Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins


Abstract
:1. Introduction
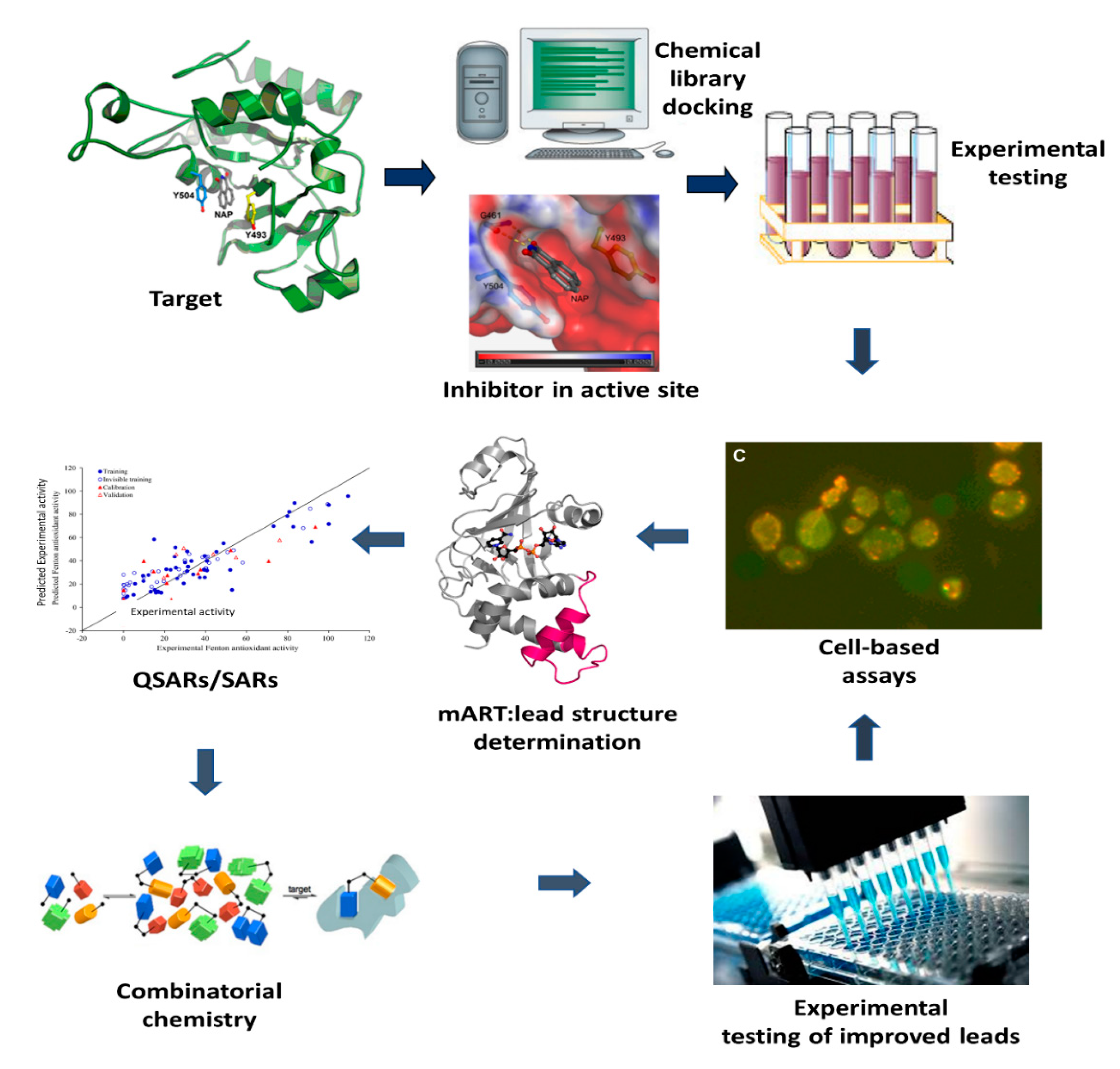
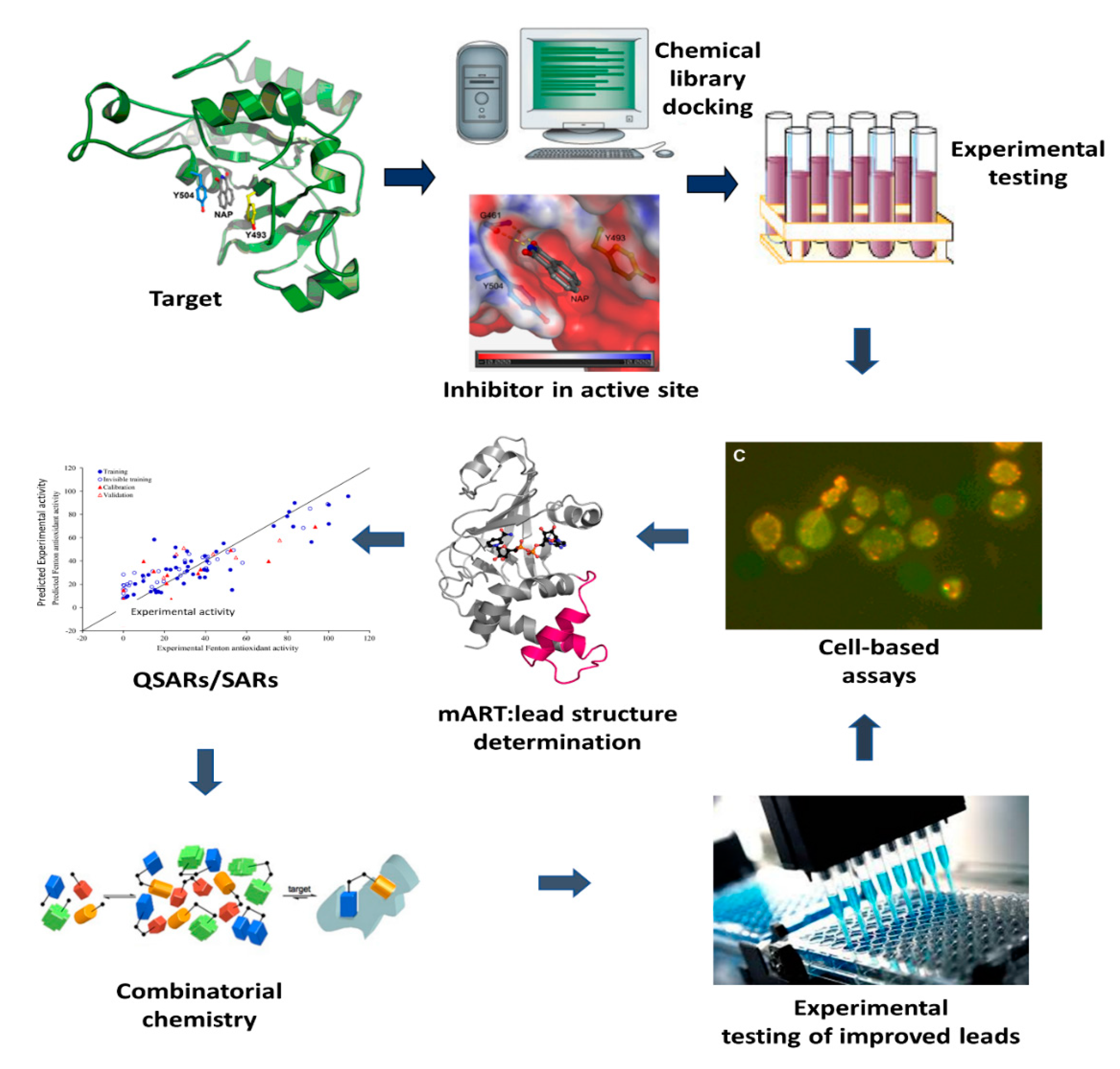
2. Anti-Virulence Approach to Inhibitor Development
3. Inhibitors against mART Toxins
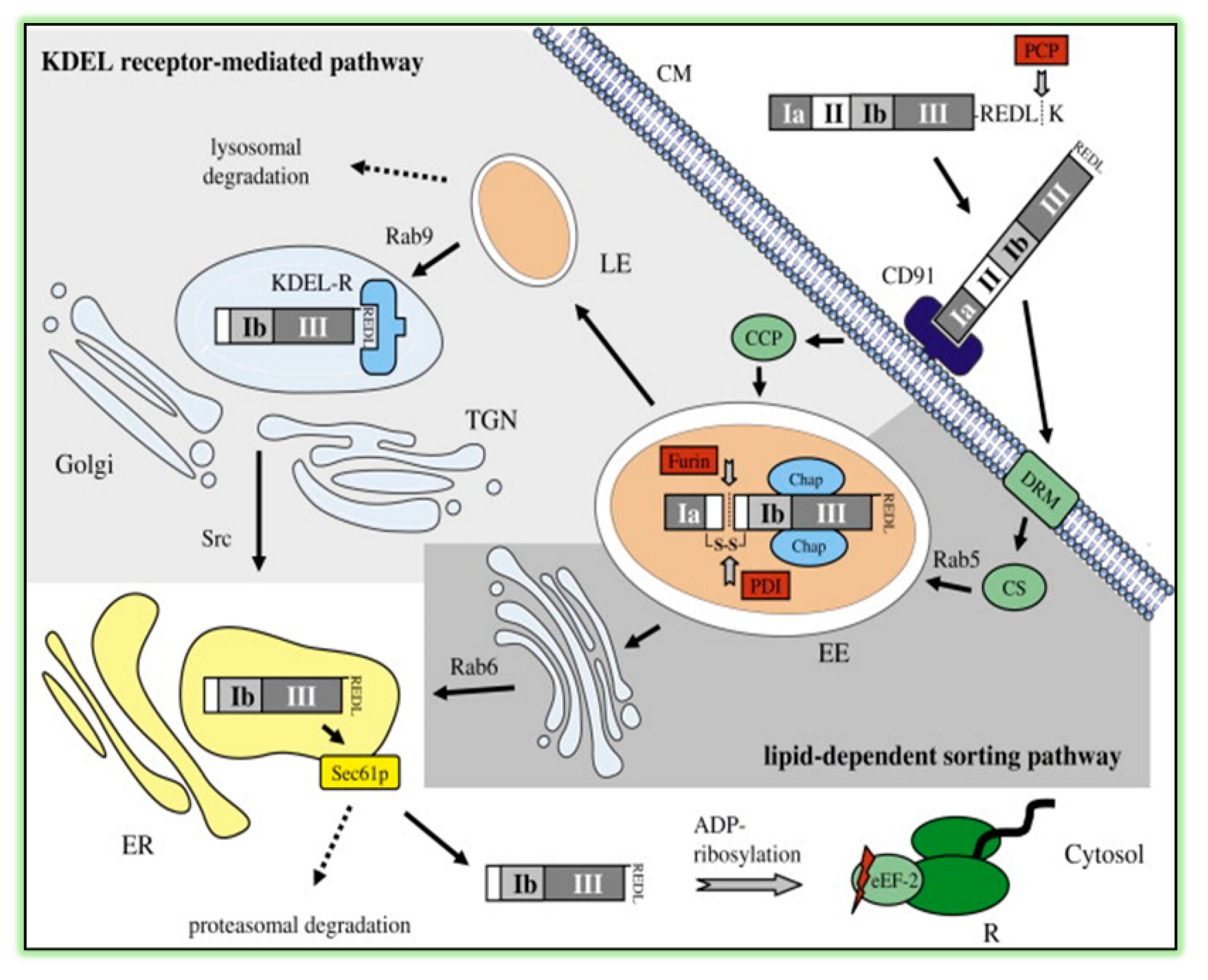
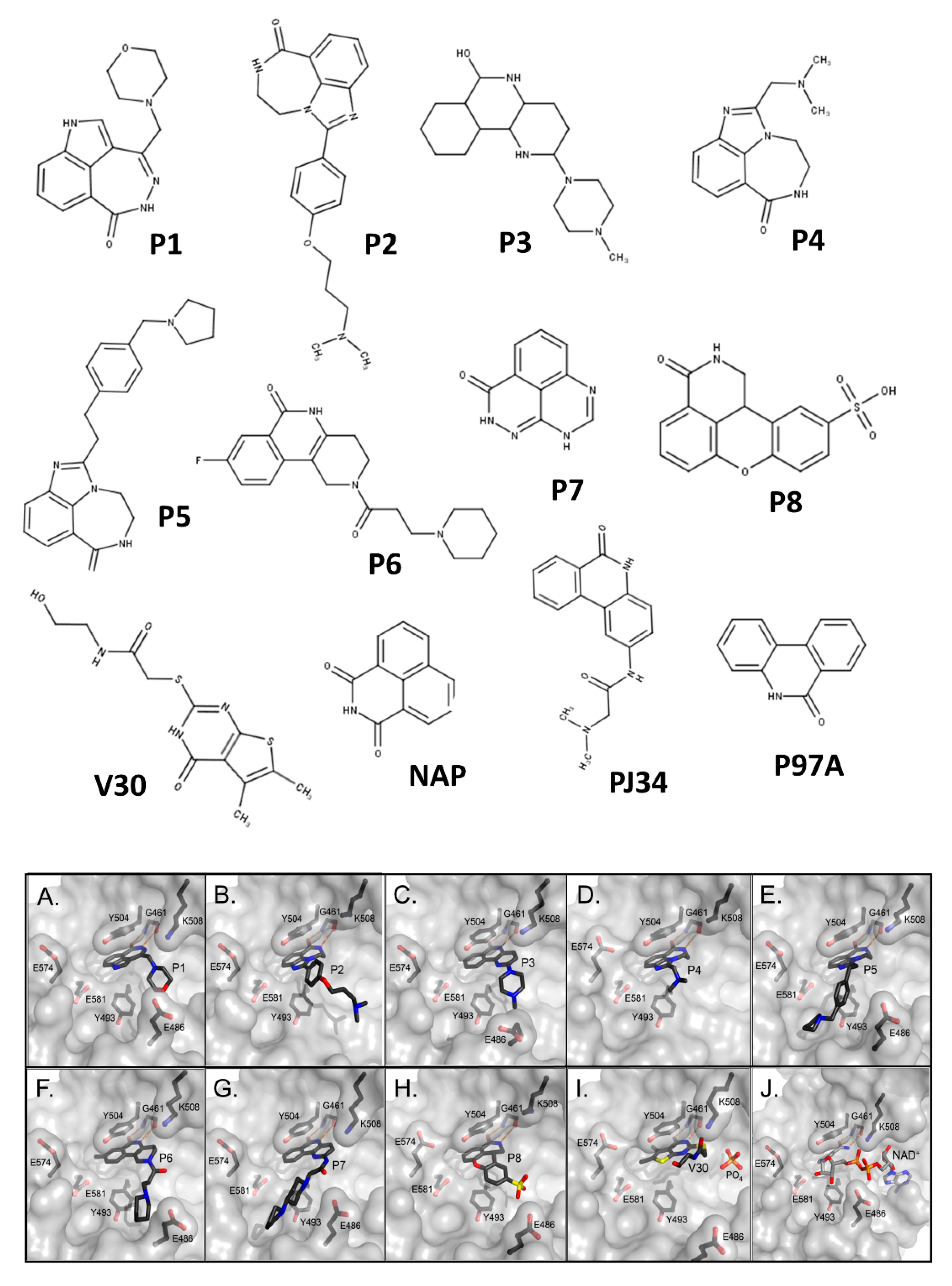
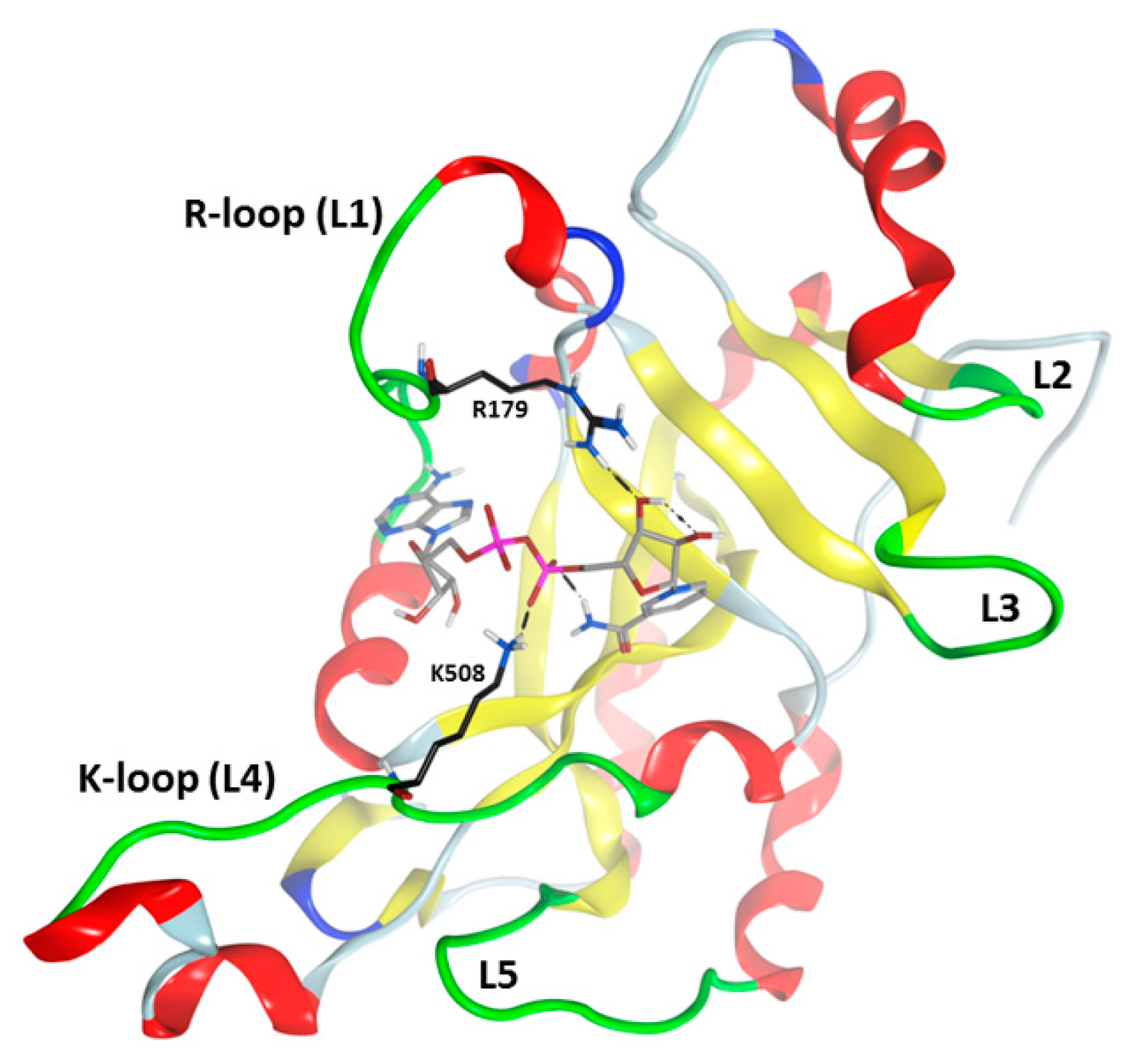
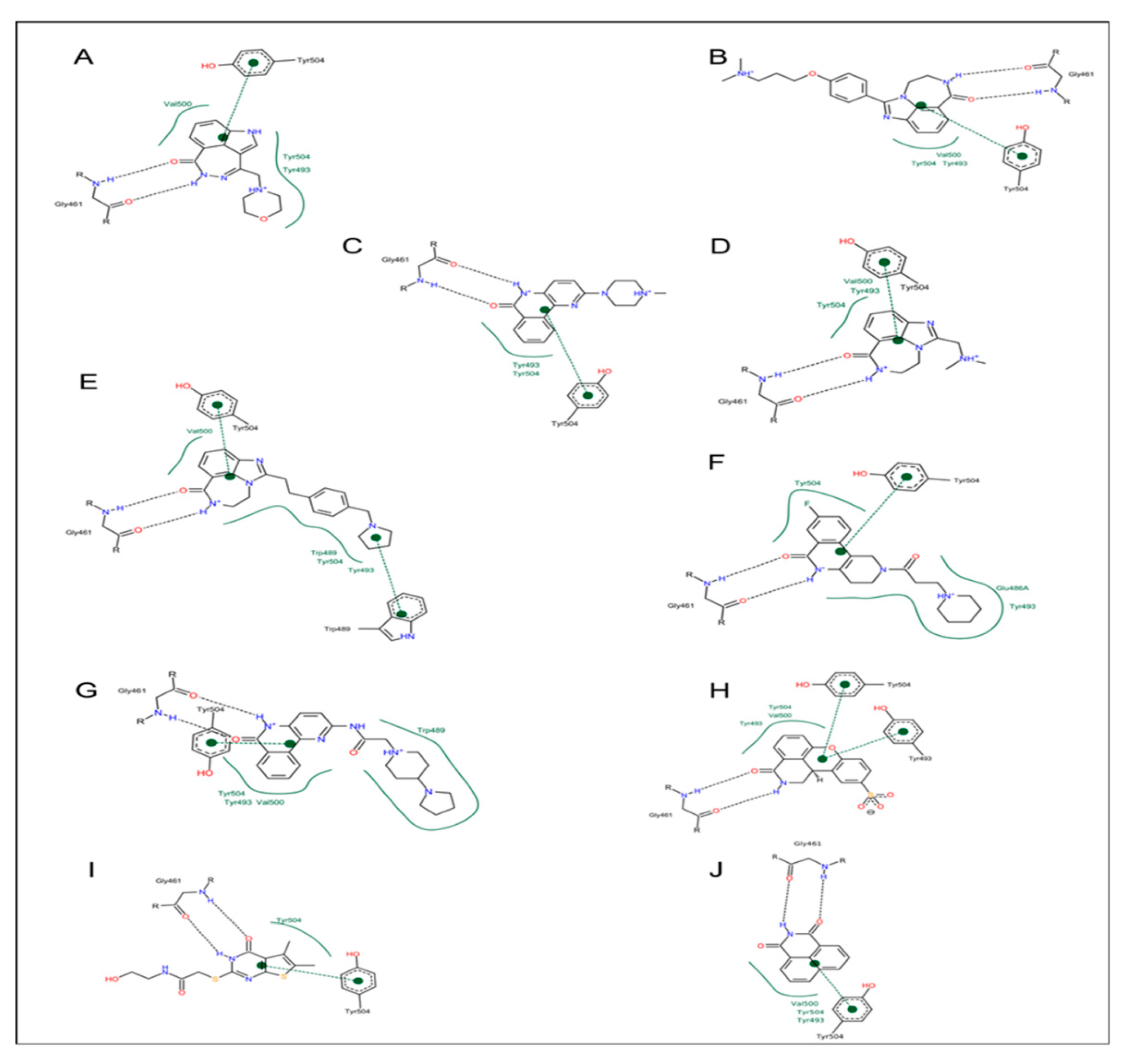
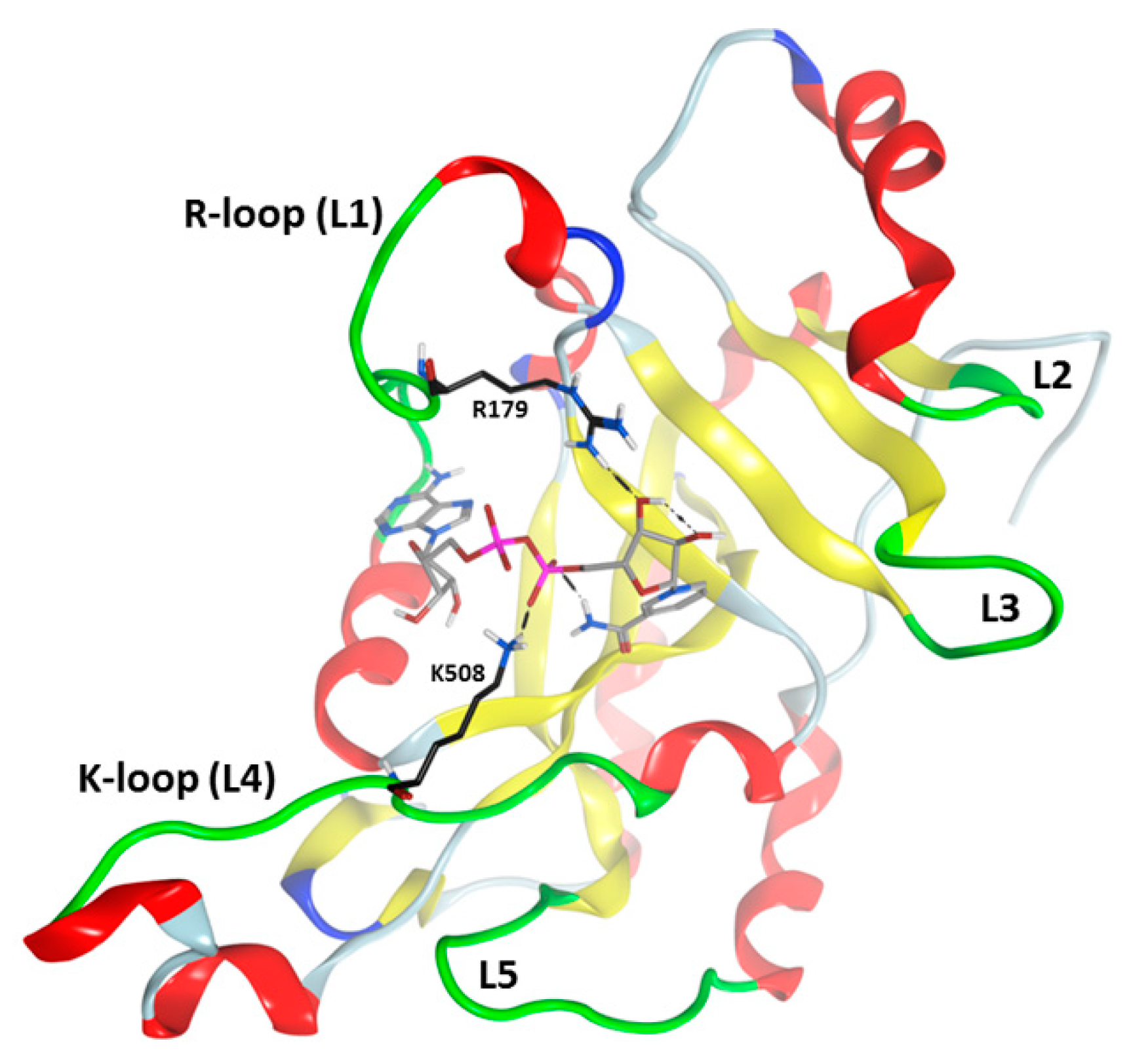
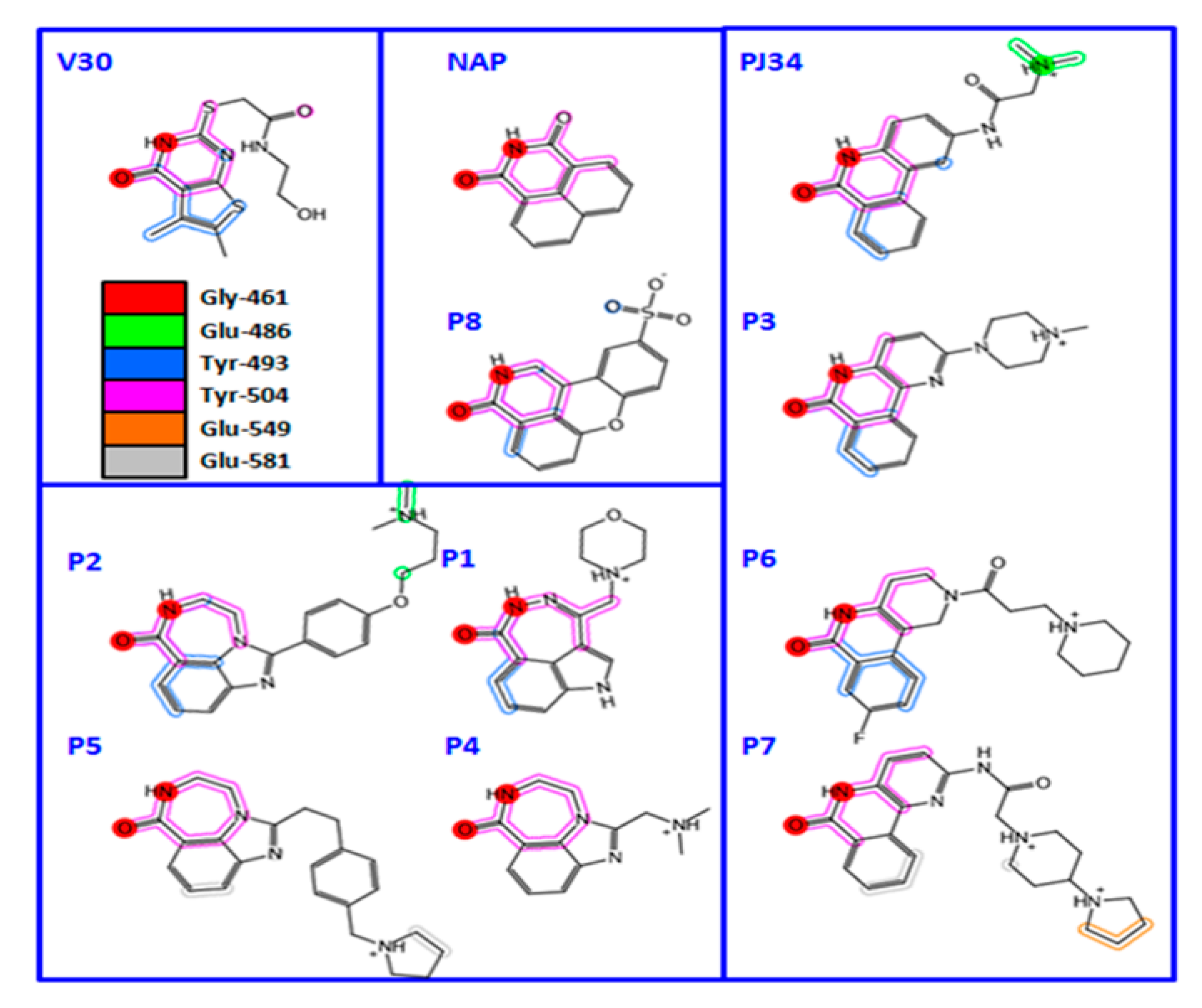
3.1. Pseudomonas aeruginosa Exotoxin A (ExoA)
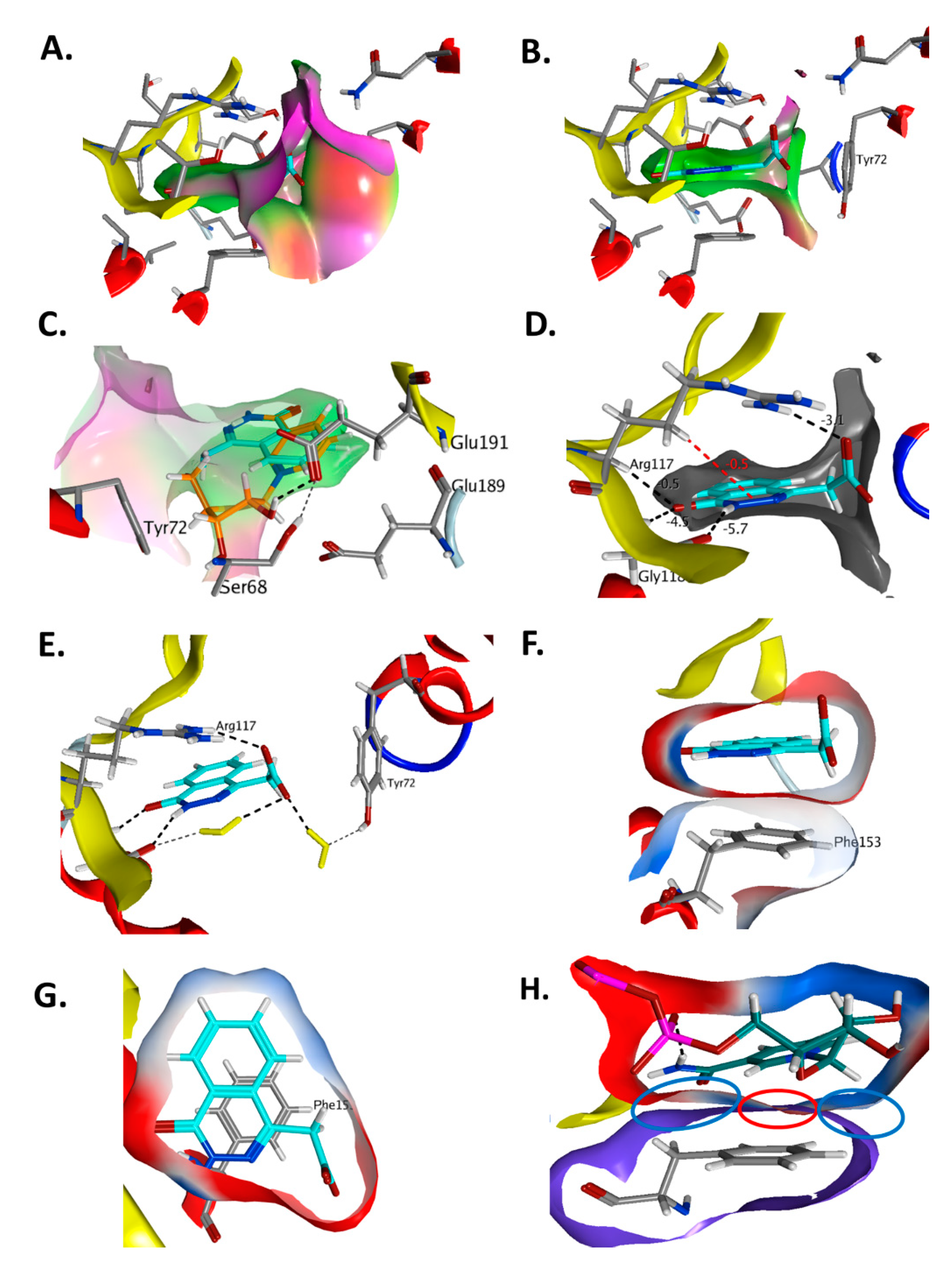
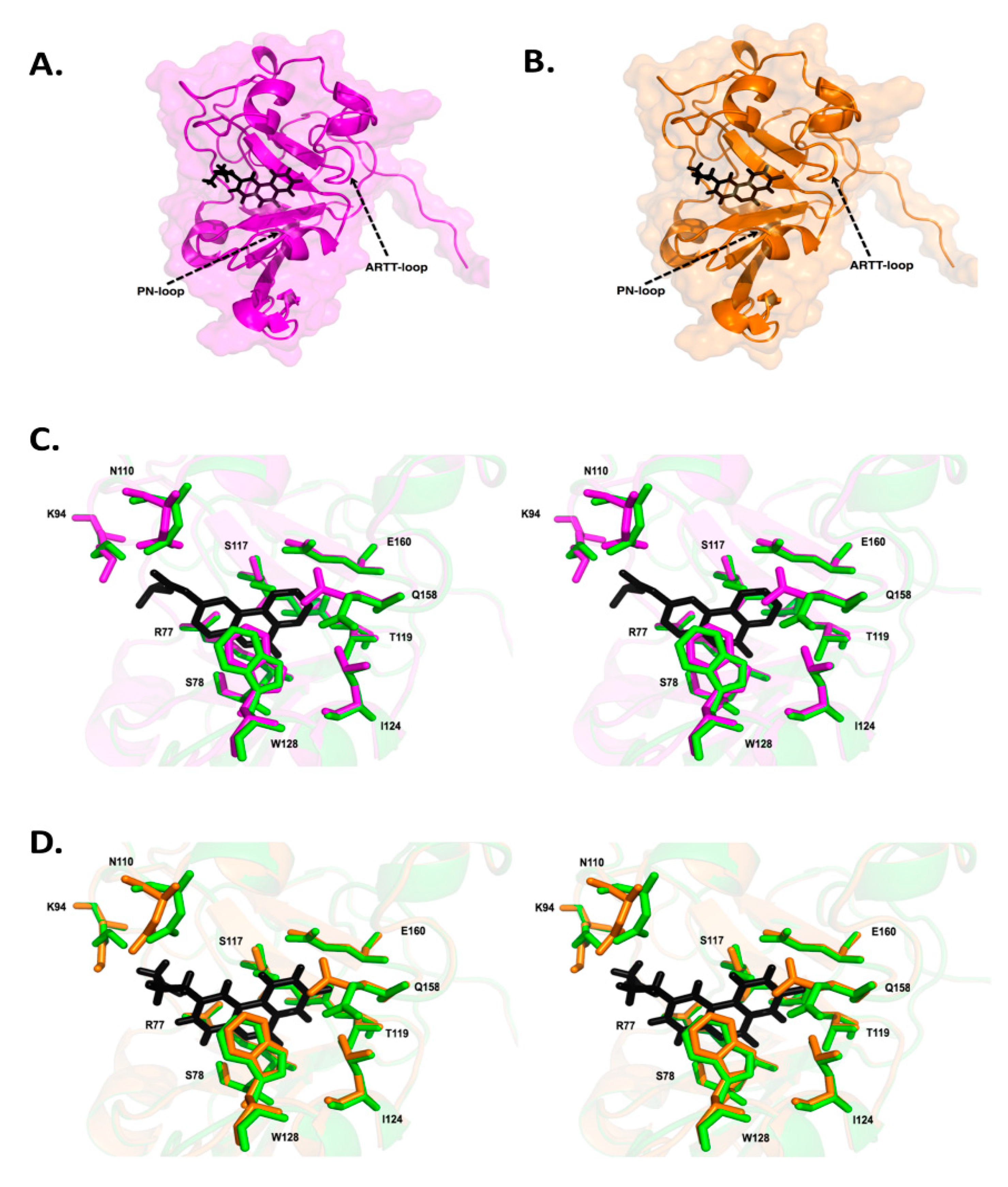
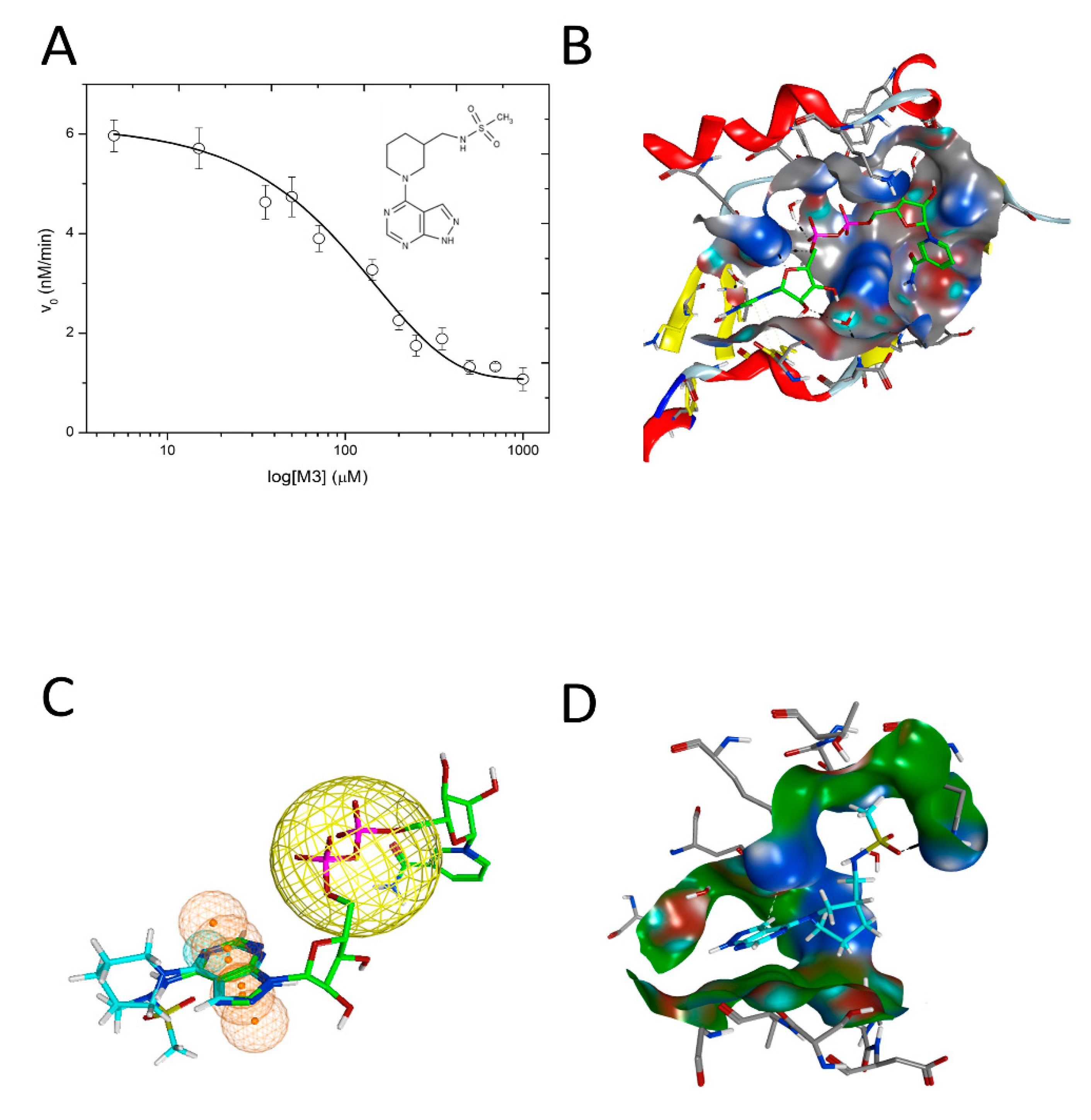
3.2. Vibrio cholerae Cholix Toxin
3.3. Vibrio splendidus Vis Toxin
3.4. Streptomyces scabies Scabin Toxin
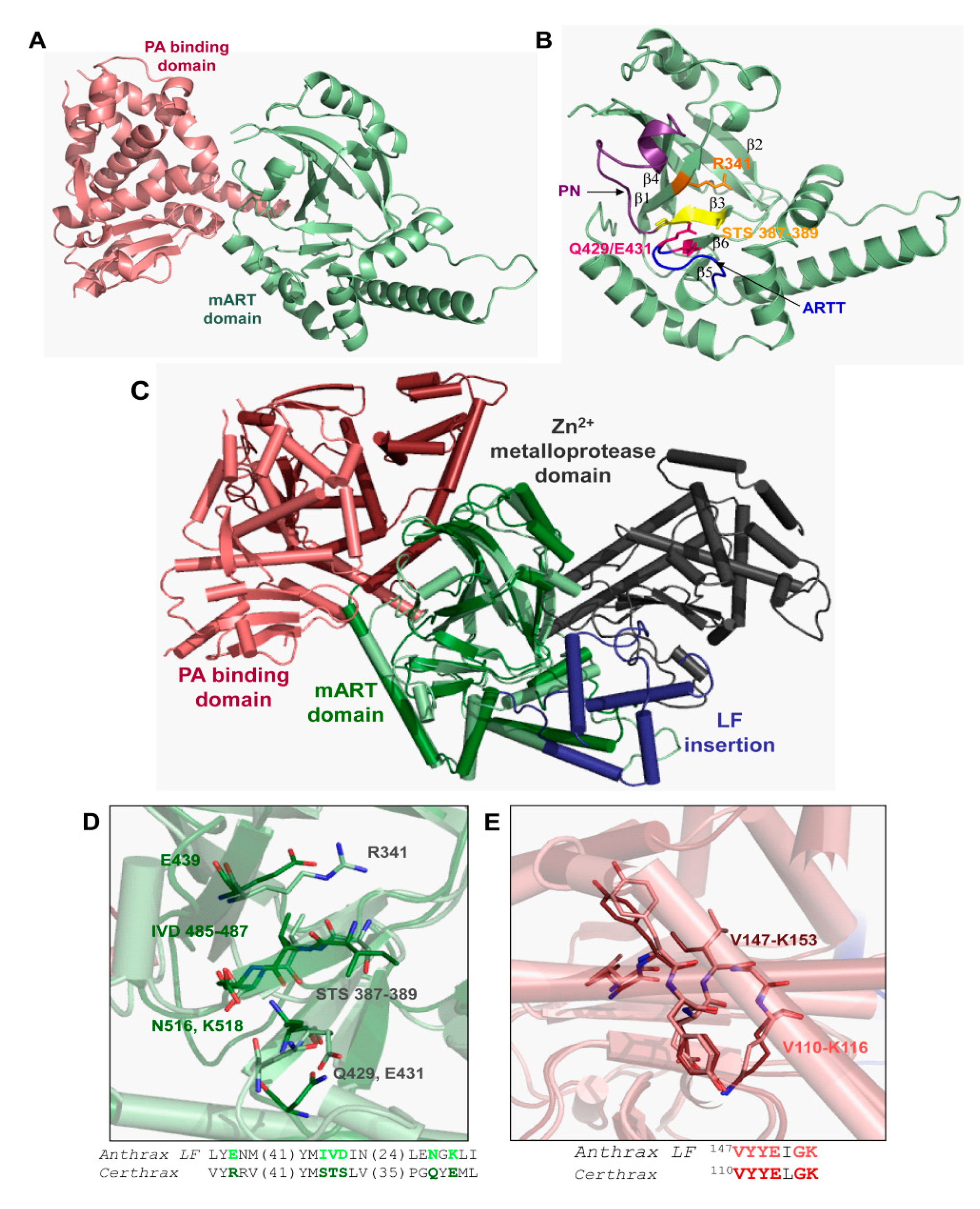
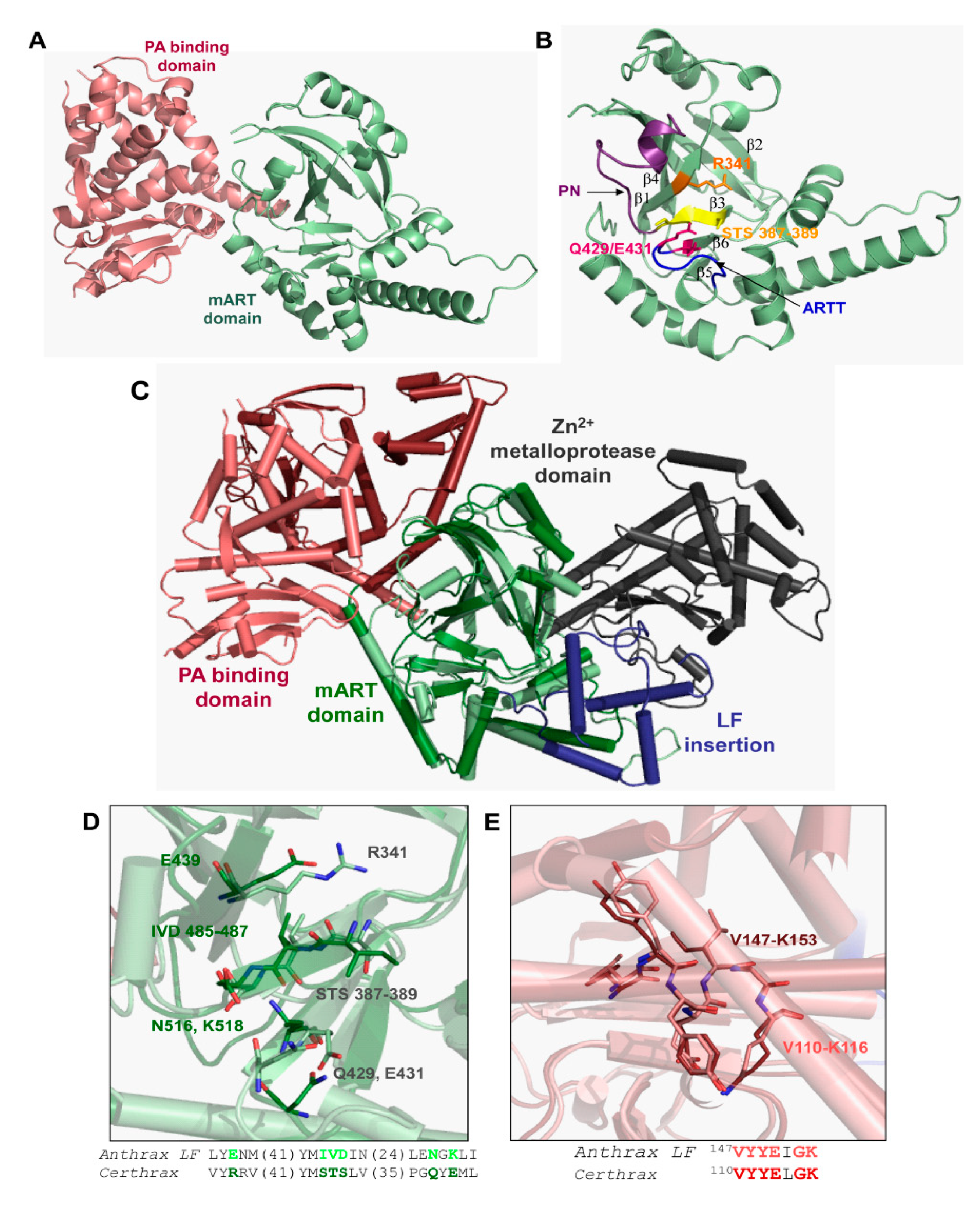
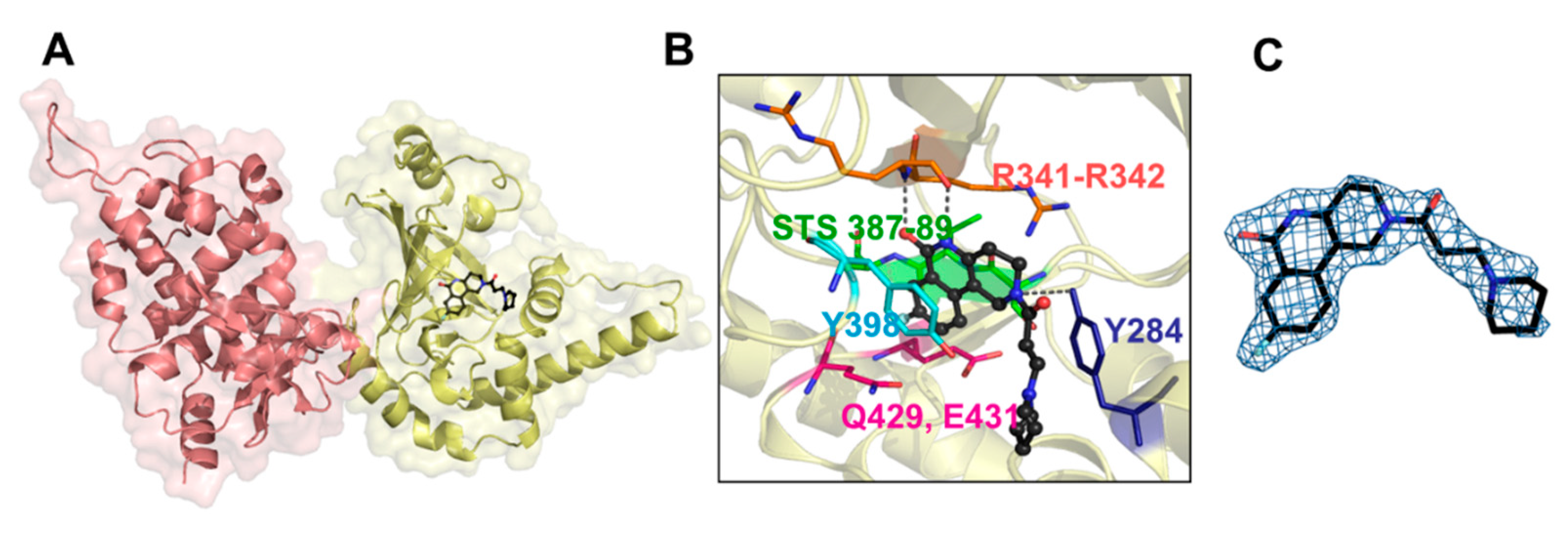
3.5. Bacillus cereus Certhrax Toxin
3.6. Paenibacillus larvae C3larvin and Plx2A
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zucca, M.; Scutera, S.; Savoia, D. New antimicrobial frontiers. Mini. Rev. Med. Chem 2011, 11, 888–900. [Google Scholar] [CrossRef]
- Lakemeyer, M.; Zhao, W.; Mandl, F.A.; Hammann, P.; Sieber, S.A. Thinking outside the box—Novel antibacterials to tackle the resistance crisis. Angew. Chem. Int. Ed. Engl. 2018, 57, 14440–14475. [Google Scholar] [CrossRef]
- Habboush, Y.; Guzman, N. Antibiotic Resistance; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Talbot, G.H.; Bradley, J.; Edwards, J.E., Jr.; Gilbert, D.; Scheld, M.; Bartlett, J.G. Bad bugs need drugs: An update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 2006, 42, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.J. Microbiology. Desperately seeking new antibiotics. Science 2008, 321, 1644–1645. [Google Scholar] [CrossRef]
- Meylan, S.; Portillo Tunon, V.; Guery, B. New antibiotics for the clinical setting, an overview. Rev. Med. Suisse 2020, 16, 713–718. [Google Scholar]
- Ghosh, D.; Veeraraghavan, B.; Elangovan, R.; Vivekanandan, P. Antibiotic Resistance and Epigenetics: More to It than Meets the Eye. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Crofts, T.S.; Gasparrini, A.J.; Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017, 15, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Spera, A.M.; Esposito, S.; Pagliano, P. Emerging antibiotic resistance: Carbapenemase-producing enterobacteria. Bad new bugs, still no new drugs. Infez. Med. 2019, 27, 357–364. [Google Scholar]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef]
- Stevens, D.L.; Bryant, A.E. Necrotizing Soft-Tissue Infections. N. Engl. J. Med. 2017, 377, 2253–2265. [Google Scholar] [CrossRef]
- Hamouda, M.A.; Jin, X.; Xu, H.; Chen, F. Quantitative microbial risk assessment and its applications in small water systems: A review. Sci. Total Environ. 2018, 645, 993–1002. [Google Scholar] [CrossRef]
- Shamloo, E.; Hosseini, H.; Abdi Moghadam, Z.; Halberg Larsen, M.; Haslberger, A.; Alebouyeh, M. Importance of Listeria monocytogenes in food safety: A review of its prevalence, detection, and antibiotic resistance. Iran. J. Vet. Res. 2019, 20, 241–254. [Google Scholar]
