1. Introduction
According to the Food and Agriculture Organization of the United Nations and a recent study by Eskola et al. in 2019, approximately 25% of food-crops worldwide are contaminated with mycotoxins, a group of secondary metabolites produced by molds [
1,
2]. Particularly, the toxins of
Aspergillus, Fusarium, and
Penicillium species are the most detrimental ones because these so-called aflatoxins, ochratoxins, trichothecenes (especially deoxynivalenol), and zearalenone have various adverse effects on the health of humans and animals [
3,
4,
5,
6,
7,
8]. From all mycotoxins, aflatoxin B1 (AFB1), mainly produced by
Aspergillus species, has the strongest adverse effects on health, as it might lead to liver cancer [
9,
10,
11,
12]. Several factors can promote the enrichment of mycotoxins within food products. Key drivers are late harvests of crops as well as elevated humidity and temperature during storage. Within the EU, but also in the USA, Brazil, and other countries, regulatory requirements regarding the maximum tolerable levels of mycotoxins in various foods have been established. Regulatory limits in the EU for the IARC group 1 carcinogen aflatoxin B1 are set to 2–8 ng/g (ppb; ng·mL
−1) in grain, corn, nuts, and fruits, as published in (EG) Nr. 1881/2006. Especially due to those harsh effects and corresponding low regulatory limits, highly sensitive and reliable detection technologies are of primary importance.
Currently, there are basically three analytical technologies used for mycotoxin testing. On the one hand, laboratory-based liquid chromatography (LC) coupled to mass spectrometry (MS) is the most common method, as well as enzyme-linked immunosorbent assay (ELISA), are routinely used for highly sensitive mycotoxin testing. On the other hand, lateral flow assays (LFA) are used for fast but less sensitive on-field testing [
13,
14,
15,
16,
17,
18,
19]. Although LC-MS/MS methods have the great advantage of high sensitivity and simultaneous detection of currently more than 500 mycotoxins within a single run, expensive equipment, highly qualified staff, and a possible complex sample cleanup inhibit the applicability for fast on-site testing [
20]. In comparison to LC-based methods, ELISA techniques are cheaper and, according to Renauld and colleagues (2019), faster in assay procedure, but also require a lab with the corresponding equipment for sample preparation and analysis [
21].
In the field of ELISA, the most commonly used format for aflatoxin detection is the competitive assay because of the small molecular structure of the antigen that prevents the simultaneous binding of two antibodies, which would be the prerequisite for a sandwich ELISA. The basic working principle is the coating of mycotoxin-conjugate onto a microtiter plate followed by the addition and incubation of a mycotoxin-specific antibody in the presence of a liquid extract of sample material. In this step, mycotoxins in the sample compete with coated mycotoxins for antibody-binding. Subsequently, antibodies saturated with soluble mycotoxins are removed during a washing step and cannot contribute to a signal, achieved by either direct readout or indirect readout by means of a labeled secondary antibody. In this format, a low signal corresponds to a high concentration of mycotoxin within the sample, and vice versa. Most of the currently commercially available ELISA-based assay formats have a limit of detection (LOD) ranging from 1 µg·mL
−1 (equals ng·mL
−1 or ppb) up to 50 µg·mL
−1, for example, Ridascreen Aflatoxin B1 30/15 test kit (r-biopharm, Darmstadt, Germany) or AgraQuant Aflatoxin B1 ELISA test (Romer Labs, Butzbach, Germany). Nevertheless, due to the requirement for laboratory equipment, their usability for on-site testing is very limited. In such cases, commercially available LFA have the highest potential for fast and cost-effective on-site mycotoxin testing. However, most of these assay systems provide only qualitative results with LODs ranging from 4 ppb to 5 ppb, as achieved with RIDA
®Quick Aflatoxin test kit(r-biopharm, Darmstadt, Germany) or AFB1 (Aflatoxin B1) lateral flow assay kit (Elabscience, Texas, USA), respectively. Primarily due to matrix interference effects, these assays can result in false-positive detection [
22,
23].
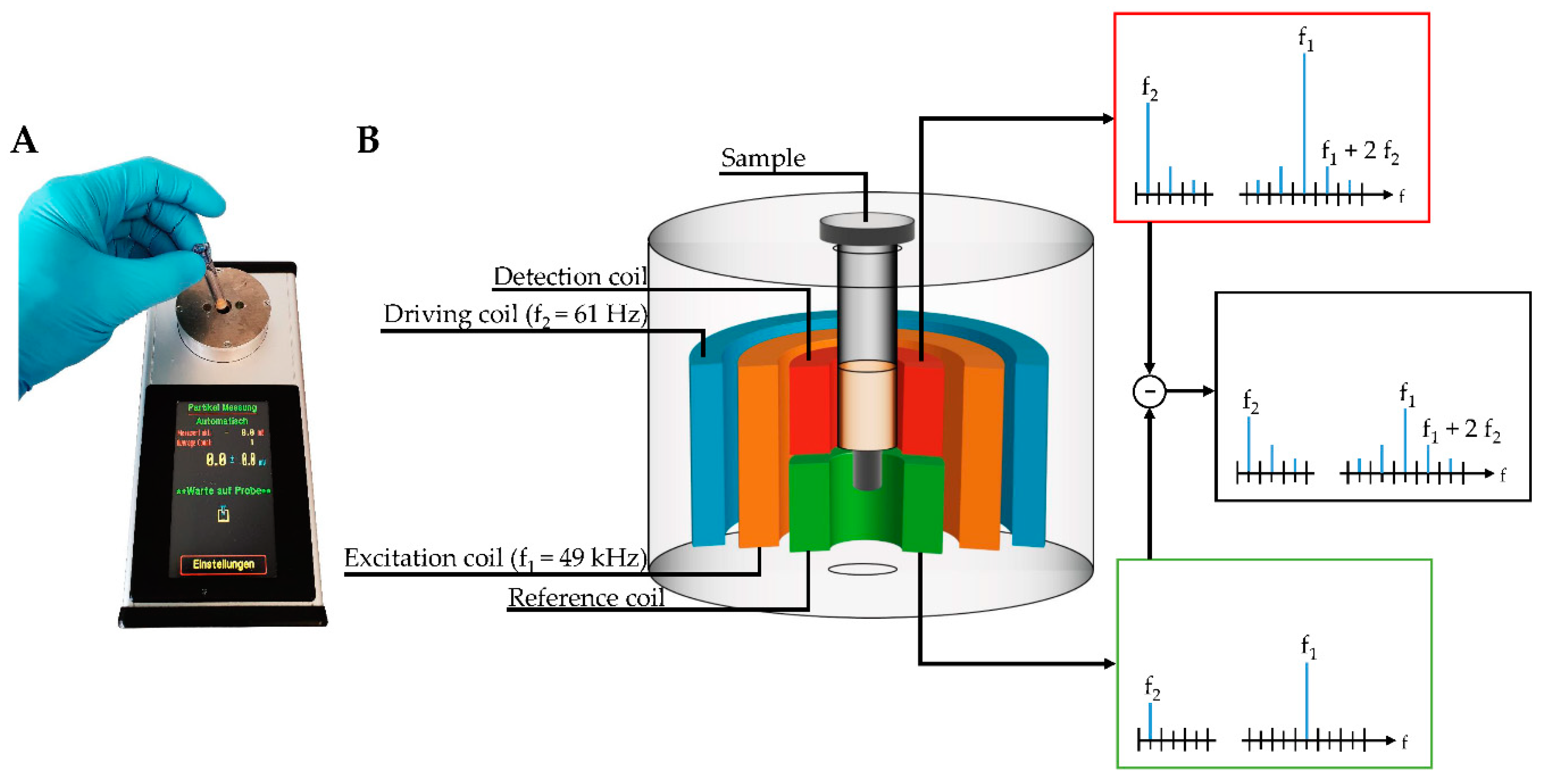
A novel, portable magnetic immunodetection approach has been described in previous studies [
24,
25,
26,
27,
28]. The detection of human and plant pathogens, as well as various proteins by sensing and quantifying superparamagnetic particles (MP) with the help of a portable magnetic reader (
Figure 1A), have been successfully demonstrated. This device can be operated using a conventional external power adapter or a portable battery, allowing an on-site readout without electrical infrastructure. For this immunomagnetic detection approach, MPs were functionalized with monoclonal antibodies directed against target molecules, retained in a sandwich-based manner within an immunofiltration column, and can be detected by means of frequency mixing magnetic detection (FMMD) technology [
29].
