Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization


Abstract
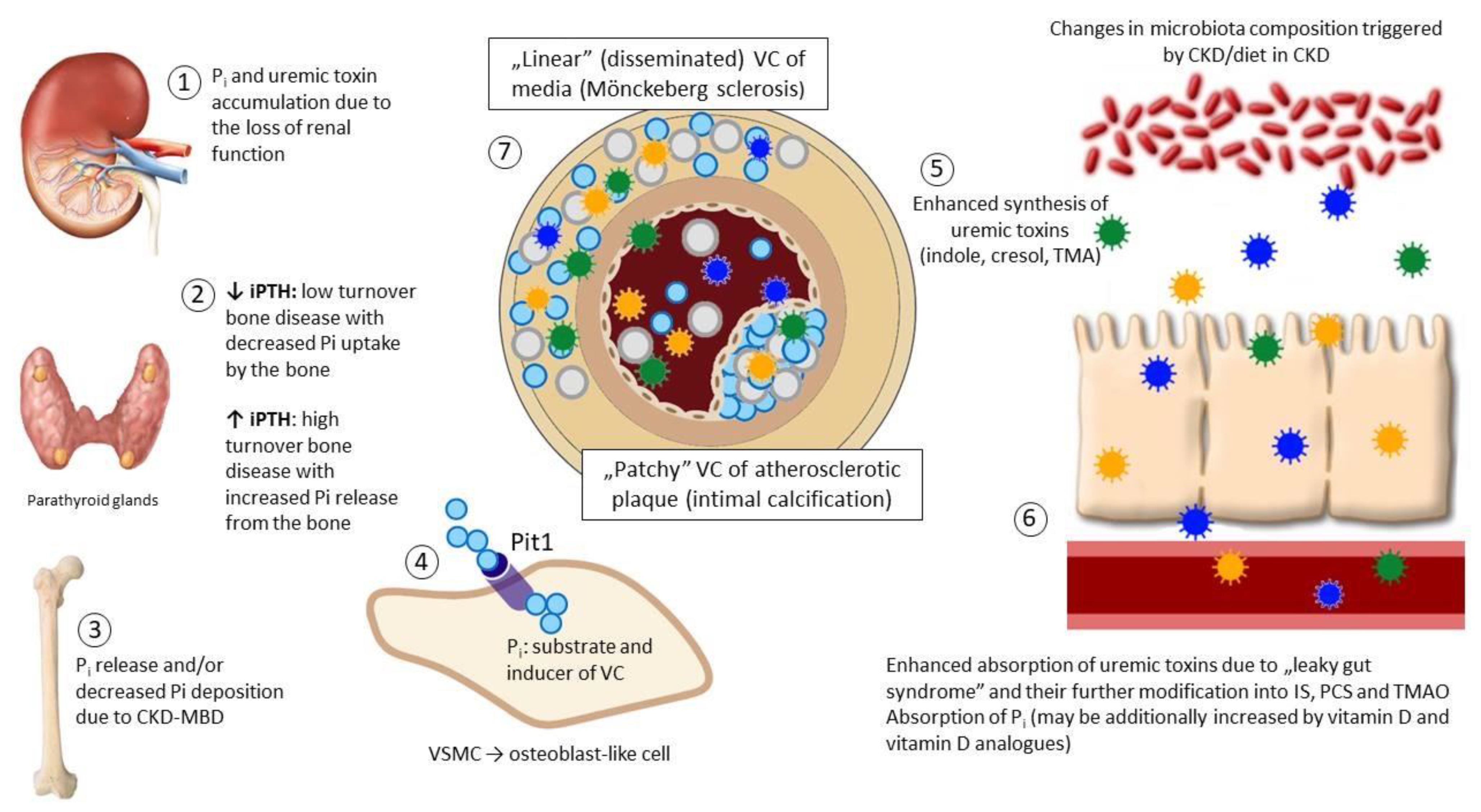
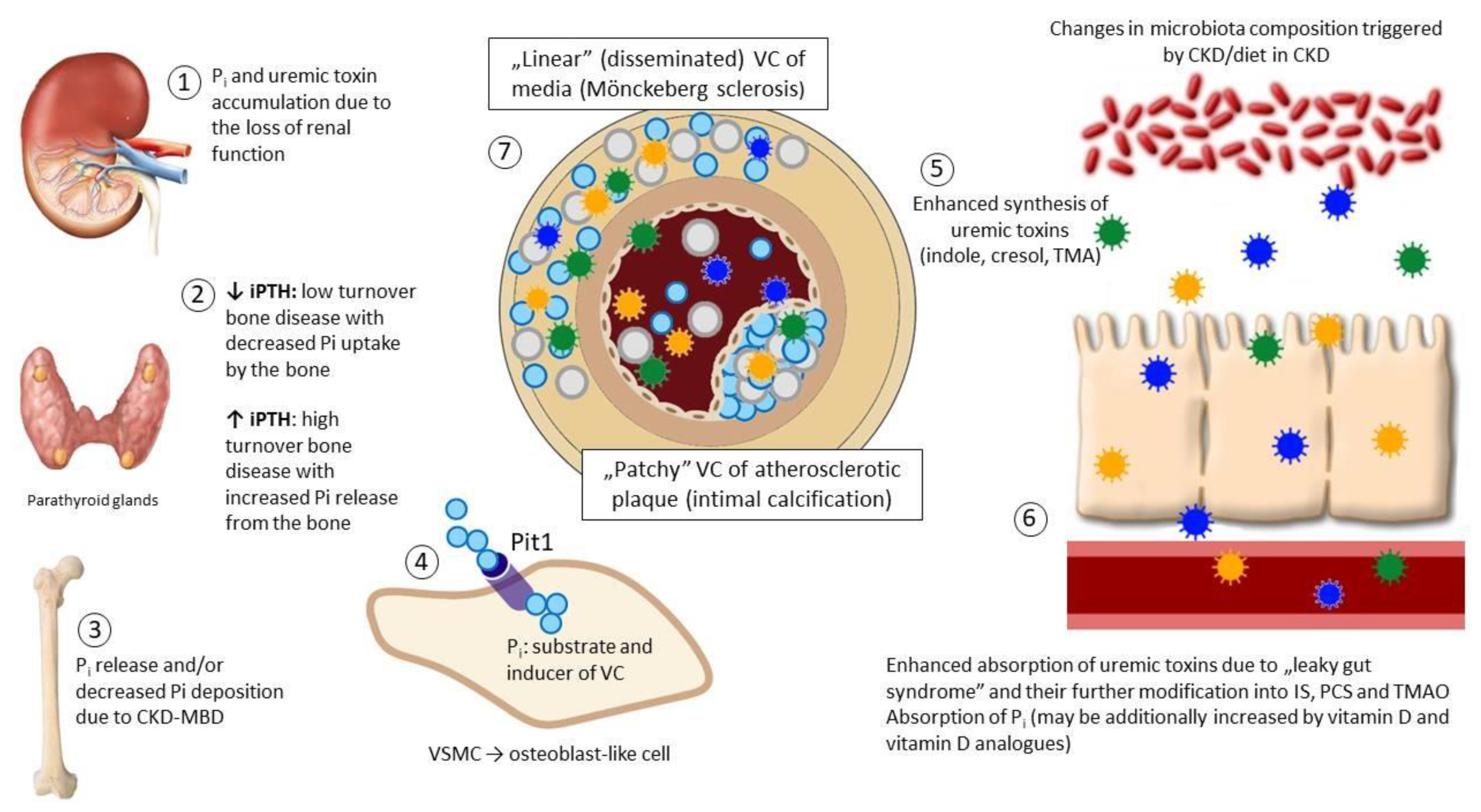
:1. Introduction: Pathologic Calcification in Cardiovascular System
2. Selected Mechanisms of VC Regulation
3. Gut Microbiota as a Source of Uremic Toxins. Impact of CKD on the Gut Microbiota
4. Uremic Toxins Involved in the Vascular Calcification
4.1. Indoxyl Sulphate (IS)
4.2. p-Cresyl Sulphate (PCS)
4.3. Trimethylamine-N-Oxide (TMAO)
5. Modification of Gut Microbiota to Prevent VC in CKD?
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicoll, R.; Henein, M.Y. The predictive value of arterial and valvular calcification for mortality and cardiovascular events. Int. J. Cardiol. Heart Vessel. 2014, 7, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of statins on serial coronary calcification during atheroma progression and regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.; Rabkin, S.W. The relationship between arterial stiffness and heart failure with preserved ejection fraction: A systemic meta-analysis. Heart Fail. Rev. 2015, 20, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, A.; Wei, F.; Chen, H. Cardiac valve calcification and risk of cardiovascular or all-cause mortality in dialysis patients: A meta-analysis. BMC Cardiovasc. Disord. 2018, 18, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stompór, T.P.; Pasowicz, M.; Sułowicz, W.; Dembińska-Kieć, A.; Janda, K.; Wójcik, K.; Tracz, W.; Zdzienicka, A.; Konieczyńska, M.; Klimeczek, P.; et al. Trends and dynamics of changes in calcification score over the 1-year observation period in patients on peritoneal dialysis. Am. J. Kidney Dis. 2004, 44, 517–528. [Google Scholar] [CrossRef]
- Stompór, T.; Pasowicz, M.; Sułowicz, W.; Dembińska-Kieć, A.; Janda, K.; Wójcik, K.; Tracz, W.; Zdzienicka, A.; Klimeczek, P.; Janusz-Grzybowska, E. An association between coronary artery calcification score, lipid profile, and selected markers of chronic inflammation in ESRD patients treated with peritoneal dialysis. Am. J. Kidney Dis. 2003, 41, 203–211. [Google Scholar] [CrossRef]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.H.; Lin, L.Y.; Lin, Y.H.; Tsai, I.J.; Huang, J.W. Abdominal aorta calcification predicts cardiovascular but not non-cardiovascular outcome in patients receiving peritoneal dialysis: A prospective cohort study. Medicine 2020, 99, 21730. [Google Scholar] [CrossRef]
- Chung, W.S.; Shih, M.P.; Wu, P.Y.; Huang, J.C.; Chen, S.C.; Chiu, Y.W.; Chang, J.M.; Chen, H.C. Progression of Aortic Arch Calcification Is Associated with Overall and Cardiovascular Mortality in Hemodialysis. Dis. Markers 2020, 2020, 6293185. [Google Scholar] [CrossRef] [PubMed]
- Su, W.Y.; Wu, P.Y.; Huang, J.C.; Chen, S.C.; Chang, J.M. Increased Proteinuria is Associated with Increased Aortic Arch Calcification, Cardio-Thoracic Ratio, Rapid Renal Progression and Increased Overall and Cardiovascular Mortality in Chronic Kidney Disease. Int. J. Med. Sci. 2020, 17, 1102–1111. [Google Scholar] [CrossRef]
- Tomiyama, C.; Higa, A.; Dalboni, M.A.; Cendoroglo, M.; Draibe, S.A.; Cuppari, L.; Carvalho, A.B.; Neto, E.; Canziani, M.E. The impact of traditional and non-traditional risk factors on coronary calcification in pre-dialysis patients. Nephrol. Dial. Transplant. 2006, 21, 2464–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens Rep. 2012, 14, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stompór, T. An overview of the pathophysiology of vascular calcification in chronic kidney disease. Perit. Dial. Int. 2007, 27, S215–S222. [Google Scholar] [CrossRef] [PubMed]