- Ameer, M.A.; Wasey, A.; Salen, P. Escherichia Coli (E Coli 0157 H7); StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Aliakbar Ahovan, Z.; Hashemi, A.; De Plano, L.M.; Gholipourmalekabadi, M.; Seifalian, A. Bacteriophage Based Biosensors: Trends, Outcomes and Challenges. Nanomaterials 2020, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.; Cerceo, E. Trends, Epidemiology, and Management of Multi-Drug Resistant Gram-Negative Bacterial Infections in the Hospitalized Setting. Antibiotics 2020, 9, 196. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, G.S.; Vijay, A.K.; Stapleton, F.; Henriquez, F.L.; Carnt, N. Understanding clinical and immunological features associated with Pseudomonas and Staphylococcus keratitis. Cont. Lens. Anterior. Eye 2020. [Google Scholar] [CrossRef]
- Garcia-Clemente, M.; de la Rosa, D.; Maiz, L.; Giron, R.; Blanco, M.; Olveira, C.; Canton, R.; Martinez-Garcia, M.A. Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases. J. Clin. Med. 2020, 9, 3800. [Google Scholar] [CrossRef]
- de Lacerda Coriolano, D.; de Souza, J.B.; Bueno, E.V.; Medeiros, S.; Cavalcanti, I.D.L.; Cavalcanti, I.M.F. Antibacterial and antibiofilm potential of silver nanoparticles against antibiotic-sensitive and multidrug-resistant Pseudomonas aeruginosa strains. Braz. J. Microbiol. 2020. [Google Scholar] [CrossRef]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H.A. Pseudomonas aeruginosa Biofilms. Int. J. Mol. Sci. 2020, 21, 8671. [Google Scholar] [CrossRef]
- Sawa, T.; Momiyama, K.; Mihara, T.; Kainuma, A.; Kinoshita, M.; Moriyama, K. Molecular epidemiology of clinically high-risk Pseudomonas aeruginosa strains: Practical overview. Microbiol. Immunol. 2020, 64, 331–344. [Google Scholar] [CrossRef]
- Keating, C.L.; Zuckerman, J.B.; Singh, P.K.; McKevitt, M.; Gurtovaya, O.; Bresnik, M.; Marshall, B.C.; Saiman, L.; Group, C.-A.S. Pseudomonas aeruginosa Susceptibility Patterns and Associated Clinical Outcomes in People with Cystic Fibrosis following Approval of Aztreonam Lysine for Inhalation. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Lyons, B.; Ravulapalli, R.; Lanoue, J.; Lugo, M.R.; Dutta, D.; Carlin, S.; Merrill, A.R. Scabin, a Novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem. 2016, 291, 11198–11215. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Hirabayashi, H.; Shikauchi, G.; Takamura, R.; Hiraga, K.; Minami, H.; Hashimoto, H.; Yamamoto, M.; Wakabayashi, K.; Shimizu, T.; et al. Structural basis of autoinhibition and activation of the DNA-targeting ADP-ribosyltransferase pierisin-1. J. Biol. Chem. 2017, 292, 15445–15455. [Google Scholar] [CrossRef] [Green Version]
- Rominski, A.; Roditscheff, A.; Selchow, P.; Bottger, E.C.; Sander, P. Intrinsic rifamycin resistance of Mycobacterium abscessus is mediated by ADP-ribosyltransferase MAB_0591. J. Antimicrob. Chemother. 2017, 72, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Luscher, B.; Butepage, M.; Eckei, L.; Krieg, S.; Verheugd, P.; Shilton, B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. [Google Scholar] [CrossRef]
