Therapeutic Uses of Bacterial Subunit Toxins

Abstract
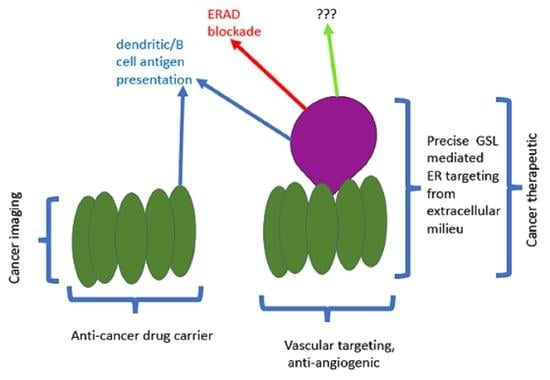
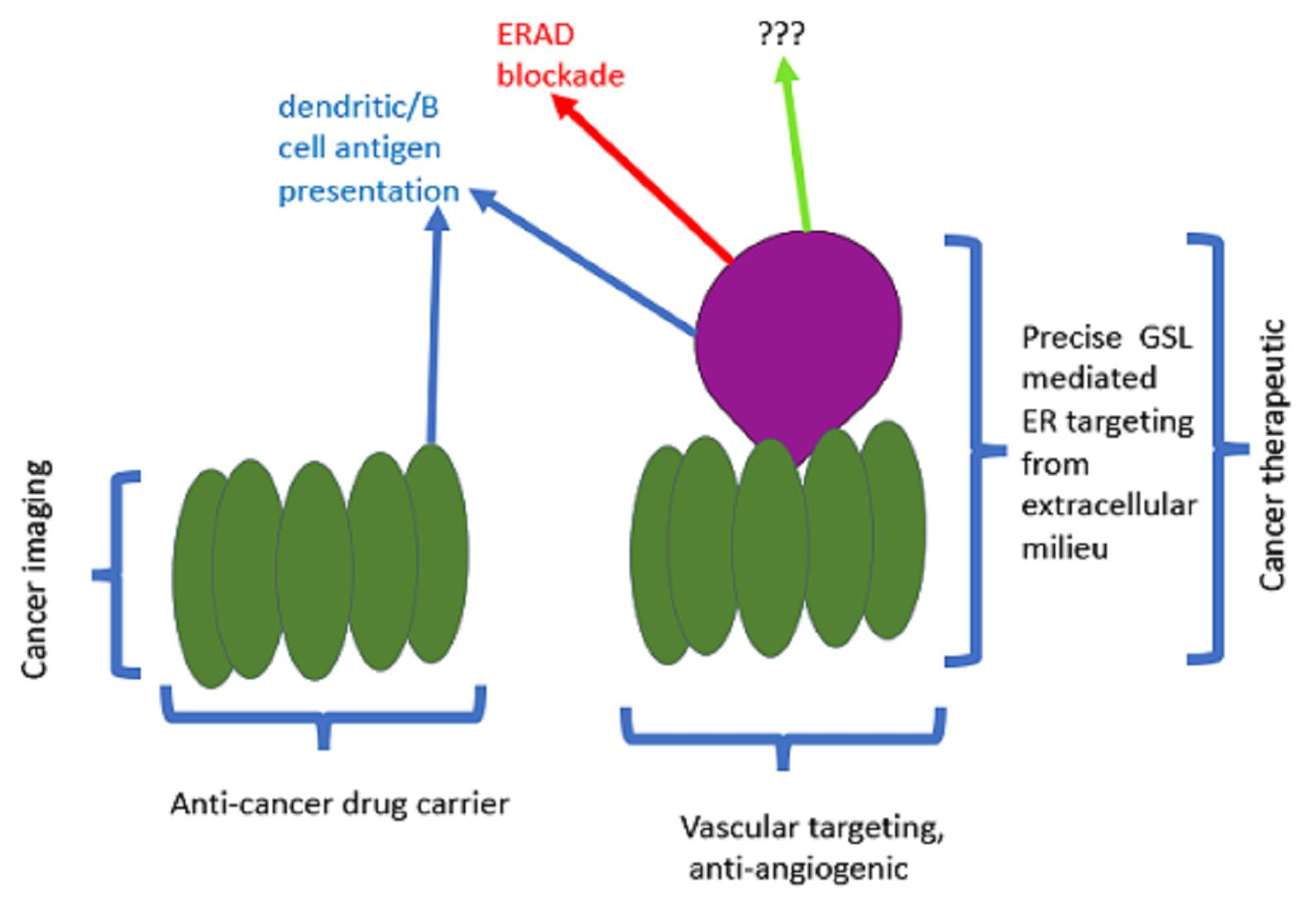
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Gb3 Is a Cancer Marker
2.1. Is This the Limit of Gb3 Detection?
2.2. Cancer Stem Cells
3. Verotoxin as a Cancer Targeting Tool
3.1. B Subunit Conjugates
3.2. Native VT1 in Cancer Therapy
3.2.1. Potency
3.2.2. Risk
3.2.3. Efficacy
4. Verotoxin Interaction with Lymphoid Cells
4.1. As an Immunogen Carrier
4.2. Effects on Lymphoid Cells
5. Verotoxin A Subunit Redirection of Intracellular Traffic