- Stompór, T. Coronary artery calcification in chronic kidney disease: An update. World J. Cardiol. 2014, 6, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Zununi Vahed, S.; Mostafavi, S.; Hosseiniyan Khatibi, S.M.; Shoja, M.M.; Ardalan, M. Vascular Calcification: An Important Understanding in Nephrology. Vasc. Health Risk Manag. 2020, 16, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R. Phosphate is a vascular toxin. Pediatr. Nephrol. 2013, 28, 583–593. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Candellier, A.; Boudot, C.; Grissi, M.; Mentaverri, R.; Choukroun, G.; Brazier, M.; Kamel, S.; Massy, Z.A. New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease. Toxins 2019, 11, 529. [Google Scholar] [CrossRef] [Green Version]
- Atoh, K.; Itoh, H.; Haneda, M. Serum indoxyl sulfate levels in patients with diabetic nephropathy: Relation to renal function. Diabetes Res. Clin. Pract. 2009, 83, 220–226. [Google Scholar] [CrossRef]
- Fan, P.C.; Chang, J.C.; Lin, C.N.; Lee, C.C.; Chen, Y.T.; Chu, P.H.; Kou, G.; Lu, Y.A.; Yang, C.W.; Chen, Y.C. Serum indoxyl sulfate predicts adverse cardiovascular events in patients with chronic kidney disease. J. Formos. Med. Assoc. 2019, 118, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Metz, R.P.; Patterson, J.L.; Wilson, E. Vascular smooth muscle cells: Isolation, culture, and characterization. Methods Mol. Biol. 2012, 843, 169–176. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Tang, J.; Zhang, L.; Li, X.; Hao, L. The Crosstalk between Calcium Ions and Aldosterone Contributes to Inflammation, Apoptosis, and Calcification of VSMC via the AIF-1/NF-κB Pathway in Uremia. Oxid Med. Cell Longev. 2020, 2020, 3431597. [Google Scholar] [CrossRef]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, F.; Carmona, A.; Obrero, T.; Jiménez, M.J.; Soriano, S.; Moreno, J.Á.; Martín-Malo, A.; Aljama, P. Role of endothelial microvesicles released by p-cresol on endothelial dysfunction. Sci. Rep. 2020, 10, 10657. [Google Scholar] [CrossRef] [PubMed]
- Alique, M.; Bodega, G.; Corchete, E.; García-Menéndez, E.; de Sequera, P.; Luque, R.; Rodríguez-Padrón, D.; Marqués, M.; Portolés, J.; Carracedo, J.; et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification in vitro. Comput. Struct. Biotechnol. J. 2020, 18, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Duque, E.J.; Elias, R.M.; Moysés, R.M.A. Parathyroid Hormone: A Uremic Toxin. Toxins 2020, 12, 189. [Google Scholar] [CrossRef] [Green Version]
- Kuczera, P.; Adamczak, M.; Wiecek, A. Fibroblast Growth Factor-23-A Potential Uremic Toxin. Toxins 2016, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.K. Phosphate is a uremic toxin. J. Ren. Nutr. 2008, 18, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Chang, Y.; Vaziri, N.D. The consequences of altered microbiota in immune-related chronic kidney disease. Nephrol. Dial. Transplant. 2020, gfaa087. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Li, X.; Ghosh, S.; Xie, C.; Chen, J.; Huang, H. Role of gut microbiota-derived metabolites on vascular calcification in CKD. J. Cell Mol. Med. 2021, 25, 1332–1341. [Google Scholar] [CrossRef]
- Metzinger-Le Meuth, V.; Burtey, S.; Maitrias, P.; Massy, Z.A.; Metzinger, L. microRNAs in the pathophysiology of CKD-MBD: Biomarkers and innovative drugs. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 337–345. [Google Scholar] [CrossRef]
- Massy, Z.A.; Metzinger-Le Meuth, V.; Metzinger, L. MicroRNAs Are Associated with Uremic Toxicity, Cardiovascular Calcification, and Disease. Contrib. Nephrol. 2017, 189, 160–168. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef]
- Al Khodor, S.; Shatat, I.F. Gut microbiome and kidney disease: A bidirectional relationship. Pediatr. Nephrol. 2017, 32, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Fiaccadori, E.; Cosola, C.; Sabatino, A. Targeting the Gut for Early Diagnosis, Prevention, and Cure of Diabetic Kidney Disease: Is the Phenyl Sulfate Story Another Step Forward? Am. J. Kidney Dis. 2020, 75, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Vaziri, N.D. Gut microbial short-chain fatty acids and the risk of diabetes. Nat. Rev. Nephrol. 2019, 15, 389–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovich, A.; Isakova, T.; Stubbs, J. Microbiome and Cardiovascular Disease in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1598–1604. [Google Scholar] [CrossRef] [Green Version]