- Fieldhouse, R.J.; Turgeon, Z.; White, D.; Merrill, A.R. Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput. Biol. 2010, 6, e1001029. [Google Scholar] [CrossRef]
- Fieldhouse, R.J.; Merrill, A.R. Needle in the haystack: Structure-based toxin discovery. Trends Biochem. Sci. 2008, 33, 546–556. [Google Scholar] [CrossRef]
- Jorgensen, R.; Purdy, A.E.; Fieldhouse, R.J.; Kimber, M.S.; Bartlett, D.H.; Merrill, A.R. Cholix Toxin, a Novel ADP-ribosylating Factor from Vibrio cholerae. J. Biol. Chem. 2008, 283, 10671–10678. [Google Scholar] [CrossRef] [Green Version]
- Visschedyk, D.; Rochon, A.; Tempel, W.; Dimov, S.; Park, H.W.; Merrill, A.R. Certhrax toxin, an anthrax-related ADP-ribosyltransferase from Bacillus cereus. J. Biol. Chem. 2012, 287, 41089–41102. [Google Scholar] [CrossRef] [Green Version]
- Ravulapalli, R.; Lugo, M.R.; Pfoh, R.; Visschedyk, D.; Poole, A.; Fieldhouse, R.J.; Pai, E.F.; Merrill, A.R. Characterization of Vis Toxin, a Novel ADP-Ribosyltransferase from Vibrio splendidus. Biochemistry 2015, 54, 5920–5936. [Google Scholar] [CrossRef]
- Krska, D.; Ravulapalli, R.; Fieldhouse, R.J.; Lugo, M.R.; Merrill, A.R. C3larvin Toxin, an ADP-ribosyltransferase from Paenibacillus larvae. J. Biol. Chem. 2015, 290, 1639–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, B.; Lugo, M.R.; Carlin, S.; Lidster, T.; Merrill, A.R. Characterization of the catalytic signature of Scabin toxin, a DNA-targeting ADP-ribosyltransferase. Biochem. J. 2018, 475, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Visschedyk, D.D.; Perieteanu, A.A.; Turgeon, Z.J.; Fieldhouse, R.J.; Dawson, J.F.; Merrill, A.R. Photox, a novel actin-targeting mono-ADP-ribosyltransferase from Photorhabdus luminescens. J. Biol. Chem. 2010, 285, 13525–13534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shniffer, A.; Visschedyk, D.D.; Ravulapalli, R.; Suarez, G.; Turgeon, Z.J.; Petrie, A.A.; Chopra, A.K.; Merrill, A.R. Characterization of an actin-targeting ADP-ribosyltransferase from Aeromonas hydrophila. J. Biol. Chem. 2012, 287, 37030–37041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebeling, J.; Funfhaus, A.; Knispel, H.; Krska, D.; Ravulapalli, R.; Heney, K.A.; Lugo, M.R.; Merrill, A.R.; Genersch, E. Characterization of the toxin Plx2A, a RhoA-targeting ADP-ribosyltransferase produced by Paenibacillus larvae. Environ. Microbiol. 2017, 19, 5100–5116. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Tremblay, O.; Heney, K.A.; Lugo, M.R.; Ebeling, J.; Genersch, E.; Merrill, A.R. Characterization of C3larvinA, a novel RhoA-targeting ADP-ribosyltransferase toxin produced by the honey bee pathogen, Paenibacillus larvae. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Turgeon, Z.; Jorgensen, R.; Visschedyk, D.; Edwards, P.R.; Legree, S.; McGregor, C.; Fieldhouse, R.J.; Mangroo, D.; Schapira, M.; Merrill, A.R. Newly discovered and characterized antivirulence compounds inhibit bacterial mono-ADP-ribosyltransferase toxins. Antimicrob. Agents Chemother. 2011, 55, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, Z.; White, D.; Jorgensen, R.; Visschedyk, D.; Fieldhouse, R.J.; Mangroo, D.; Merrill, A.R. Yeast as a tool for characterizing mono-ADP-ribosyltransferase toxins. FEMS Microbiol. Lett. 2009, 300, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Brannon, J.R.; Hadjifrangiskou, M. The arsenal of pathogens and antivirulence therapeutic strategies for disarming them. Drug Des. Devel. Ther. 2016, 10, 1795–1806. [Google Scholar] [CrossRef] [Green Version]