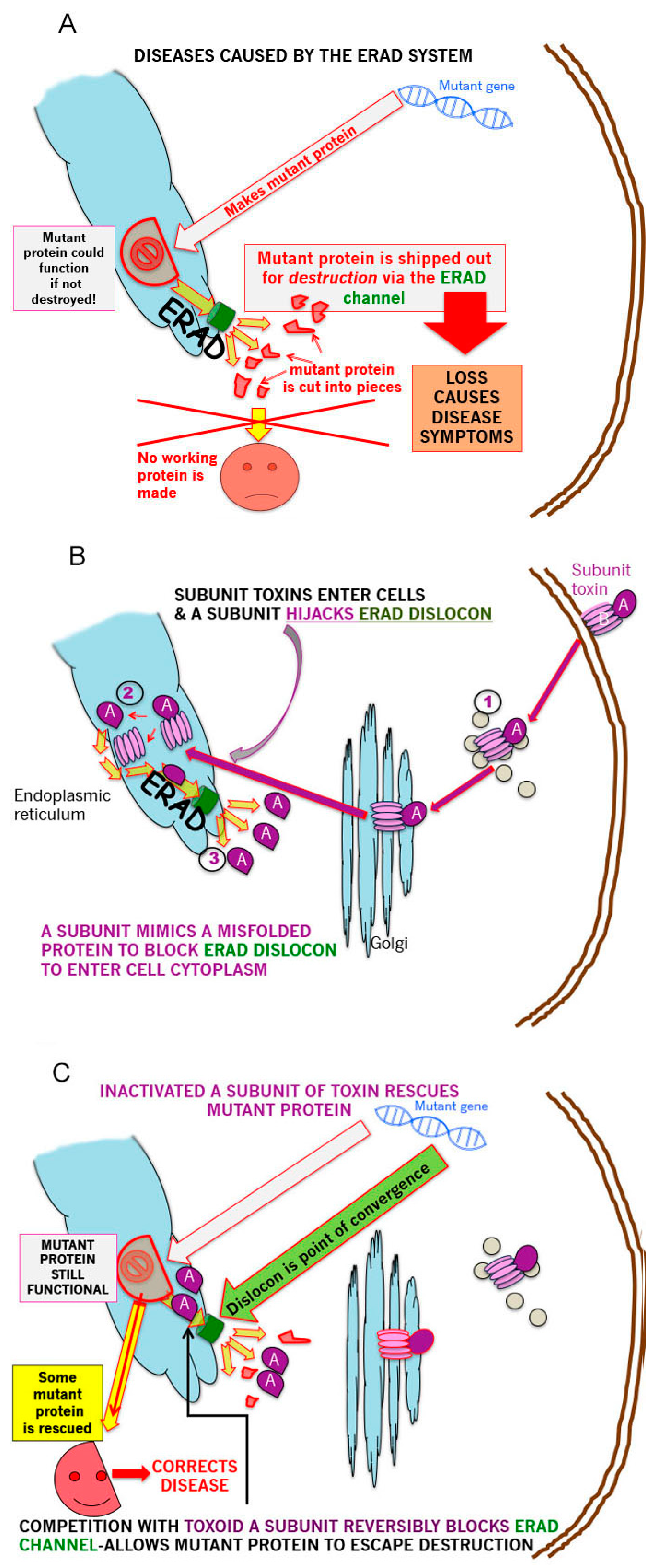
6. Cholera Toxin as a Targeted ER Associated Degradation (ERAD) Blockade
Intracellular Plumbing
7. Treatment for Protein Misfolding Diseases
7.1. F508delta CFTR
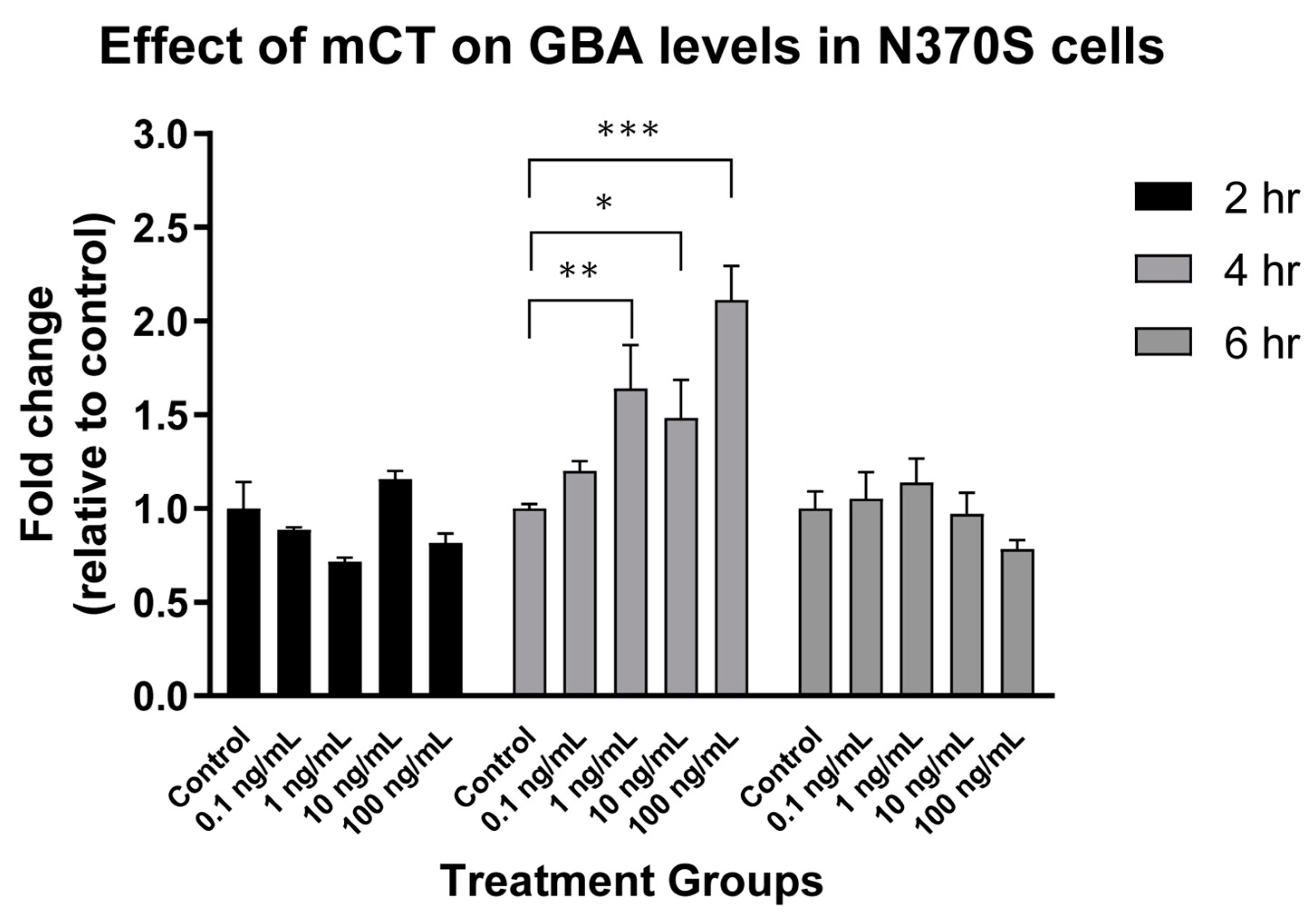
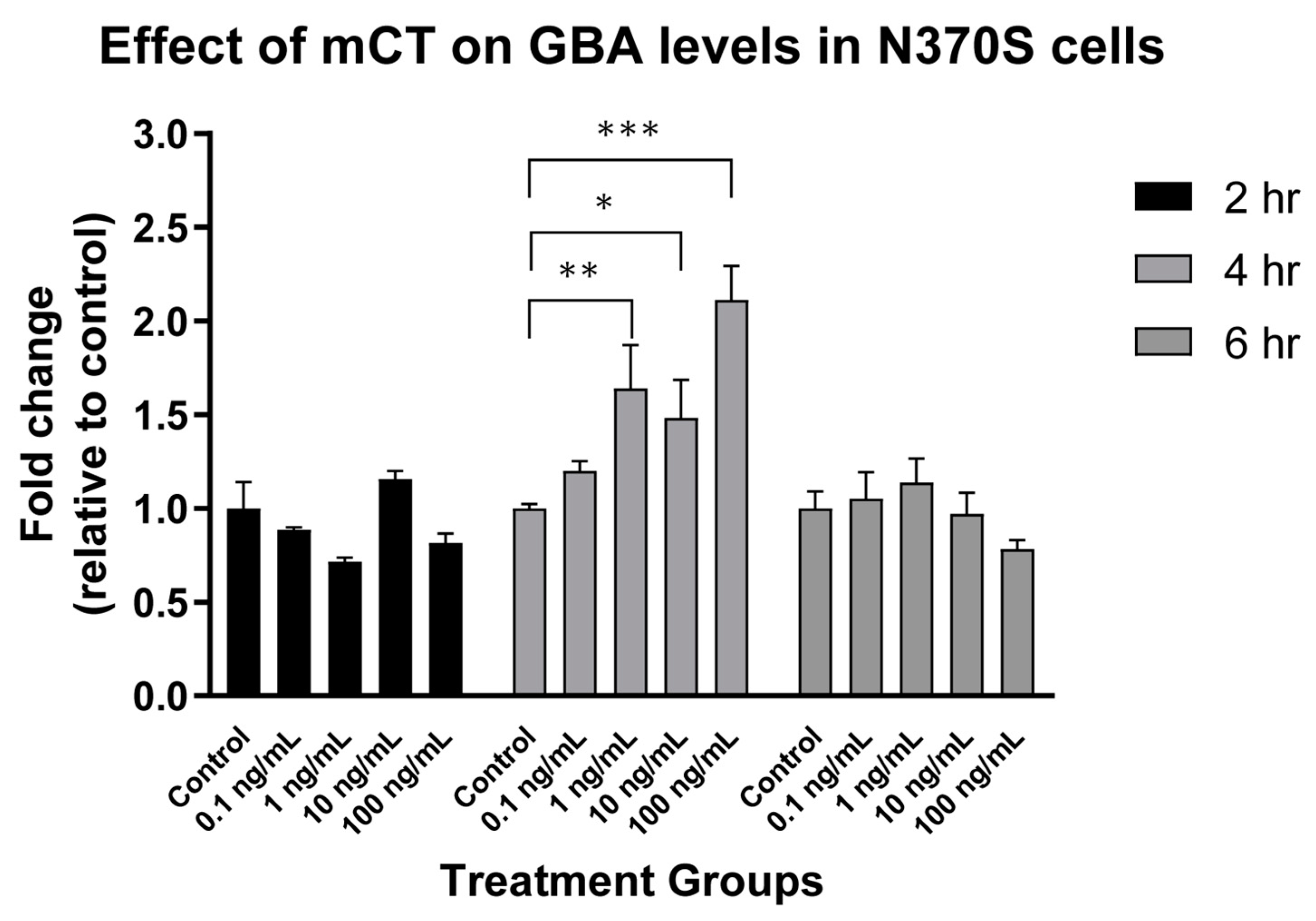
7.2. N370S Glucocerebrosidase
7.3. Cystic Fibrosis Animal Model
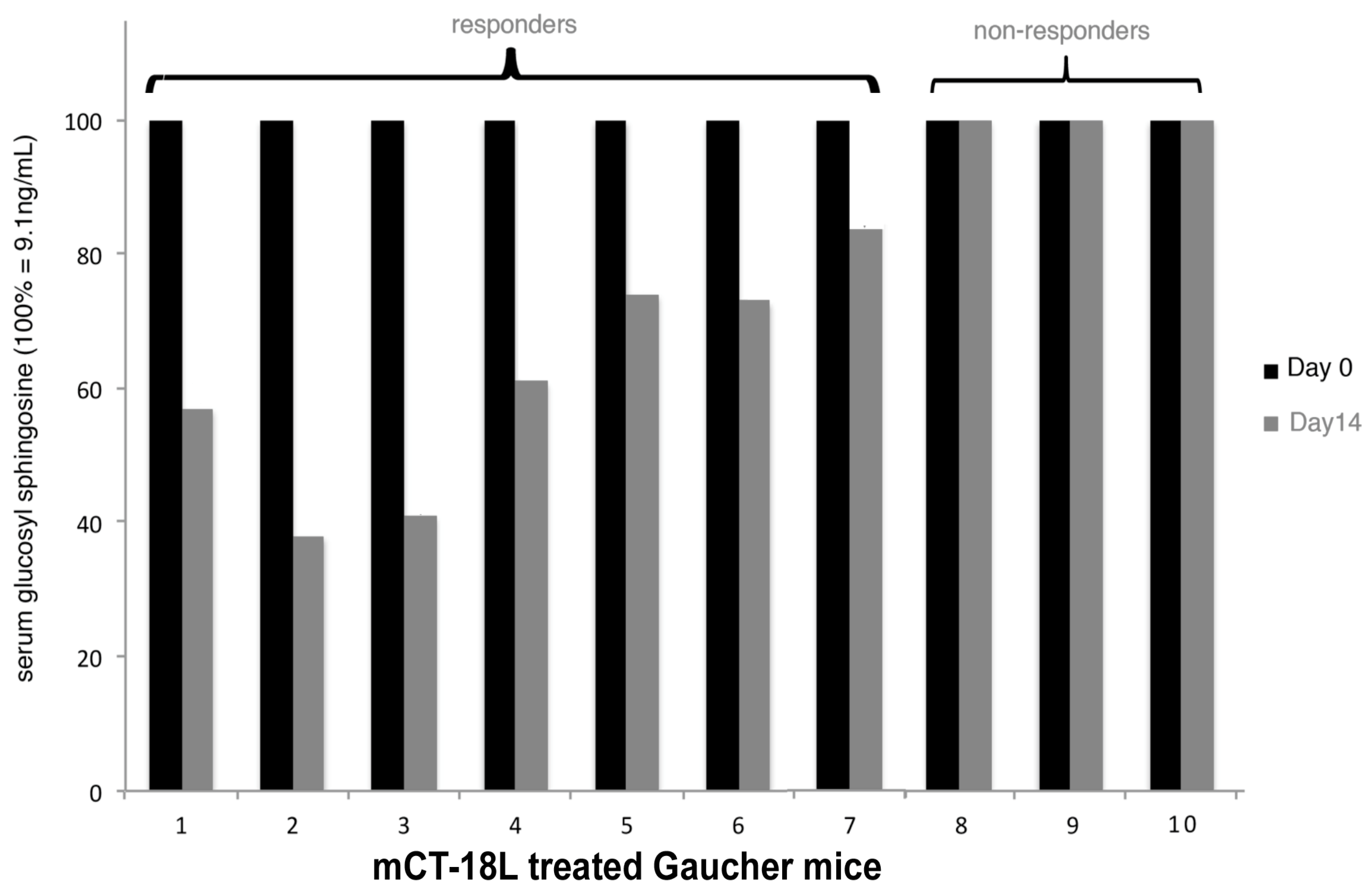
8. Gaucher Disease Animal Model
9. Future Studies
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karmali, M.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The Association between Idiopathic Hemolytic Uremic