- Canyelles, M.; Tondo, M.; Cedó, L.; Farràs, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 2018, 19, 3228. [Google Scholar] [CrossRef] [Green Version]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, W.; Nie, J. Gut Microbiota and Renal Injury. Adv. Exp. Med. Biol. 2020, 1238, 93–106. [Google Scholar] [CrossRef]
- Zhao, J.; Ning, X.; Liu, B.; Dong, R.; Bai, M.; Sun, S. Specific alterations in gut microbiota in patients with chronic kidney disease: An updated systematic review. Ren. Fail. 2021, 43, 102–112. [Google Scholar] [CrossRef]
- Clegg, D.J.; Hill Gallant, K.M. Plant-Based Diets in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 141–143. [Google Scholar] [CrossRef]
- Kim, H.; Caulfield, L.E.; Garcia-Larsen, V.; Steffen, L.M.; Grams, M.E.; Coresh, J.; Rebholz, C.M. Plant-Based Diets and Incident CKD and Kidney Function. Clin. J. Am. Soc. Nephrol. 2019, 14, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.F.; Clegg, D.J. Physiology and Pathophysiology of Potassium Homeostasis: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Appel, L.J.; Grams, M.E.; Gutekunst, L.; McCullough, P.A.; Palmer, B.F.; Pitt, B.; Sica, D.A.; Townsend, R.R. Potassium Homeostasis in Health and Disease: A Scientific Workshop Cosponsored by the National Kidney Foundation and the American Society of Hypertension. Am. J. Kidney Dis. 2017, 70, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, L.S.; Go, A.S. Epidemiology of acute infections among patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1487–1493. [Google Scholar] [CrossRef] [Green Version]
- Eyler, R.F.; Shvets, K. Clinical Pharmacology of Antibiotics. Clin. J. Am. Soc. Nephrol. 2019, 14, 1080–1090. [Google Scholar] [CrossRef] [Green Version]
- Chavers, B.M.; Solid, C.; Gilbertson, D.T.; Collins, A.J. Infection-related hospitalization rates in pediatric versus adult patients with end-stage renal disease in the United States. J. Am. Soc. Nephrol. 2007, 18, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Eddi, R.; Malik, M.N.; Shakov, R.; Baddoura, W.J.; Chandran, C.; Debari, V.A. Chronic kidney disease as a risk factor for Clostridium difficile infection. Nephrology 2010, 15, 471–475. [Google Scholar] [CrossRef]
- Wang, T.Z.; Kodiyanplakkal, R.P.L.; Calfee, D.P. Antimicrobial resistance in nephrology. Nat. Rev. Nephrol. 2019, 15, 463–481. [Google Scholar] [CrossRef]
- Rahbar Saadat, Y.; Niknafs, B.; Hosseiniyan Khatibi, S.M.; Ardalan, M.; Majdi, H.; Bahmanpoor, Z.; Abediazar, S.; Zununi Vahed, S. Gut microbiota; an overlooked effect of phosphate binders. Eur. J. Pharmacol. 2020, 868, 172892. [Google Scholar] [CrossRef]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Lopez Manosalva, A.G.; Koonen, D.P.Y.; Fu, J.; Wijmenga, C.; Zhernakova, A.; Weersma, R.K. The influence of proton pump inhibitors and other commonly used medication on the gut microbiota. Gut. Microbes. 2017, 8, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Rusu, I.G.; Suharoschi, R.; Vodnar, D.C.; Pop, C.R.; Socaci, S.A.; Vulturar, R.; Istrati, M.; Moroșan, I.; Fărcaș, A.C.; Kerezsi, A.D.; et al. Iron Supplementation Influence on the Gut Microbiota and Probiotic Intake Effect in Iron Deficiency-A Literature-Based Review. Nutrients 2020, 12, 1993. [Google Scholar] [CrossRef]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-κB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, C.; Rauner, M.; Pacyna, N.; Hempel, U.; Bornstein, S.R.; Hofbauer, L.C. miR-125b regulates calcification of vascular smooth muscle cells. Am. J. Pathol. 