In this technique, magnetic particles are subjected to two sinusoidal magnetic fields of different frequencies generated by two excitation coils, which are schematically shown within the measurement head in
Figure 1B. Here, MPs are exposed to a low- and high-frequency magnetic field, so-called driving frequency, generated by the outermost coil, and excitation frequency, generated by the middle-positioned coil. The low frequency with 61 Hz (f
2) has an amplitude of a few millitesla, resulting in alternating positive and negative magnetic saturation of superparamagnetic particles oscillating with a frequency of 2f
2 of 122 Hz [
29]. The high-frequency magnetic excitation field (f
1) with 49 kHz probes the magnetization state of the superparamagnetic particles and yields an iron oxide dose-dependent signal when the low-frequency driving field is close to zero. Finally, the resulting mixing frequency signal of f
1 + 2f
2 can be demodulated and detected by the innermost coil, composed of two adjacent sections, so-called detection coil (upper one) and reference coil (lower one). Those sections differ only in the winding-orientation of coils. With this clock- and counterclockwise orientation yielding induced voltages of opposite sign, the directly induced excitation field can be canceled out. By placing the sample carrying the MPs in the detection head, the resulting signal is amplified, measured, and directly visualized at the touchscreen of the handheld, portable magnetic reader (
Figure 1). Based on a calibration curve, the detected signal can be attributed to the amount of analyte within the sample.
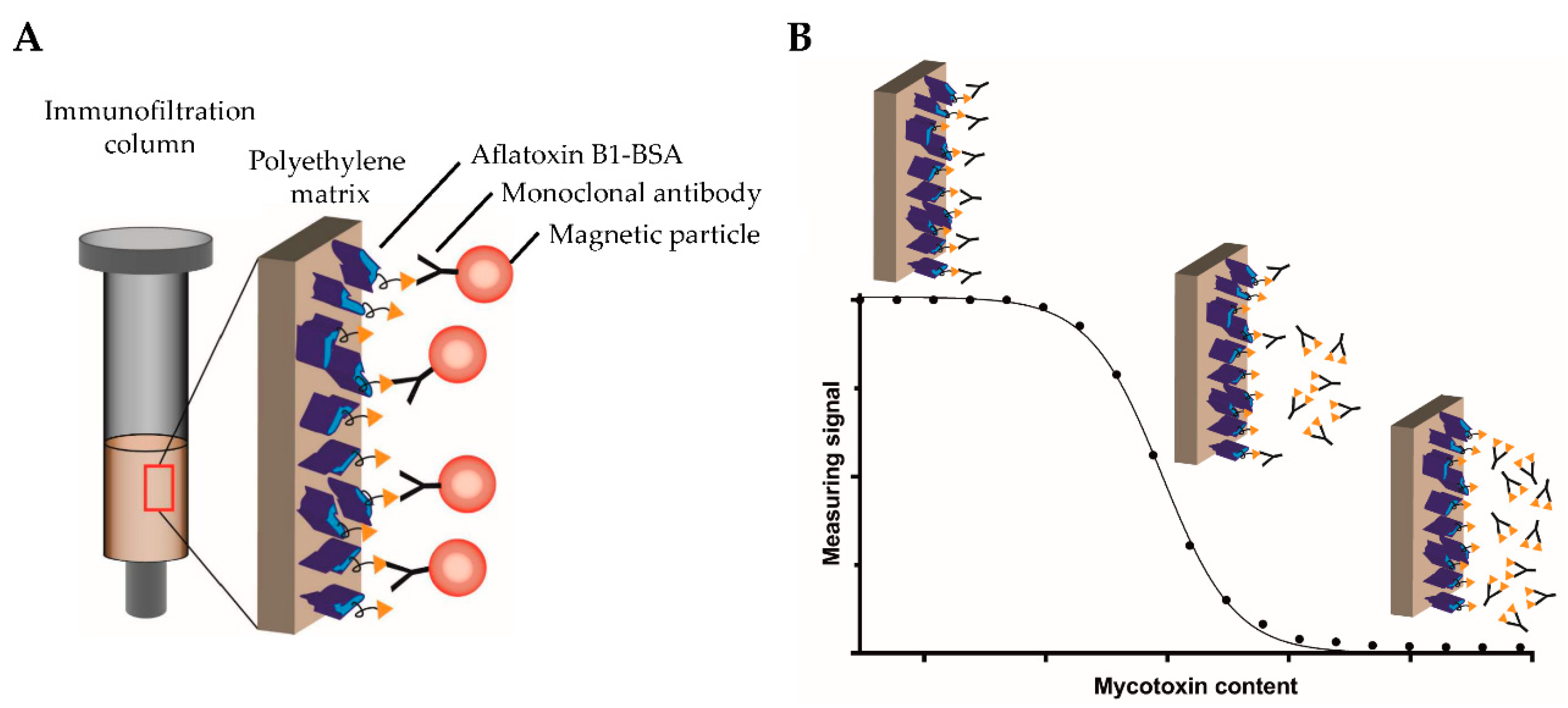
Motivated by the above-described drawbacks of currently used analytical methods for sensitive on-field testing, the aim of this study was to develop a novel, highly sensitive, and portable assay based on competitive magnetic immunodetection (cMID) and FMMD. The sensitivity should be comparable to a laboratory-based ELISA. Hence, initially, a competitive ELISA (cELISA) was established, serving as a reference method. Assay parameters, such as the used coating and antibody concentrations, were optimized to reach a sufficient sensitivity for the detection of aflatoxin B1. Afterward, the cMID assay was established using the same optimization strategy in combination with further evaluation of the required amount of nanoparticles. The basic principle of cMID is the use of biotinylated antibodies, which can be enriched within the coated immunofiltration column by a competitive binding reaction depending on the amount of pre-captured mycotoxin. By flushing magnetic particles functionalized with streptavidin through the column by gravity flow, particles can bind to retained antibodies and subsequently be detected using FMMD.