- Muhlen, S.; Dersch, P. Anti-virulence Strategies to Target Bacterial Infections. Curr. Top. Microbiol. Immunol. 2016, 398, 147–183. [Google Scholar] [CrossRef] [PubMed]
- Zambelloni, R.; Marquez, R.; Roe, A.J. Development of antivirulence compounds: A biochemical review. Chem. Biol. Drug Des. 2015, 85, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Arnoldo, A.; Curak, J.; Kittanakom, S.; Chevelev, I.; Lee, V.T.; Sahebol-Amri, M.; Koscik, B.; Ljuma, L.; Roy, P.J.; Bedalov, A.; et al. Identification of small molecule inhibitors of Pseudomonas aeruginosa exoenzyme S using a yeast phenotypic screen. PLoS Genet. 2008, 4, e1000005. [Google Scholar] [CrossRef]
- Wick, M.J.; Frank, D.W.; Storey, D.G.; Iglewski, B.H. Structure, function, and regulation of Pseudomonas aeruginosa exotoxin A. Annu. Rev. Microbiol. 1990, 44, 335–363. [Google Scholar] [CrossRef]
- Iglewski, B.H.; Kabat, D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc. Natl. Acad. Sci. USA 1975, 72, 2284–2288. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Walter, H.; Haug, M.; Meisner, C.; Grasemann, H.; Doring, G. Diagnostic value of serum antibodies in early Pseudomonas aeruginosa infection in cystic fibrosis patients. Pediatr. Pulmonol. 2007, 42, 249–255. [Google Scholar] [CrossRef]
- Du, X.; Youle, R.J.; FitzGerald, D.J.; Pastan, I. Pseudomonas exotoxin A-mediated apoptosis is Bak dependent and preceded by the degradation of Mcl-1. Mol. Cell Biol. 2010, 30, 3444–3452. [Google Scholar] [CrossRef] [Green Version]
- Carroll, S.F.; Collier, R.J. Active site of Pseudomonas aeruginosa exotoxin A. Glutamic acid 553 is photolabeled by NAD and shows functional homology with glutamic acid 148 of diphtheria toxin. J. Biol. Chem. 1987, 262, 8707–8711. [Google Scholar]
- Hessler, J.L.; Kreitman, R.J. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry 1997, 36, 14577–14582. [Google Scholar] [CrossRef]
- Smith, D.C.; Spooner, R.A.; Watson, P.D.; Murray, J.L.; Hodge, T.W.; Amessou, M.; Johannes, L.; Lord, J.M.; Roberts, L.M. Internalized Pseudomonas exotoxin A can exploit multiple pathways to reach the endoplasmic reticulum. Traffic 2006, 7, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Koopmann, J.O.; Albring, J.; Huter, E.; Bulbuc, N.; Spee, P.; Neefjes, J.; Hammerling, G.J.; Momburg, F. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 2000, 13, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Yates, S.P.; Jorgensen, R.; Andersen, G.R.; Merrill, A.R. Stealth, and mimicry by deadly bacterial toxins. Trends Biochem. Sci. 2006, 31, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Kwon, H.Y. Expression of 14-3-3delta, cdc2 and cyclin B proteins related to exotoxin A-induced apoptosis in HeLa S3 cells. Int. Immunopharmacol. 2007, 7, 1185–1191. [Google Scholar] [CrossRef]