2011, 179, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/β-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osako, M.K.; Nakagami, H.; Shimamura, M.; Koriyama, H.; Nakagami, F.; Shimizu, H.; Miyake, T.; Yoshizumi, M.; Rakugi, H.; Morishita, R. Cross-talk of receptor activator of nuclear factor-κB ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Hirose, Y.; Goto, S.; Nishijima, F.; Zrelli, H.; Zghonda, N.; Niwa, T.; Miyazaki, H. Indoxyl sulfate enhances angiotensin II signaling through upregulation of epidermal growth factor receptor expression in vascular smooth muscle cells. Life Sci. 2012, 91, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Hirose, Y.; Nishijima, F.; Tsubakihara, Y.; Miyazaki, H. ROS and PDGF-beta [corrected] receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am. J. Physiol. Cell Physiol. 2009, 297, C389–C396. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Toyama, O.; Shiota, M.; Kim-Mitsuyama, S.; Miyazaki, H. Protein tyrosine phosphatase LMW-PTP exhibits distinct roles between vascular endothelial and smooth muscle cells. J. Recept. Signal. Transduct. Res. 2005, 25, 19–33. [Google Scholar] [CrossRef]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Han, X.; Wang, L.; Diao, Z.; Liu, W. Indoxyl sulfate promotes vascular smooth muscle cell calcification via the JNK/Pit-1 pathway. Ren. Fail. 2016, 38, 1702–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Maré, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Chen, Y.; Zhu, Z.; Su, X.; Ni, J.; Du, R.; Zhang, R.; Jin, W. p-Cresyl sulfate promotes the formation of atherosclerotic lesions and induces plaque instability by targeting vascular smooth muscle cells. Front. Med. 2016, 10, 320–329. [Google Scholar] [CrossRef]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Kaysen, G.A.; Johansen, K.L.; Chertow, G.M.; Dalrymple, L.S.; Kornak, J.; Grimes, B.; Dwyer, T.; Chassy, A.W.; Fiehn, O. Associations of Trimethylamine N-Oxide With Nutritional and Inflammatory Biomarkers and Cardiovascular Outcomes in Patients New to Dialysis. J. Ren. Nutr. 2015, 25, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-κB (Nuclear Factor κB) Signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Yazdekhasti, N.; Brandsch, C.; Schmidt, N.; Schloesser, A.; Huebbe, P.; Rimbach, G.; Stangl, G.I. Fish protein increases circulating levels of trimethylamine-N-oxide and accelerates aortic lesion formation in apoE null mice. Mol. Nutr. Food Res. 2016, 60, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.A.; Benton, T.Z.; Bennett, B.J.; Jacobs, D.R., Jr.; Lloyd-Jones, D.M.; Gross, M.D.; Carr, J.J.; Gordon-Larsen, P.; Zeisel, S.H. Microbiota-Dependent Metabolite Trimethylamine N-Oxide and Coronary Artery Calcium in the Coronary Artery Risk Development in Young Adults Study (CARDIA). J. Am. Heart Assoc. 2016, 5, e003970. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, J.R.; Stedman, M.R.; Liu, S.; Long, J.; Franchetti, Y.; West, R.E., 3rd; Prokopienko, A.J.; Mahnken, J.D.; Chertow, G.M.; Nolin, T.D. Trimethylamine N-Oxide and Cardiovascular Outcomes in Patients with ESKD Receiving Maintenance Hemodialysis. Clin. J. Am. Soc. Nephrol. 2019, 14, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Glorieux, G.; Meijers, B. p-cresol sulfate and indoxyl sulfate: Some clouds are gathering in the uremic toxin sky. Kidney Int. 2017, 92, 1323–1324. [Google Scholar] [CrossRef]
- Shafi, T.; Sirich, T.L.; Meyer, T.W.; Hostetter, T.H.; Plummer, N.S.; Hwang, S.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R. Results of the HEMO Study suggest that p-cresol sulfate and indoxyl sulfate are not associated with cardiovascular outcomes. Kidney Int. 2017, 92, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef]