Figure 2 visualizes the basic cMID principle (
Figure 2A) and the competitive binding reaction within the column with the corresponding measuring signal (
Figure 2B).
3. Discussion
In this study, we demonstrated for the first time, a competitive magnetic immunodetection assay for efficient detection and quantification of aflatoxin B1 with comparable sensitivity to described laboratory-based cELISA. Especially due to the handheld reader device which can be operated using a portable battery and without additional laboratory equipment, the method described here is suitable for sensitive on-site aflatoxin B1 testing. Commonly used analytical methods for detection of mycotoxins either exhibit high sensitivity in a laboratory-based setting or can be used on-site with reduced sensitivity [
21].
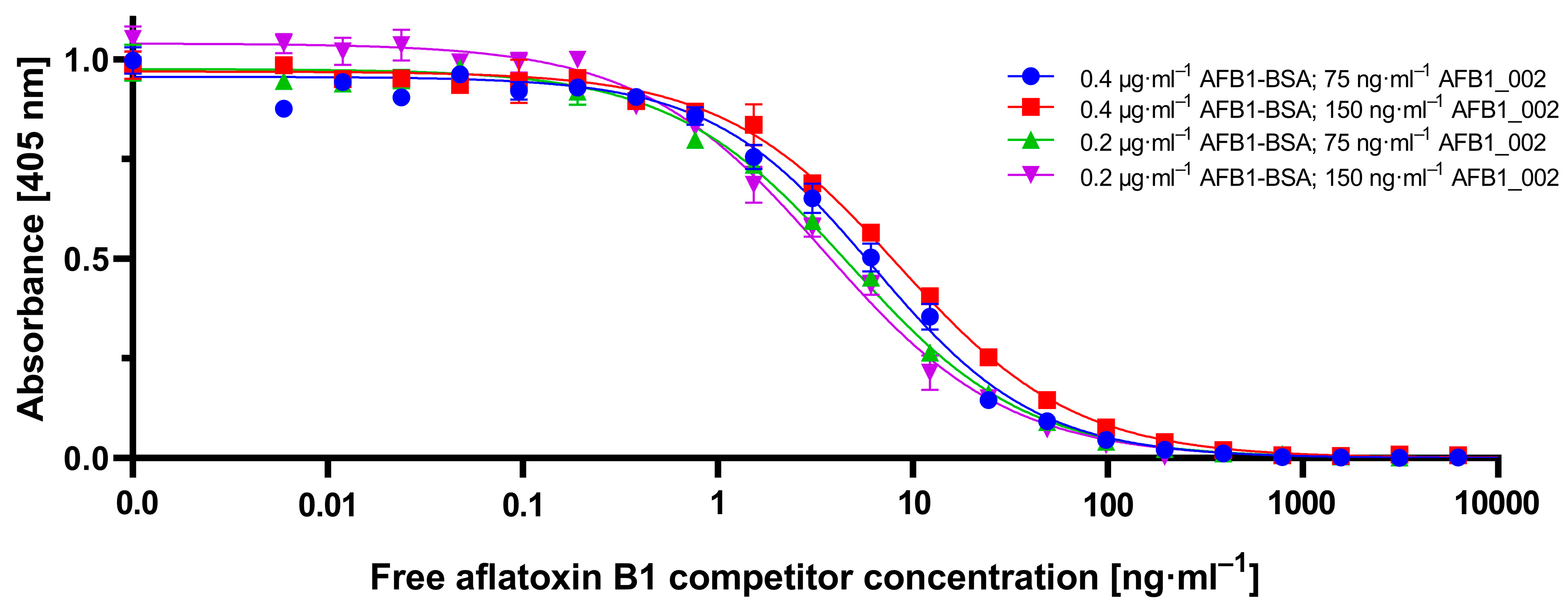
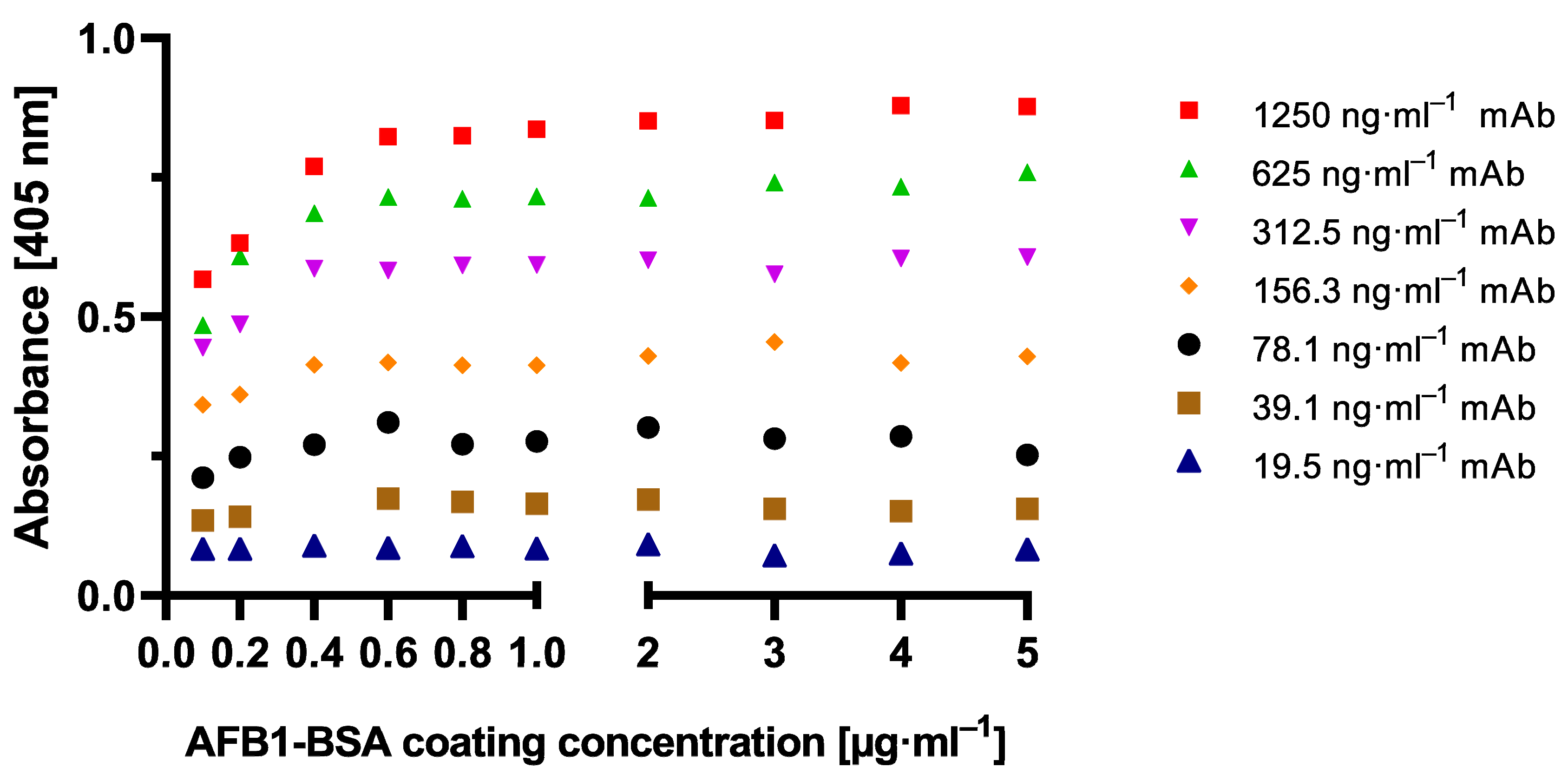
To establish the novel cMID method, several concentration settings of a cELISA as reference method were tested and optimized using checkerboard titration (