- Sharma, A.K.; FitzGerald, D. Pseudomonas exotoxin kills Drosophila S2 cells via apoptosis. Toxicon 2010, 56, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, R.; Merrill, A.R.; Yates, S.P.; Marquez, V.E.; Schwan, A.L.; Boesen, T.; Andersen, G.R. Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature 2005, 436, 979–984. [Google Scholar] [CrossRef]
- Spahn, C.M.; Gomez-Lorenzo, M.G.; Grassucci, R.A.; Jorgensen, R.; Andersen, G.R.; Beckmann, R.; Penczek, P.A.; Ballesta, J.P.; Frank, J. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 2004, 23, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef]
- Armstrong, S.; Li, J.H.; Zhang, J.; Merrill, A.R. Characterization of competitive inhibitors for the transferase activity of Pseudomonas aeruginosa exotoxin A. J. Enzym. Inhib. Med. Chem. 2002, 17, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Yates, S.P.; Taylor, P.L.; Jorgensen, R.; Ferraris, D.; Zhang, J.; Andersen, G.R.; Merrill, A.R. Structure-function analysis of water-soluble inhibitors of the catalytic domain of exotoxin A from Pseudomonas aeruginosa. Biochem. J. 2005, 385, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Purdy, A.; Rohwer, F.; Edwards, R.; Azam, F.; Bartlett, D.H. A Glimpse into the Expanded Genome Content of Vibrio cholerae through Identification of Genes Present in Environmental Strains. J. Bacteriol. 2005, 187, 2992–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, R.; Wang, Y.; Visschedyk, D.; Merrill, A.R. The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 2008, 9, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdy, A.E.; Balch, D.; Lizarraga-Partida, M.L.; Islam, M.S.; Martinez-Urtaza, J.; Huq, A.; Colwell, R.R.; Bartlett, D.H. Diversity, and distribution of cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. Environ. Microbiol. Rep. 2010, 2, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.P.; Asakura, M.; Chowdhury, N.; Neogi, S.B.; Hinenoya, A.; Golbar, H.M.; Yamate, J.; Arakawa, E.; Tada, T.; Ramamurthy, T.; et al. Novel cholix toxin variants, ADP-ribosylating toxins in Vibrio cholerae non-O1/non-O139 strains, and their pathogenicity. Infect. Immun. 2013, 81, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Saidi, S.M.; Chowdhury, N.; Awasthi, S.P.; Asakura, M.; Hinenoya, A.; Iijima, Y.; Yamasaki, S. Prevalence of Vibrio cholerae O1 El Tor variant in a cholera-endemic zone of Kenya. J. Med. Microbiol. 2014, 63, 415–420. [Google Scholar] [CrossRef]
- Dalsgaard, A.; Albert, M.J.; Taylor, D.N.; Shimada, T.; Meza, R.; Serichantalergs, O.; Echeverria, P. Characterization of Vibrio cgolerae non-O1 serogroups obtained from an outbreak of diarrhea in Lima, Peru. J. Clin. Microbiol. 1995, 33, 2715–2722. [Google Scholar] [CrossRef] [Green Version]
- Dalsgaard, A.; Forslund, A.; Hesselbjerg, A.; Bruun, B. Clinical manifestations and characterization of extra-intestinal Vibrio cholerae non-O1, non-O139 infections in Denmark. Clin. Microbiol. Infect. 2000, 6, 625–627. [Google Scholar] [CrossRef] [Green Version]
- Yahiro, K.; Ogura, K.; Terasaki, Y.; Satoh, M.; Miyagi, S.; Terasaki, M.; Yamasaki, E.; Moss, J. Cholix toxin, an eukaryotic elongation factor 2 ADP-ribosyltransferase, interacts with Prohibitins and induces apoptosis with mitochondrial dysfunction in human hepatocytes. Cell Microbiol. 2019, 21, e13033. [Google Scholar] [CrossRef]