- Meijers, B.K.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, T.; Powe, N.R.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Hostetter, T.H. Trimethylamine N-Oxide and Cardiovascular Events in Hemodialysis Patients. J. Am. Soc. Nephrol. 2017, 28, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.T.; Lin, S.H. Uremic Vascular Calcification: The Pathogenic Roles and Gastrointestinal Decontamination of Uremic Toxins. Toxins 2020, 12, 812. [Google Scholar] [CrossRef]
- Zhao, Y.; Cai, Y.; Cui, L.Y.; Tang, W.; Liu, B.; Zheng, J.J.; Si, W.Z.; Wang, X.; Xu, M.J. Suppression of Gut Bacterial Translocation Ameliorates Vascular Calcification through Inhibiting Toll-Like Receptor 9-Mediated BMP-2 Expression. Oxid Med. Cell Longev. 2019, 17, 3415682. [Google Scholar] [CrossRef] [Green Version]
- Knöpfel, T.; Pastor-Arroyo, E.M.; Schnitzbauer, U.; Kratschmar, D.V.; Odermatt, A.; Pellegrini, G.; Hernando, N.; Wagner, C.A. The intestinal phosphate transporter NaPi-IIb (Slc34a2) is required to protect bone during dietary phosphate restriction. Sci. Rep. 2017, 7, 11018. [Google Scholar] [CrossRef] [Green Version]
- Fouque, D.; Vervloet, M.; Ketteler, M. Targeting Gastrointestinal Transport Proteins to Control Hyperphosphatemia in Chronic Kidney Disease. Drugs 2018, 78, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Block, G.A.; Rosenbaum, D.P.; Yan, A.; Chertow, G.M. Efficacy and Safety of Tenapanor in Patients with Hyperphosphatemia Receiving Maintenance Hemodialysis: A Randomized Phase 3 Trial. J. Am. Soc. Nephrol. 2019, 30, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Peter, W.L.; Wazny, L.D.; Weinhandl, E.; Cardone, K.E.; Hudson, J.Q. A Review of Phosphate Binders in Chronic Kidney Disease: Incremental Progress or Just Higher Costs? Drugs 2017, 77, 1155–1186. [Google Scholar] [CrossRef] [PubMed]
- Ruospo, M.; Palmer, S.C.; Natale, P.; Craig, J.C.; Vecchio, M.; Elder, G.J.; Strippoli, G.F. Phosphate binders for preventing and treating chronic kidney disease-mineral and bone disorder (CKD-MBD). Cochrane Database Syst. Rev. 2018, 8, CD006023. [Google Scholar] [CrossRef] [PubMed]
- Stompór, T.; Rajzer, M.; Kawecka-Jaszcz, K.; Dembińska-Kieć, A.; Janda, K.; Wójcik, K.; Tabor, B.; Zdzienicka, A.; Grzybowska, E.J.; Sulowicz, W. Renal transplantation ameliorates the progression of arterial stiffness in patients treated with peritoneal dialysis. Perit. Dial. Int. 2005, 25, 492–496. [Google Scholar] [CrossRef]
- Alappan, H.R.; Vasanth, P.; Manzoor, S.; O’Neill, W.C. Vascular Calcification Slows But Does Not Regress After Kidney Transplantation. Kidney Int. Rep. 2020, 5, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Mursawa, H.; Hatakeyama, S.; Yamamoto, H.; Tanaka, Y.; Soma, O.; Matsumoto, T.; Yoneyama, T.; Hashimoto, Y.; Koie, T.; Fujita, T.; et al. Slow Progression of Aortic Calcification Is a Potential Benefit of Pre-emptive Kidney Transplantation. Transplant. Proc. 2018, 50, 145–149. [Google Scholar] [CrossRef] [PubMed]
- D’Marco, L.; Bellasi, A.; Mazzaferro, S.; Raggi, P. Vascular calcification, bone and mineral metabolism after kidney transplantation. World J. Transplant. 2015, 5, 222–230. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| IS 1 | ||
|---|---|---|
| Model | Design and Methods | Key Findings |
| In vitro [75] | Medium containing IS at concentrations ranging between 100 and 1000 μmol/L added to cultured VSMC for 24 h or at a concentration of 500 μmol/L added to cultured VSMC for various durations (12–72 h) | Dose- and time-dependent osteoblastic differentiation of VSMC Enhanced calcium deposition within culture Increased expression of Pit-1 mRNA and protein in VSMC Activation of c-Jun N-terminal kinase implicated in IS-induced Pit-1 expression and activation |
