E.; Barker, P.; Gatzy, J.; Beermann, F.; Verdumo, C.; Schmidt, A.; Boucher, R.; Rossier, B.C. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat. Genet. 1996, 12, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Czikora, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial sodium channel-alpha mediates the protective effect of the TNF-derived TIP peptide in pneumolysin-induced endothelial barrier dysfunction. Front. Immunol 2017, 8, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagata, T.; Yamagata, Y.; Masse, C.; Tessier, M.C.; Brochiero, E.; Dagenais, A.; Berthiaume, Y. Modulation of Na+ transport and epithelial sodium channel expression by protein kinase C in rat alveolar epithelial cells. Can. J. Physiol. Pharmacol. 2005, 83, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. Cancer 2007, 7, 554–562. [Google Scholar] [CrossRef]
- La Pietra, L.; Hudel, M.; Pillich, H.; Abu Mraheil, M.; Berisha, B.; Aden, S.; Hodnik, V.; Lochnit, G.; Rafiq, A.; Perniss, A.; et al. Phosphocholine antagonizes listeriolysin o-induced host cell responses of Listeria monocytogenes. J. Infect. Dis. 2020, jiaa022. [Google Scholar] [CrossRef]
- Patterson, M. Streptococcus. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. Available online: https://www.ncbi.nlm.nih.gov/books/NBK7611/ (accessed on 31 March 2020).
- Domon, H.; Oda, M.; Maekawa, T.; Nagai, K.; Takeda, W.; Terao, Y. Streptococcus pneumoniae disrupts pulmonary immune defense via elastase release following pneumolysin-dependent neutrophil lysis. Sci. Rep. 2016, 6, 38013. [Google Scholar] [CrossRef]
- Hirst, R.A.; Kadioglu, A.; O’Callaghan, C.; Andrew, P.W. The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin. Exp. Immunol. 2004, 138, 195–201. [Google Scholar] [CrossRef]
- Cockeran, R.; Anderson, R.; Feldman, C. Pneumolysin as a vaccine and drug target in the prevention and treatment of invasive pneumococcal disease. Arch. Immunol. Et Ther. Exp. 2005, 53, 189–198. [Google Scholar]
- Henriques-Normark, B.; Tuomanen, E.I. The pneumococcus: Epidemiology, microbiology, and pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a010215. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Mitchell, T.J. Streptococcus pneumoniae: Virulence factors and variation. Clin. Microbiol. Infect. 2010, 16, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Smythe, C.J.; Duncan, J.L. Thiol activated (oxygen labile) cytolysins. In Bacterial Toxins and Cell Membranes; Jeljaszewicz, J., Waldstrom, T., Eds.; Academic Press: London, UK, 1978; pp. 129–183. [Google Scholar]