Figure A1). Here, the most suitable combination of antigen coating concentration and the amount of used antibody was determined. As explained in
Section 2.1.1, in all tested antibody concentrations, for a coating of 0.4 µg·mL
−1 and onwards, a saturation in measuring signal could be detected (
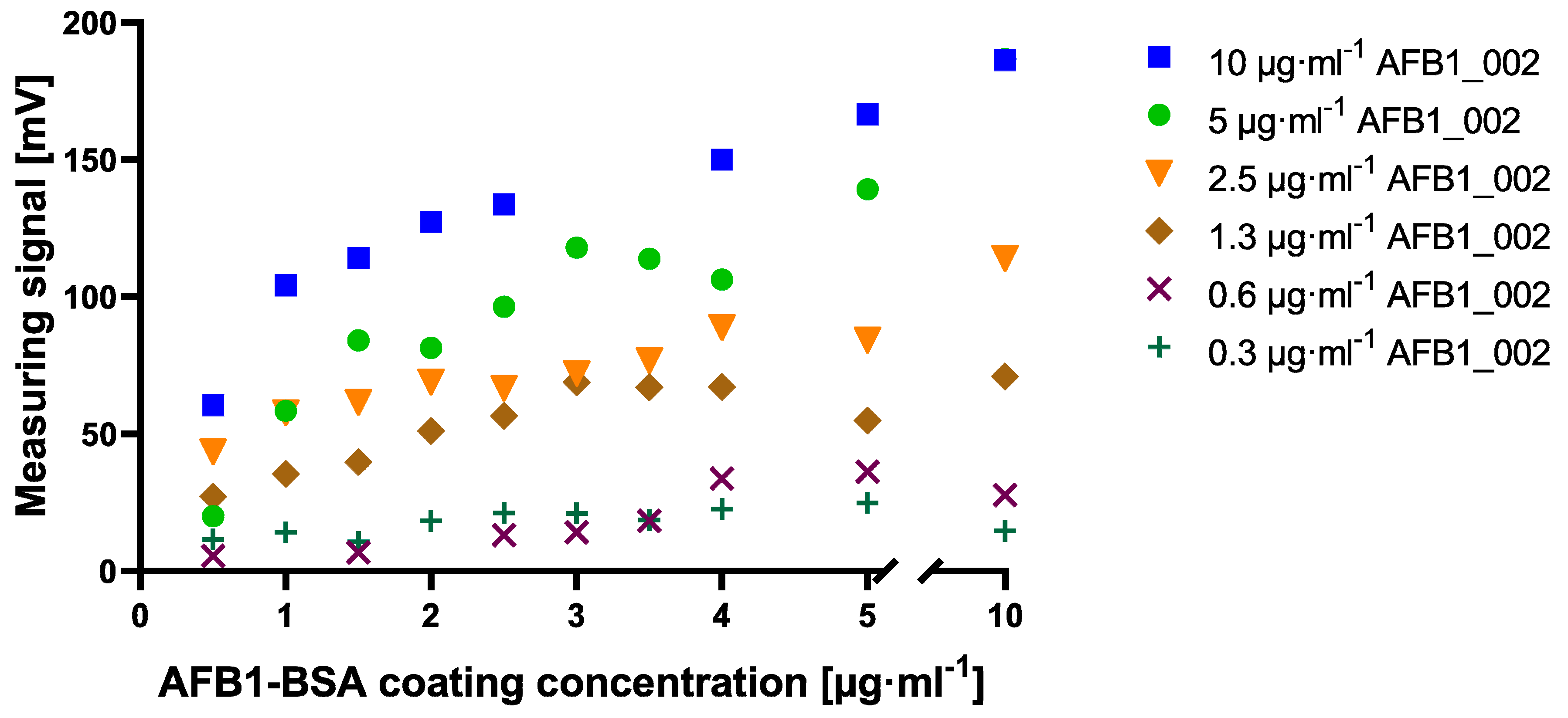
Figure A1). A clear difference to this can be seen within the checkerboard optimization experiment for cMID (
Figure A2). Here, a saturation-like performance can either be seen when using antibody concentrations up to 2.5 µg·mL
−1 or, if further increasing antibody concentrations, saturation cannot be reached. This could be due to the more than 40-fold higher protein binding surface of immunofiltration columns in comparison to the binding surface of an ELISA microtiter plate well. Especially due to the highly porous membrane of the immunofiltration column, a higher protein binding capacity can be reached. High binding 96-well microtiter plates (Greiner Bio-One, article number 655061) have a maximum binding capacity of 600 ng·cm