| In vitro [25] | Endothelial progenitor cells cultured with extracelluar matrix vesicles (EMV) derived from IS-treated endothelial cells human umbilical vein endothelial cells (HUVEC) incubated with IS at the concentration of 256 μg/mL to obtain EMV | IS activated oxidative stress, apoptosis and EMV release from humen umbilical vascular endothelial cells (HUVEC) IS activated increased expression of adhesion molecules by HUVEC EMV induced by IS characterized with upregulation of the cluster of miRNA promoting inflammation, apoptosis and cellular senescence EMV derived from IS-treated HUVEC induced oxidative stress and decreased angiogenic potential of endothelial progenitor cells EMV derived from IS-treated HUVEC increased NFκB expression in endothelial progenitor cells |
| In vitro [27] | HUVEC incubated with IS at the concentration of 250 μmol/L to obtain matrix vesciles (MV) VSMC incubated with MV from IS-treated HUVEC | IS induced senescent phenotype of HUVEC and MV release MV obtained from IS-treated HUVEC induced calcium deposit accumulation in VSMC in culture MV obtained from IS-treated HUVEC induced expression of inflammatory chemokines and cytokines in VSMC in culture |
| In vitro [28] | EC, VSMC and rat aortic rings exposed to IS at the concentration of 250 μmol/L, phosphate 3 mmol/L or both | Conditioned media from phosphate- and IS-treated EC induced calcium deposition in cultured VSMC Treatment of EC with phosphate and IS increased IL8 mRNA and protein expression in these cells Phosphate and IS induced osteoblastic differentiation of VSMC Phosphate and IS induced vascular calcification in rat aortic rings and increased mRNA expression of rat IL8 homologues in aortic rings Antagonizing IL8 partially prevented calcification in cultured VSMC and rat aortic rings treated with IS |
| In vitro [68] | Human aortic VSMC incubated with IS at concentrations ranging between 250 and 1000 μmol/L | In cultured VSMC IS downregulates miR-29b, suppressor of the Wnt/β-catenin pathway (Wnt/β-catenin pathway stimulates VC) miR-29b expression decreased and RUNX and osteopontin (OPN; markers of osteoblastic differentiation) increased in human radial artery samples obtained from end-stage kidney disease (ESRD) patients during arterio-venous fistula surgery treated with IS |
| In vitro and in vivo rat model [71] | Medium containing IS at a concentration of 250 μmol/L added to cultured VSMC IS added to drinking water Serum concentration 13.66 ± 1.19 mg/dL | IS potentiated Ang II-mediated signaling in VSMC IS augmented Ang II-mediated phosphorylation of extracellular signal-regulated kinase (ERK) and epidermal growth factor receptor (EGFR) IS increased ROS production and reactive oxygen species (ROS)-mediated EGFR expression in aortas from uremic rats |
| In vivo rat model [76] | IS added to drinking water Serum TAC 69.4 ± 5.7 μmol/L | Increased serum glucose and lower expression of glucose transporter 1(GLUT1) in the aorta (prodiabetic milieu) following exposure to IS 10-fold higher aortic calcium content following exposure to IS as compared to control Activation of acute phase, inflammatory and procoagulation pathways implicated in IS-induced aortic calcification |
| Clinical [88] | Observational study; analysis of outcome in a cohort of 1273 ESRD patients treated with hemodialysis, depending on the baseline serum IS level (sub-study of the longitudinal HEMO study) Mean baseline serum IS: 2.5 ± 1.2 mg/dL (median 2.4 mg/dL) | No association between serum IS and any of the analyzed outcomes, including: Cardiac death, sudden cardiac death, first CV event, death from any cause |
| PCS 1 | ||
| Model | Designand Methods | Key findings |
| In vivo rat model [76] | PCS added to drinking water Time-averaged concentration 151.0 ± 40.7 μmol/L | Increased serum glucose and lower expression of GLUT1 in the aorta (prodiabetic milieu) following exposure to PCS 10-fold higher aortic calcium content following exposure to PCS as compared to control Activation of acute phase, inflammatory and procoagulation pathways as implicated in PCS-induced aortic calcification |