- Lugo, M.R.; Merrill, A.R. The Father, Son and Cholix Toxin: The Third Member of the DT Group Mono-ADP-Ribosyltransferase Toxin Family. Toxins 2015, 7, 2757–2772. [Google Scholar] [CrossRef] [Green Version]
- Fieldhouse, R.J.; Jorgensen, R.; Lugo, M.R.; Merrill, A.R. The 1.8 Å cholix toxin crystal structure in complex with NAD+ and evidence for a new kinetic model. J. Biol. Chem. 2012, 287, 21176–21188. [Google Scholar] [CrossRef] [Green Version]
- Lugo, M.R.; Merrill, A.R. A comparative structure-function analysis of active-site inhibitors of Vibrio cholerae cholix toxin. J. Mol. Recognit. 2015, 28, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.E.; Yeates, T.O.; Eisenberg, D. Unusual conformation of nicotinamide adenine dinucleotide (NAD) bound to diphtheria toxin: A comparison with NAD bound to the oxidoreductase enzymes. Protein Sci. 1997, 6, 2084–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugo, M.R.; Merrill, A.R. Pocket analysis of the full-length cholix toxin. An assessment of the structure-dynamics of the apo catalytic domain. J. Biomol. Struct. Dyn. 2015, 33, 2452–2468. [Google Scholar] [CrossRef] [PubMed]
- Gay, M.; Berthe, F.C.; Le, R.F. Screening of Vibrio isolates to develop an experimental infection model in the Pacific oyster Crassostrea gigas. Dis. Aquat. Organ. 2004, 59, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Armijos-Jaramillo, V.; Santander-Gordon, D.; Soria, R.; Pazmino-Betancourth, M.; Echeverria, M.C. A whole genome analysis reveals the presence of a plant PR1 sequence in the potato pathogen Streptomyces scabies and other Streptomyces species. Mol. Phylogenet. Evol. 2017, 114, 346–352. [Google Scholar] [CrossRef]
- Loria, R.; Kers, J.; Joshi, M. Evolution of plant pathogenicity in Streptomyces. Annu Rev. Phytopathol. 2006, 44, 469–487. [Google Scholar] [CrossRef]
- Bignell, D.R.; Fyans, J.K.; Cheng, Z. Phytotoxins produced by plant pathogenic Streptomyces species. J. Appl. Microbiol. 2014, 116, 223–235. [Google Scholar] [CrossRef]
- Wanner, L.A.; Kirk, W.W.; Qu, X.S. Field efficacy of nonpathogenic Streptomyces species against potato common scab. J. Appl. Microbiol. 2014, 116, 123–133. [Google Scholar] [CrossRef]
- St-Onge, R.; Gadkar, V.J.; Arseneault, T.; Goyer, C.; Filion, M. The ability of Pseudomonas sp. LBUM 223 to produce phenazine-1-carboxylic acid affects the growth of Streptomyces scabies, the expression of thaxtomin biosynthesis genes and the biological control potential against common scab of potato. FEMS Microbiol. Ecol. 2011, 75, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.V.; Mann, S.G.; Antelmann, H.; Widdick, D.A.; Fyans, J.K.; Chandra, G.; Hutchings, M.I.; Toth, I.; Hecker, M.; Loria, R.; et al. The twin arginine protein transport pathway exports multiple virulence proteins in the plant pathogen Streptomyces scabies. Mol. Microbiol. 2010, 77, 252–271. [Google Scholar] [CrossRef] [Green Version]