- Jefferies, J.M.; Tocheva, A.S.; Rubery, H.; Bennett, J.; Garland, J.; Christodoulides, M.; Faust, S.N.; Smith, A.; Mitchell, T.J.; Clarke, S.C. Identification of novel pneumolysin alleles from paediatric carriage isolates of streptococcus pneumoniae. J. Med. Microbiol. 2010, 59, 808–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonev, B.B.; Gilbert, R.J.; Andrew, P.W.; Byron, O.; Watts, A. Structural analysis of the protein/lipid complexes associated with pore formation by the bacterial toxin pneumolysin. J. Biol. Chem. 2001, 276, 5714–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.J.; Dalziel, C.E. The biology of pneumolysin. In Macpf/cdc Proteins—Agents of Defense, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 145–160. [Google Scholar] [CrossRef]
- Cassidy, S.K.; O’Riordan, M.X. More than a pore: The cellular response to cholesterol-dependent cytolysins. Toxins 2013, 5, 618–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.; Steel, H.C.; Cockeran, R.; von Gottberg, A.; de Gouveia, L.; Klugman, K.P.; Mitchell, T.J.; Feldman, C. Comparison of the effects of macrolides, amoxicillin, ceftriaxone, doxycycline, tobramycin and fluoroquinolones, on the production of pneumolysin by Streptococcus pneumoniae in vitro. J. Antimicrob. Chemother. 2007, 60, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Rubins, J.B.; Charboneau, D.; Fasching, C.; Berry, A.M.; Paton, J.C.; Alexander, J.E.; Andrew, P.W.; Mitchell, T.J.; Janoff, E.N. Distinct roles for pneumolysin’s cytotoxic and complement activities in the pathogenesis of pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 1996, 153, 1339–1346. [Google Scholar] [CrossRef]
- Rubins, J.B.; Charboneau, D.; Paton, J.C.; Mitchell, T.J.; Andrew, P.W.; Janoff, E.N. Dual function of pneumolysin in the early pathogenesis of murine pneumococcal pneumonia. J. Clin. Investig. 1995, 95, 142–150. [Google Scholar] [CrossRef]
- Witzenrath, M.; Gutbier, B.; Hocke, A.C.; Schmeck, B.; Hippenstiel, S.; Berger, K.; Mitchell, T.J.; de los Toyos, J.R.; Rosseau, S.; Suttorp, N.; et al. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit. Care Med. 2006, 34, 1947–1954. [Google Scholar] [CrossRef]
- Cockeran, R.; Steel, H.C.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin potentiates production of prostaglandin e(2) and leukotriene b(4) by human neutrophils. Infect. Immun. 2001, 69, 3494–3496. [Google Scholar] [CrossRef] [Green Version]
- Nel, J.G.; Durandt, C.; Theron, A.J.; Tintinger, G.R.; Pool, R.; Richards, G.A.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin mediates heterotypic aggregation of neutrophils and platelets in vitro. J. Infect. 2017, 74, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Adams, W.; Bhowmick, R.; Bou Ghanem, E.N.; Wade, K.; Shchepetov, M.; Weiser, J.N.; McCormick, B.A.; Tweten, R.K.; Leong, J.M. Pneumolysin induces 12-lipoxygenase-dependent neutrophil migration during Streptococcus pneumoniae infection. J. Immunol. 2020, 204, 101–111. [Google Scholar] [CrossRef]