| In vitro [26] | HUVEC incubated with PCS at a concentration of 25 μg/mL to obtain EMV Endothelial cells cultured with EMV derived from PCS-treated endothelial cells | PCS induced EMV release from HUVEC EMV induced by PCS characterized with changes in miRNA profile towards promotion of cellular senescence and reduction in proliferative and angiogenic capacity of endothelial cells EMV derived from PCS-treated HUVEC decreased endothelial cell migration and induced cell senescence |
| In vitro, in vivo [78] | HUVEC and HVSMC incubated with PCS at a concentration ranging between 0.02 and 0.5 mmol/L Vascular contractility of mouse aortic rings tested with PCS at a concentration of 0.15 mmol/L | PCS activated oxidative stress in endothelial cells (HUVEC) and VSMC (maximum effect at a concentration of 0.15 mmol/L) PCS promoted phenylephrine-induced contraction of smooth muscles within aortic wall PCS promoted aortic wall remodeling |
| Clinical [88] | Observational study; analysis of outcome in a cohort of 1273 ESRD patients treated with hemodialysis, depending on the baseline serum PCS level (substudy of the longitudinal HEMO study) Mean baseline serum PCS: 3.3 ± 1.7 mg/dL (median 3.3 mg/dL) | No association between serum PCS and any of the analyzed outcomes, including: Cardiac death, sudden cardiac death, first CV event, death from any cause |
| Clinical [89,90] Same study, two publications | Observational study; analysis of outcome in a cohort of 175 ESRD patients treated with hemodialysis, depending on the baseline serum PCS level Mean baseline total serum PCS: 19.0 ± 0.9 mg/L (median 18.9 mg/L) Mean baseline free serum PCS: 2.59 ± 0.17 mg/L (median 1.97 mg/L) | Baseline concentration of free but not total PCS predicted all-cause mortality over median follow-up period of 30 months Baseline concentration of free but not total PCS predicted new CV event defined as composite of death from cardiac causes, non-fatal myocardial infarction, myocardial ischemia, ischemic stroke or new peripheral vascular disease Free serum PCS concentration predicted new CV events in the whole cohort and in non-diabetic, but not in diabetic patients |
| TMAO 1 | ||
| Model | Design and Methods | Key findings |
| In vitro, ex vivo, in vivo, clinical [50] | Rat aortic VSMC incubated with TMAO at concentrations of 100, 300 and 600μmol/L Human VSMC incubated with TMAO at concentrations of 100, 300 and 600μmol/L Rat aortic rings incubated with TMAO at concentrations of 100 and 300 μmol/L Human tibial arterial rings (obtained from amputated limbs) incubated with TMAO at concentrations of 100 and 300 μmol/LRats treated with TMAO injected intraperitoneally (3.3 μmol/kg daily) (serum TMAO level between 23 and 35μmol/L; exact values not provided in the text, deduced from the figure) CKD patients with serum TMAO concentration measured and VC assessed using coronary artery calcification (CAC) (serum TMAO level ranging between 40 and 200 μmol/L; exact values not provided in the text, deduced from the figure) Effect of gut decontamination on vascular lesions in experimental animals | TMAO induced mRNA expression of Runx2 and BMP2 in cultured rat and human VSMC, isolated rat aortic rings and isolated fragments of human tibial arteries TMAO induced VC in isolated rat aortic rings and isolated fragments of human tibial arteries TMAO injected intraperitoneally increased VC in subtotally nephrectomized (CKD) by means of CT assessment and histomorphometric analysis TMAO increased serum IL-1β in CKD rats TMAO activated NLRP3 inflammasome and upegulated NF-κB Patients with aortic arch calcification (AAC) were characterized with significantly higher serum TMAO level as compared with those without AAC; the advancement of AAC correlated with serum TMAO Gut decontamination with antibiotics led to significantly decreased serum TMAO and lowered Runx2 and BMP protein expression in CKD animals, and almost completely abolished calcium and phosphate accumulation in their aortas (otherwise having no influence on the development of CKD) |