- Lugo, M.R.; Lyons, B.L.; Lento, C.; Wilson, D.J.; Merrill, A.R. Dynamics of Scabin toxin. A proposal for the binding mode of the DNA substrate. PLoA ONE 2018, 13, e0194425. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.C.; Barbieri, J.T. Bacillus cereus Certhrax ADP-ribosylates vinculin to disrupt focal adhesion complexes and cell adhesion. J. Biol. Chem. 2014, 289, 10650–10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.C.; Vergis, J.M.; Ebrahimi, A.V.; Ventura, C.L.; O’Brien, A.D.; Barbieri, J.T. Host cell cytotoxicity and cytoskeleton disruption by CerADPr, an ADP-ribosyltransferase of Bacillus cereus G9241. Biochemistry 2013, 52, 2309–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genersch, E. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 2010, 103, S10–S19. [Google Scholar] [CrossRef]
- Tauber, J.P.; Collins, W.R.; Schwarz, R.S.; Chen, Y.; Grubbs, K.; Huang, Q.; Lopez, D.; Peterson, R.; Evans, J.D. Natural Product Medicines for Honey Bees: Perspective and Protocols. Insects 2019, 10, 356. [Google Scholar] [CrossRef] [Green Version]
- Funfhaus, A.; Poppinga, L.; Genersch, E. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ. Microbiol. 2013, 15, 2951–2965. [Google Scholar] [CrossRef]
- Lugo, M.R.; Ravulapalli, R.; Dutta, D.; Merrill, A.R. Structural variability of C3larvin toxin. Intrinsic dynamics of the alpha/beta fold of the C3-like group of mono-ADP-ribosyltransferase toxins. J. Biomol. Struct. Dyn. 2016, 34, 2537–2560. [Google Scholar] [CrossRef]
- Galdino, A.C.M.; de Oliveira, M.P.; Ramalho, T.C.; de Castro, A.A.; Branquinha, M.H.; Santos, A.L.S. Anti-Virulence Strategy against the Multidrug-Resistant Bacterial Pathogen Pseudomonas aeruginosa: Pseudolysin (Elastase B) as a Potential Druggable Target. Curr. Protein. Pept. Sci. 2019, 20, 471–487. [Google Scholar] [CrossRef]
- Kongkham, B.; Prabakaran, D.; Puttaswamy, H. Opportunities and challenges in managing antibiotic resistance in bacteria using plant secondary metabolites. Fitoterapia 2020, 147, 104762. [Google Scholar] [CrossRef]
- Marini, E.; Magi, G.; Ferretti, G.; Bacchetti, T.; Giuliani, A.; Pugnaloni, A.; Rippo, M.R.; Facinelli, B. Attenuation of Listeria monocytogenes Virulence by Cannabis sativa L. Essential Oil. Front. Cell Infect. Microbiol. 2018, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Liu, F.; Zhu, K.; Shen, J.Z. Natural Products That Target Virulence Factors in Antibiotic-Resistant Staphylococcus aureus. J. Agric. Food Chem. 2019, 67, 13195–13211. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.G.; El-Dougdoug, N.K. Controlling foodborne pathogens with natural antimicrobials by biological control and antivirulence strategies. Heliyon 2020, 6, e05020. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
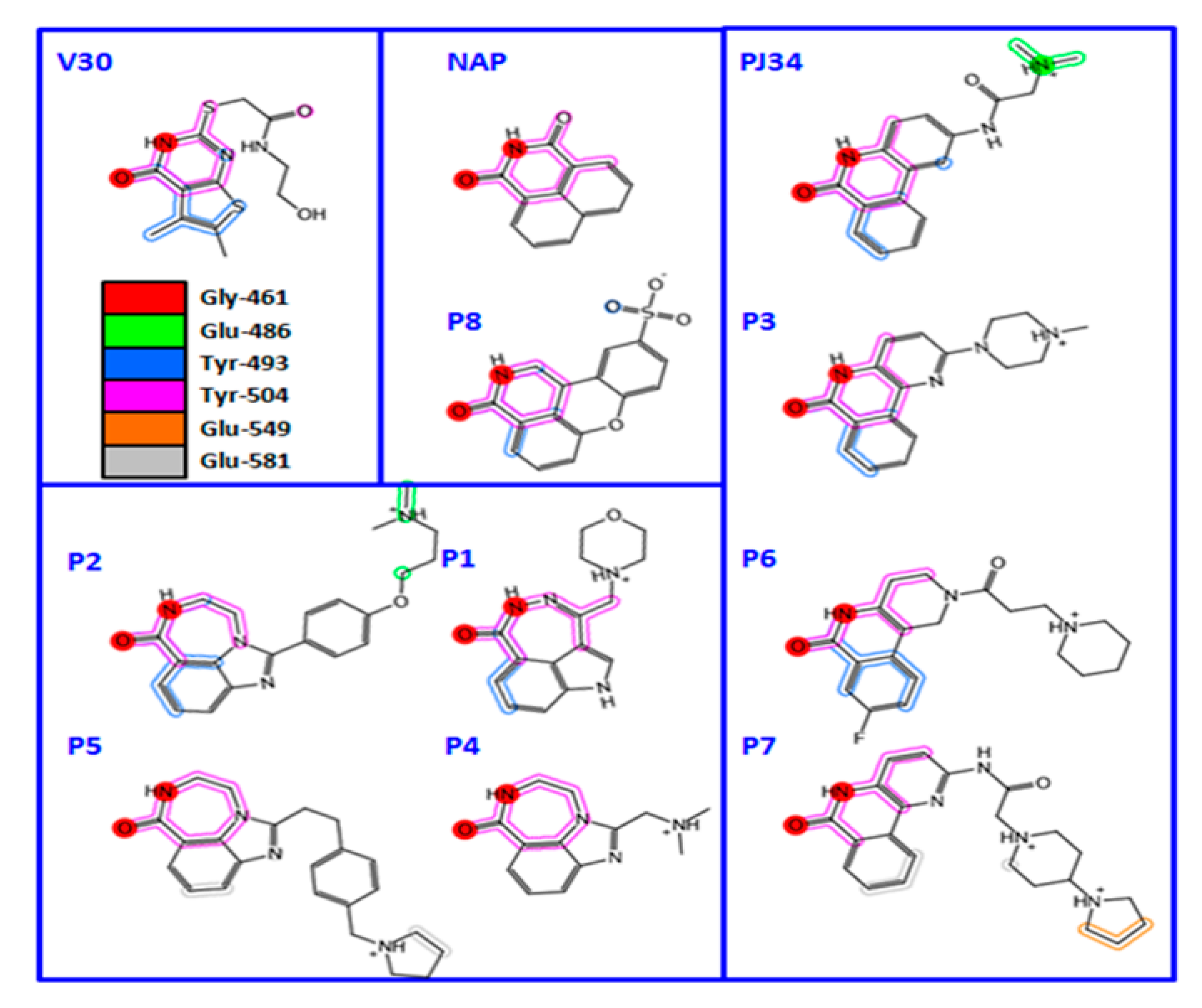
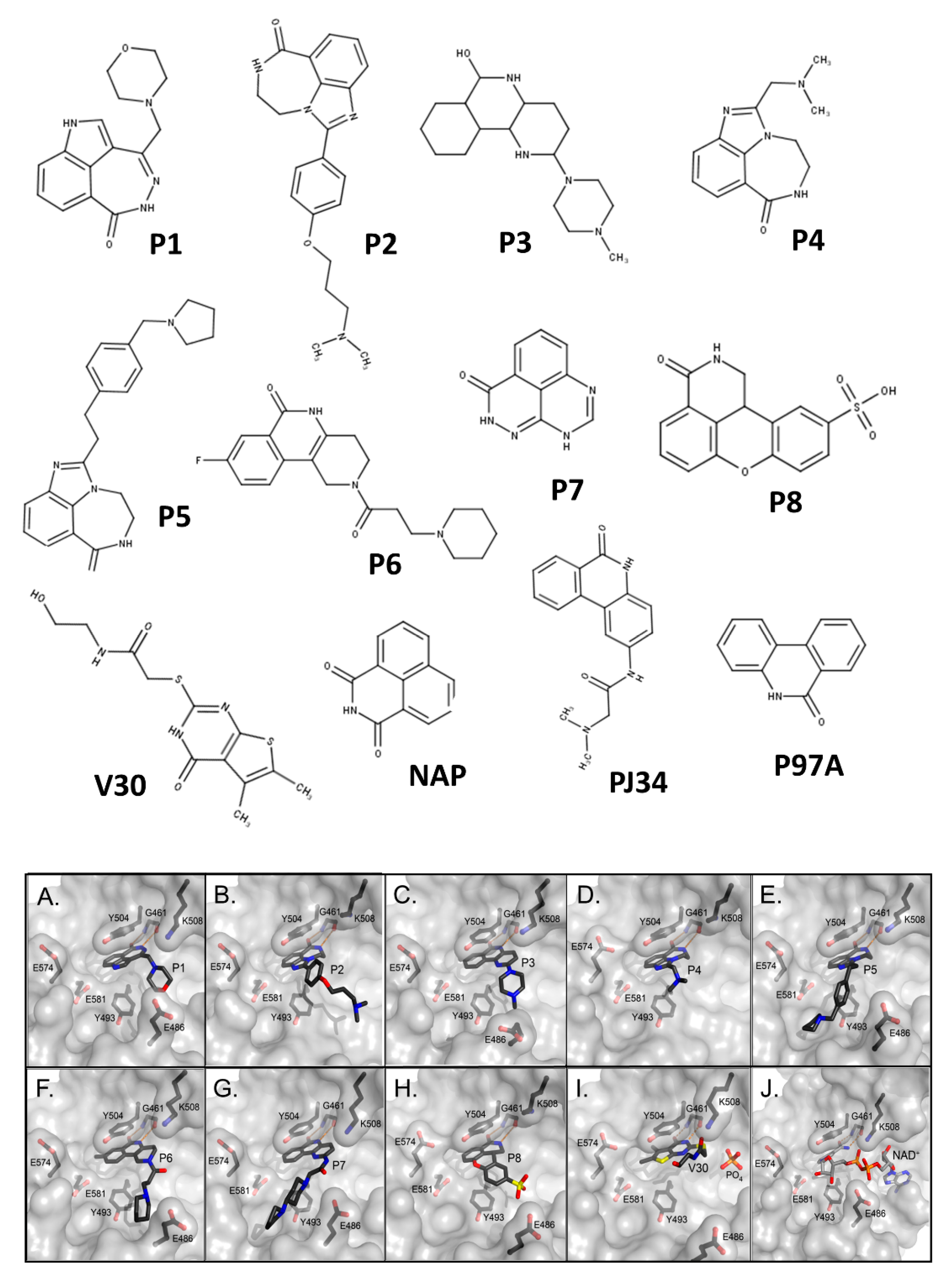
| Compound | Inhibitor Name | Structure | IC50 (μM) |
|---|---|---|---|
| PJ-34 | N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide | 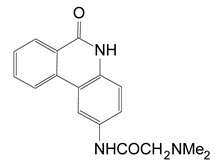 | 0.296 ± 0.080 |
| D | 3-(morpholin-4-ylmethyl)-1,5-dihydro-6H-[1,2]diazepino[4,5,6-cd]indol-6-one | 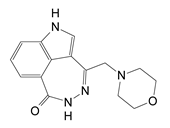 | 0.617 ± 0.023 |
| F | 1-[4-(3-dimethylamino-propoxy)-phenyl]-8,9-dihydro-7H-2,7,9a-triaza-benzo[cd]azulen-6-one HCl |  | 0.495 ± 0.090 |