- Rubins, J.B.; Duane, P.G.; Charboneau, D.; Janoff, E.N. Toxicity of pneumolysin to pulmonary endothelial cells in vitro. Infect. Immun. 1992, 60, 1740–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubins, J.B.; Duane, P.G.; Clawson, D.; Charboneau, D.; Young, J.; Niewoehner, D.E. Toxicity of pneumolysin to pulmonary alveolar epithelial cells. Infect. Immun. 1993, 61, 1352–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, U.A.; Srivastava, M.; Paton, J.C.; Mack, M.; Everhart, M.B.; Blackwell, T.S.; Christman, J.W.; Schlöndorff, D.; Seeger, W.; Lohmeyer, J. Pneumolysin-induced lung injury is independent of leukocyte trafficking into the alveolar space. J. Immunol. 2004, 173, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, P.; He, F.; Kwang, J.; Engelward, B.P.; Chow, V.T. Pneumococcal pneumolysin induces DNA damage and cell cycle arrest. Sci. Rep. 2016, 6, 22972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larpin, Y.; Besançon, H.; Iacovache, M.-I.; Babiychuk, V.S.; Babiychuk, E.B.; Zuber, B.; Draeger, A.; Köffel, R. Bacterial pore-forming toxin pneumolysin: Cell membrane structure and microvesicle shedding capacity determines differential survival of cell types. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 1665–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Cernysiov, V.; Davidson, D.; Song, H.; Tang, J.; Luo, S.; Lu, Y.; Qian, J.; Gyurova, I.E.; Waggoner, S.N.; et al. Critical role of lipid scramblase tmem16f in phosphatidylserine exposure and repair of plasma membrane after pore formation. Cell Rep. 2020, 30, 1129–1140.e1125. [Google Scholar] [CrossRef] [Green Version]
- Fatykhova, D.; Rabes, A.; Machnik, C.; Guruprasad, K.; Pache, F.; Berg, J.; Toennies, M.; Bauer, T.T.; Schneider, P.; Schimek, M.; et al. Serotype 1 and 8 pneumococci evade sensing by inflammasomes in human lung tissue. PLoS ONE 2015, 10, e0137108. [Google Scholar] [CrossRef]
- Kostadinova, E.; Chaput, C.; Gutbier, B.; Lippmann, J.; Sander, L.E.; Mitchell, T.J.; Suttorp, N.; Witzenrath, M.; Opitz, B. NLRP3 protects alveolar barrier integrity by an inflammasome-independent increase of epithelial cell adherence. Sci. Rep. 2016, 6, 30943. [Google Scholar] [CrossRef] [Green Version]
- McNeela, E.A.; Burke, A.; Neill, D.R.; Baxter, C.; Fernandes, V.E.; Ferreira, D.; Smeaton, S.; El-Rachkidy, R.; McLoughlin, R.M.; Mori, A.; et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of tlr4. PLoS Pathog. 2010, 6, e1001191. [Google Scholar] [CrossRef] [Green Version]