| In vivo [83] | apo-E-null mice fed with diets with an equivalent content of protein, but originating from fish, casein or soy (identical in terms of calories, carbohydrate, fat, etc.) TMA content in experimental diets: 529 mg/kg (fish), 24.5 mg/kg (casein) and 23 mg/kg (soy) | Serum TMAO concentrations in animals fed with experimental diets: 7.0 μmol/L (fish), 1.0 μmol/L (casein) and 1.5 μmol/L (soy) More advanced atherosclerosis and more calcified atherosclerotic lesions in animals fed with fish protein Aortic wall mRNA expression of of several inflammatory markers (including CD36, IL6 and ICAM-1) significantly higher in animals fed with fish protein |
| Clinical trial [85] | Observational trial of 817 apparently healthy participants aged between 33 and 55 years with serial measurements of CAC and common carotid artery intima-media thickness (CCA-IMT) Baseline median TMAO concentration of 2.6 μmol/L (interquartile range: 1.8–4.2) | No association between baseline serum TMAO concentration and progression in CAC nor CCA-IMT value; no association between baseline serum TMAO concentration and CV events nor GFR loss (10-year follow-up) |
| Clinical [84] | 4007 patients who underwent coronary angiography and had baseline serum TMAO assessment Median baseline serum TMAO level 3.7 (interquartile range 2.4–6.2) μmol/L | Baseline serum TMAO level significantly associated with the risk of major adverse cardiovascular events (defined as: death, MI or stroke) in three-year follow-up |
| Clinical [79] | 235 ESRD patients treated with hemodialysis, with baseline serum TMAO assessment Mean baseline serum TMAO 50 ± 32 μmol/L (median 43 μmol/L) | No association between baseline serum TMAO and all cause death, CV death or CV hospitalization (median follow-up of 4 years) |
| Clinical [86] | 1242 patients treated with hemodialysis, with baseline serum TMAO assessment from the EVOLVE prospective randomized trial Baseline serum TMAO range: 2.5–1103.1 μmol/L (considered “extraordinarily wide range” by authors) | No association between baseline serum TMAO, all-cause death and vascular composite outcome defined as: Cardiovascular death, MI, peripheral vascular event, stroke and hospitalization for unstable angina |
| Clinical [91] | Observational study; analysis of outcome in a cohort of 1273 ESRD patients treated with hemodialysis, depending on the baseline serum TMAO level (sub-study of the longitudinal HEMO study) Mean baseline serum TMAO: 101.9 ± 63.9 μmol/L (median 88 μmol/L) | No association between baseline serum TMAO and any of the analyzed outcomes, including: Cardiac death, sudden cardiac death, first CV event, death from any cause for the whole group and among Black patients TMAO significantly associated with all above endpoints among Whites The difference in outcome between races despite comparable serum TMAO between respective groups |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipska, I.; Winiarska, A.; Knysak, M.; Stompór, T. Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization. Toxins 2021, 13, 274. https://doi.org/10.3390/toxins13040274
Filipska I, Winiarska A, Knysak M, Stompór T. Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization. Toxins. 2021; 13(4):274. https://doi.org/10.3390/toxins13040274
Chicago/Turabian StyleFilipska, Iwona, Agata Winiarska, Monika Knysak, and Tomasz Stompór. 2021. "Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization" Toxins 13, no. 4: 274. https://doi.org/10.3390/toxins13040274
APA StyleFilipska, I., Winiarska, A., Knysak, M., & Stompór, T. (2021). Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization. Toxins, 13(4), 274. https://doi.org/10.3390/toxins13040274