| G | 2-(4-methylpiperazin-1-yl)-5H-benzo[c][1,5]naphthyridin-6-one MsOH | 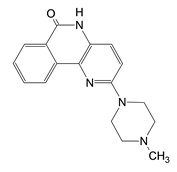 | 1.965 ± 0.032 |
| H | 1-dimethylaminomethyl-8,9-dihydro-7H-2,7,9a-triaza-benzo[cd]azulen-6-one HCl |  | 0.964 ± 0.050 |
| I | 1-[2-(4-pyrrolidin-1-ylmethyl-phenyl)-ethyl]-8,9-dihydro-7H-2,7,9a-triaza-benzo[cd]azulen-6-one hydrochloride HCl |  | 6.52 ± 0.39 |
| L | 8-fluoro-2-(3-piperidin-1-ylpropanoyl)-1,3,4,5-2H-tetrahydrobenzo[c]-1,6-naphthyridin-6-one MsOH |  | 1.542 ± 0.044 |
| M | N-(6-oxo-5,6-dihydrobenzo[c][1,5]naphthyridin-2-yl)-2-(4-pyrrolidin-1-ylpiperidin-1-yl)acetamide·HCl | 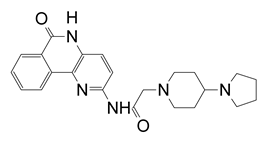 | 0.375 |
| N | 2-(4-isopropylpiperazin-1-yl)-5H-benzo[c][1,5]naphthyridin-6-one MsOH |  | 1.07 |
| P | 1,11b-dihydro-[1]benzopyrano[4,3,2-de]isoquinolin-3(2H)-one-10-sulfonic acid |  | 0.453 |
| Compound | a Kd (nM) | b Ki (nM) | c IC50 (nM) | d EC50 (µM) |
|---|---|---|---|---|
| P1 | 10 ± 2; 650 ± 50 | 22 ± 4 | 170 ± 30 | 2.9 ± 0.8 |
| P2 | 260 ± 10 | 63 ± 5 | 480 ± 40 | e n.d. |
| P3 | 1470 ± 30 | 90 ± 17 | 690 ± 130 | e n.d. |
| P4 | 1380 ± 30 | 132 ± 7 | 960 ± 50 | 12.6 ± 3.3 |
| P5 | 750 ± 10 | 582 ± 124 | 4460 ± 950 | 16.7 ± 1.9 |
| P6 | 1100 ± 20 | 80 ± 18 | 610 ± 140 | 3.4 ± 1.6 |
| P7 | 680 ± 40 | 118 ± 7 | 908 ± 118 | e n.d. |
| P8 | 160 ± 30; 5210 ± 1780 | 136 ± 3 | 1040 ± 136 | e n.d. |
| V30 | 931 ± 74 | 367 ± 3 | 2815 ± 22 | 8.8 ± 0.5 |
| NAP | 950 ± 30 | 12 ± 1 | 90 ± 10 | 3.8 ± 0.9 |
| PJ34 | 820 ± 54 | 37 ± 9 | 280 ± 70 | e n.d. |
| PJ97A | 393 ± 64 | 610 ± 175 | 4674 ± 175 | e n.d. |
| Inhibitor | a IC50 (μM) | b Ki (μM) | c pIC50 | d KD (μM) |
|---|---|---|---|---|
| PJ34 | 12 ± 1 | 3 ± 0.2 | 4.9 | 14 ± 0.5 |
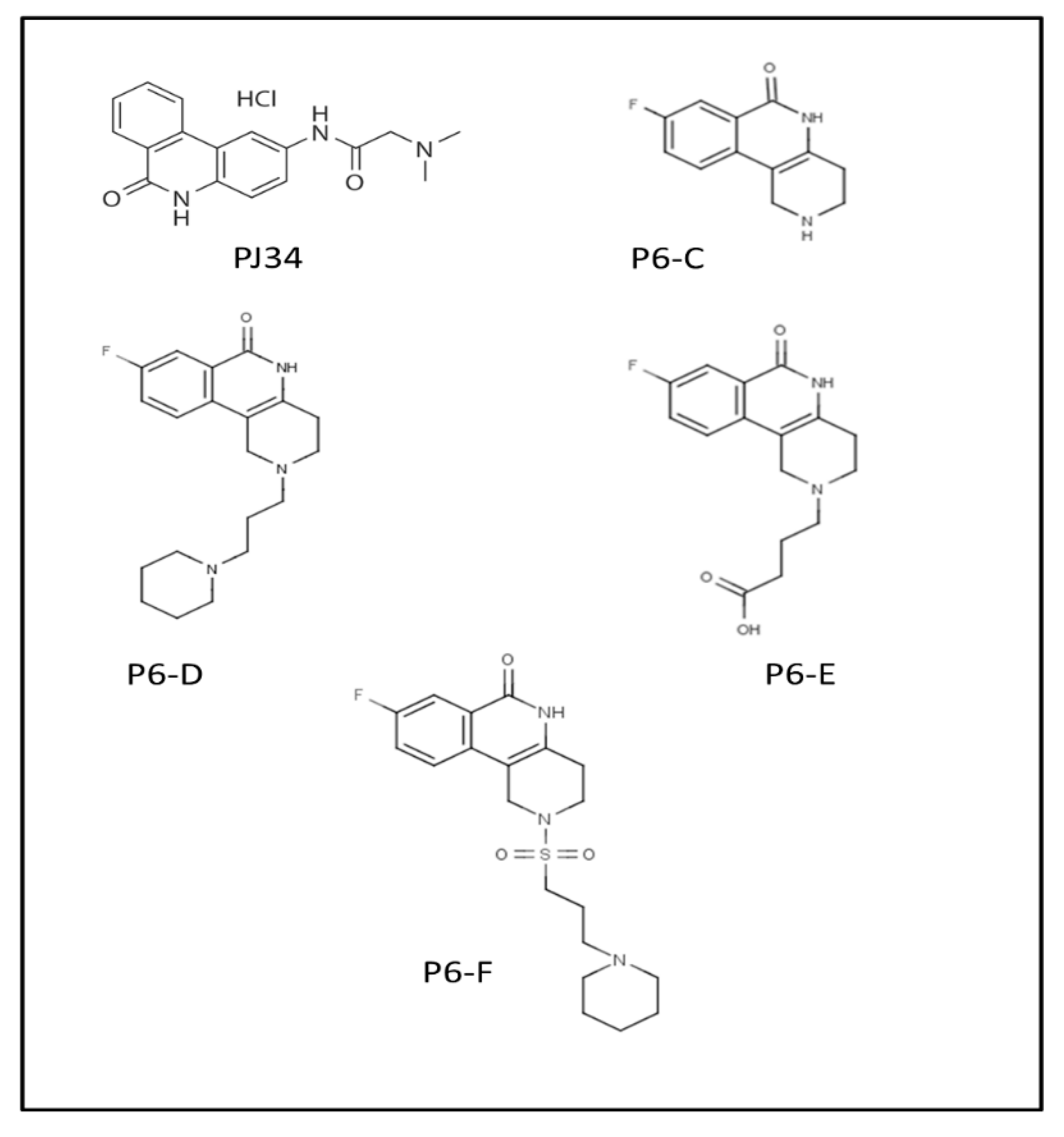
| P6-C | 89 ± 4 | 19 ± 1 | 4.1 | 25 ± 1 |
| P6-D | 97 ± 7 | 18 ± 1 | 4.0 | 42 ± 5 |
| P6-E | 119 ± 2 | 24 ± 0.3 | 3.9 | 50 ± 6 |
| P6-F | 38 ± 2 | 7 ± 0.2 | 4.4 | e ND |
| Inhibitor | KD (μM) a | IC50 (μM) b | Ki (μM) c |
|---|---|---|---|
| P6 | 1.7 ± 0.2 | 6.1 ± 1.2 | 1.8 ± 0.4 |
| P3 | 3.3 ± 0.4 | 7.2 ± 2.6 | 2.1 ± 0.8 |
| Suramin | 1.9 ± 0.7 | 10.6 ± 1.5 | 3.1 ± 0.4 |
| P6F | 3.1 ± 0.3 | 12.1 ± 1.1 | 3.6 ± 0.3 |
| P1 | 1.3 ± 0.2 | 13.0 ± 2.7 | 3.9 ± 0.8 |
| PJ97A | 1.6 ± 0.6 | 16.8 ± 1.6 | 5.0 ± 0.5 |
| PJ34 | 5.8 ± 2.6 | 32.3 ± 1.1 | 9.6 ± 0.3 |
| P6D | n.d. d | 76.1 ± 1.2 | 22.5 ± 0.4 |
| V30 | n.d. | 87.1 ± 1.3 | 25.8 ± 0.4 |
| P6C | n.d. | 121.3 ± 1.7 | 35.9 ± 0.5 |
| V23 | n.d. | 380.2 ± 3.3 | 112.4 ± 1.0 |
| P6G | n.d. | >1000 | >295 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugo, M.R.; Merrill, A.R. Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins. Toxins 2021, 13, 16. https://doi.org/10.3390/toxins13010016
Lugo MR, Merrill AR. Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins. Toxins. 2021; 13(1):16. https://doi.org/10.3390/toxins13010016
Chicago/Turabian StyleLugo, Miguel R., and Allan R. Merrill. 2021. "Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins" Toxins 13, no. 1: 16. https://doi.org/10.3390/toxins13010016
APA StyleLugo, M. R., & Merrill, A. R. (2021). Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins. Toxins, 13(1), 16. https://doi.org/10.3390/toxins13010016