2, whereas ABICAP immunofiltration columns can bind up to 24,000 ng·cm
2, which in fact explains the absence of signal saturation at the used conditions. A further difference when using checkerboard titration for assay optimization of cELISA or cMID can be seen in fluctuating readout signals, whereas in ELISA, concentration-dependent signals were obtained with almost no variability between consecutive measuring points. In contrast, signals generated in MID checkerboard titration varied, especially at high antibody concentrations. This could be caused by an overdose of magnetic particles, which might result in sterical hindrances and a partly erratic aggregation of particles within the assay matrix. Based on this hypothesis, a magnetic-particle dose adaptation was made by reducing the applied concentration towards robustly detectable saturated measuring signals (
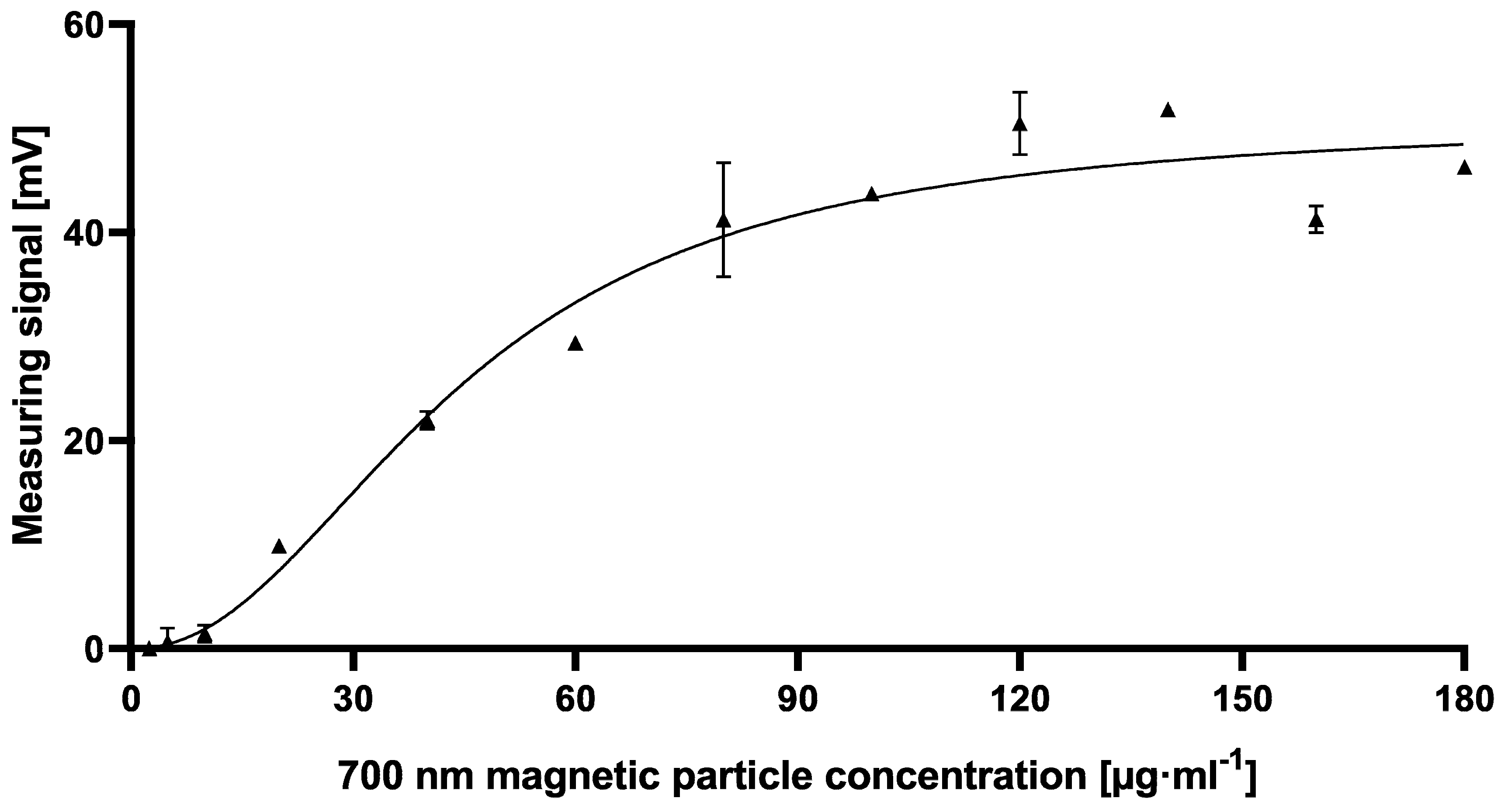
Figure 5).