- N’Guessan, P.D.; Schmeck, B.; Ayim, A.; Hocke, A.C.; Brell, B.; Hammerschmidt, S.; Rosseau, S.; Suttorp, N.; Hippenstiel, S. Streptococcus pneumoniae r6x induced p38 MAPK and JNK-mediated caspase-dependent apoptosis in human endothelial cells. Thromb. Haemost. 2005, 94, 295–303. [Google Scholar] [CrossRef]
- Ratner, A.J.; Lysenko, E.S.; Paul, M.N.; Weiser, J.N. Synergistic proinflammatory responses induced by polymicrobial colonization of epithelial surfaces. Proc. Natl. Acad. Sci. USA 2005, 102, 3429–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Kumar, S.; Yu, Y.; Aggarwal, S.; Gross, C.; Wang, Y.; Chakraborty, T.; Verin, A.D.; Catravas, J.D.; Lucas, R.; et al. PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+ -toxins. PLoS ONE 2014, 9, e99823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, K.; Neill, D.R.; Malak, H.A.; Spelmink, L.; Khandaker, S.; Dalla Libera Marchiori, G.; Dearing, E.; Kirby, A.; Yang, M.; Achour, A.; et al. Pneumolysin binds to the mannose receptor c type 1 (mrc-1) leading to anti-inflammatory responses and enhanced pneumococcal survival. Nat. Microbiol. 2019, 4, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Surana, N.K.; Kasper, D.L. Approach to the patient with an infectious disease. In Harrison’s Principles of Internal Medicine, 19th ed.; Kasper, D.L., Fauci, A.S., Hauser, S.L., Longo, D.L., Jameson, J.L., Loscalzo, J., Eds.; McGrawHill: New York, NY, USA, 2015; pp. 759–768. [Google Scholar]
- Rudkin, J.K.; McLoughlin, R.M.; Preston, A.; Massey, R.C. Bacterial toxins: Offensive, defensive, or something else altogether? PLoS Pathog. 2017, 13, e1006452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
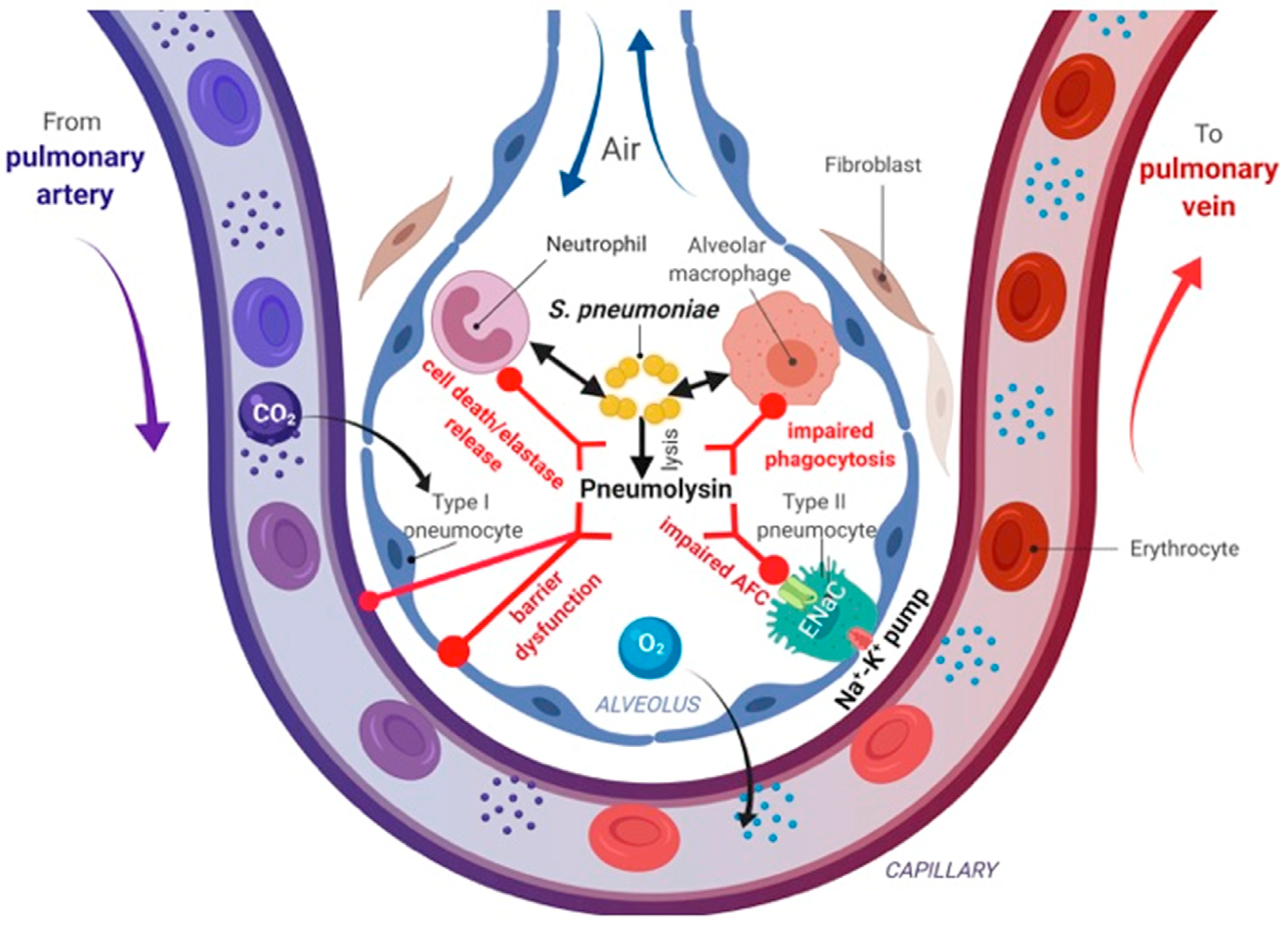
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, R.; Hadizamani, Y.; Gonzales, J.; Gorshkov, B.; Bodmer, T.; Berthiaume, Y.; Moehrlen, U.; Lode, H.; Huwer, H.; Hudel, M.; et al. Impact of Bacterial Toxins in the Lungs. Toxins 2020, 12, 223. https://doi.org/10.3390/toxins12040223
Lucas R, Hadizamani Y, Gonzales J, Gorshkov B, Bodmer T, Berthiaume Y, Moehrlen U, Lode H, Huwer H, Hudel M, et al. Impact of Bacterial Toxins in the Lungs. Toxins. 2020; 12(4):223. https://doi.org/10.3390/toxins12040223
Chicago/Turabian StyleLucas, Rudolf, Yalda Hadizamani, Joyce Gonzales, Boris Gorshkov, Thomas Bodmer, Yves Berthiaume, Ueli Moehrlen, Hartmut Lode, Hanno Huwer, Martina Hudel, and et al. 2020. "Impact of Bacterial Toxins in the Lungs" Toxins 12, no. 4: 223. https://doi.org/10.3390/toxins12040223
APA StyleLucas, R., Hadizamani, Y., Gonzales, J., Gorshkov, B., Bodmer, T., Berthiaume, Y., Moehrlen, U., Lode, H., Huwer, H., Hudel, M., Mraheil, M. A., Toque, H. A. F., Chakraborty, T., & Hamacher, J. (2020). Impact of Bacterial Toxins in the Lungs. Toxins, 12(4), 223. https://doi.org/10.3390/toxins12040223