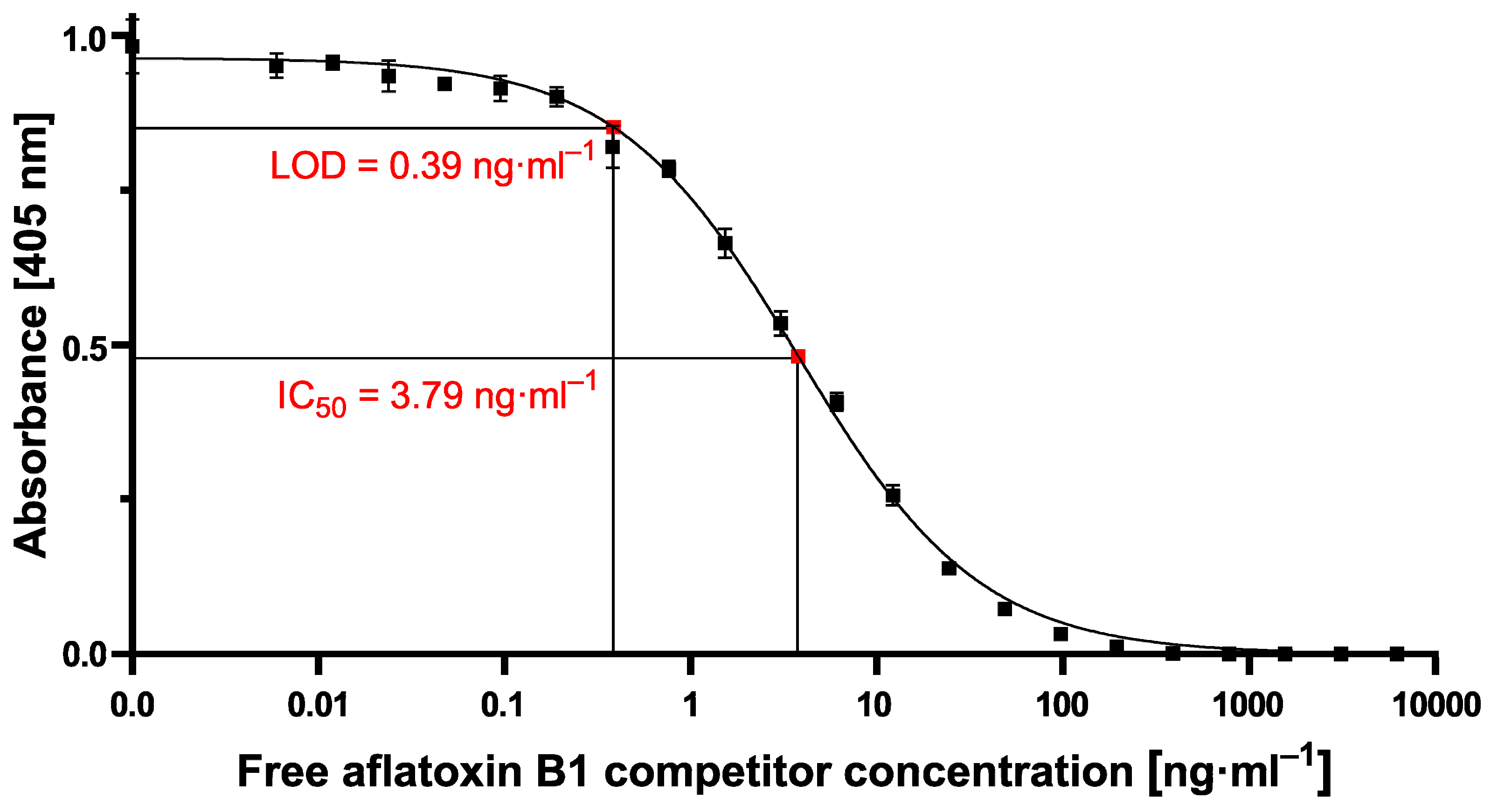
However, the resulting cMID assay did not lead to the expected results when using 700 nm magnetic beads. Switching to 70 nm magnetic particles instead resulted in a tenfold increase of measuring signal combined with reduced variability and provides a basis for a highly sensitive and reliable assay (
Figure 6). The results presented here correlate with data published by Achtsnicht et al. in 2019, where the authors used magnetic frequency mixing for the detection of cholera toxin subunit B (CTB) in a sandwich-based manner [
27]. In their experiments, using magnetic particles with hydrodynamic diameters of 75 nm and 1010 nm, they observed an eight-fold higher response signal with smaller particles compared to the larger beads when detecting a concentration of 750 ng·mL
−1 CTB. The most simple explanation for this striking difference would be the higher absolute number of small particles compared to larger particles when used at identical concentrations (80 µg·mL
−1) since the molar concentration is proportional to the molecular weight of the particles. By applying a higher ratio of small particles onto the column, a higher amount of small MPs could bind to antigens coated on the column surface. Another explanation could be the occurrence of steric hindrance within the ABICAP immunofiltration matrix when using large particles. Especially if there is a low concentration of free mycotoxin within the sample, a high amount of biotinylated antibody will be retained within the matrix in a competitive assay approach, resulting in a high antibody density on the matrix surface. When large MPs are applied, steric hindrance can occur at the surface. Either multiple, closely located biotinylated antibodies could be linked to one MP, or multiple antibodies could be blocked due to the size of large MPs. Here, especially the thousand-fold increased volume of 700 nm diameter MPs in comparison to 70 nm MPs in diameter might have the most important role. By the greatly increased size and volume, several antibodies are covered by one single MP, so that they are no longer accessible for binding of further MPs. By using smaller particles, shielding effects and sterical hindrances at the surface are reduced, resulting in a higher magnetic particle density within the column, and finally, a higher readout signal. A similar effect observed by Achtsnicht et al. (2019) when using small 75 nm instead of large 1010 nm particles in their MID experiments was also explained by the blocking of remaining binding sites [
27]. The authors also found similar high standard deviations when large particles remain in a high concentration within the column (
Figure 6A). With such a high SD, sensitive detection of either CTB or aflatoxin B1 is almost impossible. In their study, a LOD of 3.1 ng·mL
−1 was found when using the large particles, but a more than 15-fold lower LOD of 0.2 ng·mL
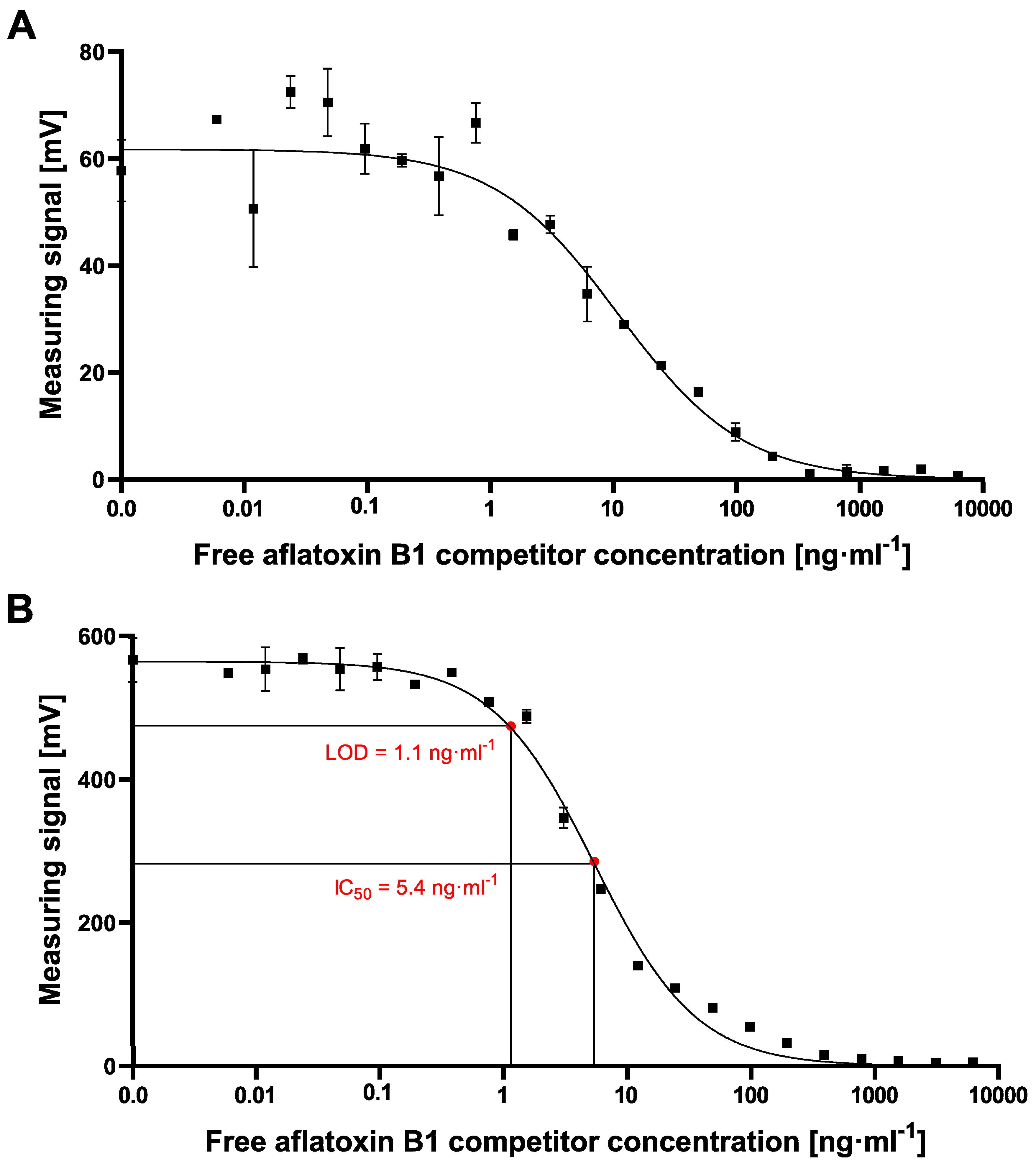
−1 could be reached using the small-sized MPs. In our study, especially due to the high standard deviations at high measuring signals when exciting the 700 nm MPs for aflatoxin B1 detection, a reliable calculation of LOD or IC
50 values was not possible. However, by using 70 nm particles, high and stable readout signals in combination with a perfectly matching nonlinear-fit (R
2 = 0.9938) enabled the reliable calculation of LOD and IC
50 values of 1.1 ng·mL
−1 and 5.4 ng·mL
−1, respectively, which are in the same sensitivity range as our comparative lab-based cELISA
Although the best performance of the cMID assay was achieved with 70 nm small magnetic particles, the general applicability of 700 nm large particles in cMID assays should not be completely neglected since, in contrast to small particles, larger particles can be separated in a gradient magnetic field. However, the possibility of magnetic separation and thereby enrichment of magnetic particles might play a crucial role in sample preparation where cleanup and concentrating of an analyte is required to achieve sufficient assay sensitivity, as it was used for example by Lee et al. (2013) or Xuan and colleagues (2019) [
30,
31]. In the case of mycotoxin detection, magnetic separation-based sample cleanup could be beneficial, especially when working with partially soluble or insoluble, or roughly homogenized, e.g., grain samples. After magnetic beads captured the mycotoxin molecules in the sample, a magnetic separation step can be applied in which magnetic particles are retained in the magnetic field, while sample debris can be discarded. Additionally, separated mycotoxin-loaded magnetic particles could be resuspended in a much smaller volume, resulting in an enrichment of analyte. As a consequence, this could lead to increased sensitivity when samples are finally analyzed by cMID.
The 70 nm particles used in this study cannot be efficiently separated in a magnetic gradient field, since their magnetic attraction is low, which was shown by Achtsnicht and colleagues (2018) [
32]. They correlated the magnetophoretic velocity of superparamagnetic particles when applying a magnetic field and found an increased velocity with increasing particle size [
32]. Typically only bigger particles above 700 nm are used for magnetic separation experiments [
30,
31]. Further testing of different beads or other strategies as combining big and small-sized particles will be addressed in further studies to enable an optimized pairing of separation and cMID.
In conclusion, we successfully demonstrated the development and implementation of a competitive magnetic immunodetection assay for the detection and quantification of aflatoxin B1 with a LOD of 1.1 ng·mL
−1. Based on our findings, it can be concluded that the competitive magnetic immunodetection is a powerful tool for portable, easy-to-use on-site monitoring of mycotoxin contamination in various matrices to reduce the risk of processing contaminated food and agricultural products. In future work, we will focus on the further optimization of the cMID procedure using the 70 nm MPs. Especially, a significant reduction of the assay time from currently approximately 4.5 h to less than one hour should be addressed, similar to that described by Rettcher et al. (2015) [
26]. There a sandwich-based MID assay lasting less than 30 min was achieved by a more than 50% reduction of initially needed assay time [
26]. By using MPs, pre-conjugated with specific anti-mycotoxin antibodies, ready-to-use coated and blocked immunofiltration columns, and testing successive reductions of incubation times of sample-pre-incubation and competitive binding reaction within the matrix, an additional increase of applicability for on-site testing should be possible. The applicability of the described cMID approach will be further studied regarding the detection and quantification of other mycotoxins, as well as a combination of those toxins within a food matrix of one sample. Especially the multiplex detection of various mycotoxins within one sample will be addressed as shown by the multiplex detection of several antibodies in stacked sample matrices by Achtsnicht et al. (2019) [
33]. Here the authors detected two different target molecules, namely antibodies, within one sample solution by stacking 3D printed immunofiltration columns coated with different capture antibodies in a sandwich-based MID approach. By adapting this procedure, multiple individually coated matrices could be used for the specific retention of corresponding anti-mycotoxin antibodies in a cMID assay, and with this, a multiplex detection in a food matrix could be obtained.
4. Materials and Methods
4.1. Material and Chemicals
Dimethyl sulfoxide, Tween-20, Aflatoxin B1, Aflatoxin B1-BSA, EZ-Link™ NHS-PEG4 Biotinylation Kit, ABTS buffer, as well as ABTS tablets were purchased from Merck KGaA, Darmstadt, Germany. NaCl, KCl, Na2HPO4 × 12 H20, KH2PO4, Na2(CO3), NaHCO3, Milk powder, and Albumin Fraction V (biotin-free) were acquired from Carl Roth, Karlsruhe, Germany.
The used coupling buffer was prepared by dissolving 15 mM Na2CO3 and 35 mM NaHCO3 in MilliQ-water, and pH was set to 9.6 with glacial acetic acid. Phosphate buffered saline (PBS) was prepared by dissolving 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4 × 12 H2O, and 1.5 mM KH2PO4 in MilliQ-water and setting pH to 7.4 with hydrochloric acid. As a washing buffer, PBS-T was produced by adding 0.05% (v/v) Tween-20 to PBS. ELISA blocking buffer (EBP) was prepared by adding 5% (w/v) milk powder to PBS. For magnetic immunodetection experiments, blocking solution consists of 1% (w/v) albumin fraction V (biotin free) in PBS, and is called MID-BP. All other chemicals, except Tween-20, were acquired from Roth.
Immunofiltration columns (ABICAP HP columns) were purchased from Senova Gesellschaft für Biowissenschaft und Technik mbH, Weimar, Germany. High binding 96-well microtiter plates (article number 655061) were purchased from Greiner Bio-One GmbH, Frickenhausen, Germany. Anti-mycotoxin monoclonal antibody AFB1_002 was purchased from fzmb GmbH, Bad Langensalza, Germany. Secondary antibody goat anti-mouse IgG-HRPO (article number 115-035-008) was purchased from Jackson ImmunoResearch Europe Ltd. UK. 700 nm streptavidin-functionalized magnetic particles (nanomag®-CLD/synomag®-CLD; article number 05-19-502 S09718) as well as 70 nm streptavidin-functionalized magnetic particles (synomag®-D, article number 104-19-701) were purchased from micromod Partikeltechnologie GmbH, Rostock, Germany.
4.2. Optimization of ELISA
The most suitable aflatoxin B1-BSA coating concentrations for a competitive ELISA protocol in combination with appropriate anti-aflatoxin B1 monoclonal antibody concentrations were determined by checkerboard titration. Throughout the following protocol, all incubation steps were performed at room temperature for one hour in the dark. For coating varying concentrations of AFB1-BSA, the antigen was diluted in a coupling buffer, and 100 µL per well was added to a 96-well highbinding microtiter plate (Greiner Bio-One) and incubated as mentioned above. After washing each well thrice with PBS-T, all wells were blocked using 200 µL of EBP and incubated. Then, the plate was washed again, and concentrations of monoclonal antibody ranging from 19.5 ng·mL−1 up to 1250 ng·mL−1, diluted in PBS, were added. After another washing step, as described above, 100 µL of secondary antibody goat anti-mouse IgG-HRPO, diluted 1:10,000 in PBS was added to each well and incubated. Prior to readout with 100 µL of 1 mg·mL−1 ATBS substrate in ABTS buffer, the plate was washed again. Absorption was measured at 405 nm after 10 min incubation in the dark.
4.3. cELISA Procedure
For competitive ELISA procedure, AFB1-BSA conjugate was diluted in coupling buffer and plated with 100 µL per well onto a high binding 96-well microtiter plate. As mentioned in
Section 4.2, all incubation steps were performed at room temperature for one hour in the dark. After the washing step with PBS-T, each well was blocked with EBP and incubated. Meanwhile, pre-incubation of free aflatoxin B1with the monoclonal anti-aflatoxin B1 antibody was prepared. For this, 75 µL of a serial dilution of aflatoxin B1 in PBS was prepared, and then 75 µL of antibody solution diluted in PBS was added and incubated. After washing the blocked assay plate three times with PBS-T, 100 µL of pre-incubated samples were transferred to each well, respectively. After incubation, the plate was washed three times with PBS-T. Subsequently, 100 µL of conjugated goat anti-mouse IgG-HRPO secondary antibody, diluted 1:10,000 in PBS was added to each well and incubated. After washing three times with PBS-T, 100 µL of 1 mg·mL
−1 ABTS substrate in ABTS buffer was added, and absorbance was measured at 405 nm after 15 min incubation in the dark.
4.4. Preparation of Immunofiltration Columns
The equilibration of immunofiltration columns was done, as described by Rettcher et al. (2015) [
26]. In brief, after degassing in 96% (v/v) ethanol in a desiccator at –0.8 bar pressure, columns were washed sequentially with 750 µL 50% (v/v) ethanol-water, 750 µL water and twice 750 µL coupling buffer. Afterward, for coupling of aflatoxin B1-BSA conjugate to the matrix, the conjugate was applied to the column in gravity flow diluted in 500 µL coupling buffer and incubated for one hour at room temperature in dark surrounding. For checkerboard titration, a coating concentration, as shown in
Figure A2, was used. For the bead-response curve, a coating concentration of 2 µg·mL
−1 was applied, as well as for cMID assays. Subsequent washing of the columns was performed twice with 750 µL PBS. Remaining binding sites were blocked by applying twice 750 µL of MID-BP. After the first 750 µL flushed through the column by gravity flow, an incubation time of 5 min was set. After the second time, columns were incubated for further 30 min after they were washed again twice with PBS.
After equilibration or blocking, columns can be stored in the coupling buffer or PBS, respectively, at 4 °C for at least 14 days. In this study, a maximum storage time of one day was used.
4.5. Biotinylation of Anti-Aflatoxin B1 Monoclonal Antibody
For biotinylation of antibodies, the EZ-Link™ NHS-PEG4 Biotinylation Kit was used as described by the manufacturer’s instruction.
4.6. Optimization of cMID
After coating and blocking of immunofiltration columns, 500 µL samples of biotinylated antibody, diluted in PBS to various final concentrations between 0.3 µg·mL
−1 and 10 µg·mL
−1, were applied and incubated for one hour at room temperature. Afterward, a washing step was performed by rinsing two times 750 µL PBS through the column. Subsequently, for checkerboard titration, 500 µL of 180 µg·mL
−1 700 nm magnetic beads suspension in PBS (pH 7.4) was added and flushed through by gravity flow. For bead response analysis, various concentrations of 700 nm magnetic beads were applied. Another washing step, as described above, was performed. For readout, columns were inserted into the portable magnetic reader and the measuring signal in mV was detected, as previously described in Rettcher et al. (2015) [
26].
4.7. cMID Calibration Curve Analysis
For cMID calibration curve experiments, a pre-incubation of free mycotoxin and biotinylated antibody was performed. For this, serially diluted aflatoxin B1 samples in PBS, with concentrations ranging from 0.006 ng·mL−1 to 100,000 ng·mL−1, were mixed 1:1 with 225 µL biotinylated antibody, also diluted in PBS. After incubation of one hour at room temperature in a dark surrounding, 500 µL of each sample was applied on coated and blocked columns and also incubated as mentioned above. After washing each column twice with 750 µL PBS, 80 µg·mL−1 of 70 nm or 700 nm magnetic particles were applied and incubated, as mentioned before. After washing twice, the readout was done as described above.
4.8. Data Analysis
For competitive ELISA as well as for competitive magnetic immunodetection and data analysis, a Hill Slope fit was done with GraphPad Prism 8.0.0. The following formulas were used to determine the LOD on the signal and on the concentration scale:


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}