Syndrome and Infection by Verotoxin-Producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef]
- Jokiranta, T.S. HUS and atypical HUS. Blood 2017, 129, 2847–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, H.; Takeda, T. Escherichia coli Shiga toxin. J. Nat. Toxins 2000, 9, 299–313. [Google Scholar] [PubMed]
- Ray, P.E.; Liu, X.-H. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr. Nephrol. 2001, 16, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Andreoli, S. Extrarenal manifestations of the hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli (STEC HUS). Pediatr. Nephrol. 2019, 34, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between Virulence Factors of Shiga Toxin-ProducingEscherichia coli and Disease in Humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werber, D.; Fruth, A.; Buchholz, U.; Prager, R.; Kramer, M.H.; Ammon, A.; Tschäpe, H. Strong Association Between Shiga Toxin-Producing Escherichia coli O157 and Virulence Genes stx 2 and eae as Possible Explanation for Predominance of Serogroup O157 in Patients with Haemolytic Uraemic Syndrome. Eur. J. Clin. Microbiol. Infect. Dis. 2003, 22, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Watahiki, M.; Isobe, J.; Kimata, K.; Shima, T.; Kanatani, J.-I.; Shimizu, M.; Nagata, A.; Kawakami, K.; Yamada, M.; Izumiya, H.; et al. Characterization of Enterohemorrhagic Escherichia coli O111 and O157 Strains Isolated from Outbreak Patients in Japan. J. Clin. Microbiol. 2014, 52, 2757–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [CrossRef]
- Okuda, T.; Numata, S.-I.; Ito, M.; Ohta, M.; Kawamura, K.; Wiels, J.; Urano, T.; Tajima, O.; Furukawa, K.; Furukawa, K. Targeted Disruption of Gb3/CD77 Synthase Gene Resulted in the Complete Deletion of Globo-series Glycosphingolipids and Loss of Sensitivity to Verotoxins. J. Biol. Chem. 2006, 281, 10230–10235. [Google Scholar] [CrossRef] [Green Version]
- Porubsky, S.; Luckow, B.; Bonrouhi, M.; Speak, A.; Cerundolo, V.; Platt, F.; Gröne, H.J. Glycosphingolipids Gb3 and iGb3. In vivo roles in hemolytic-uremic syndrome and iNKT cell function. Pathologe 2008, 29 (Suppl. 2), 297–302. [Google Scholar]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H.; et al. Kinetic Analysis of Binding between Shiga Toxin and Receptor Glycolipid Gb3Cer by Surface Plasmon Resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.; Proulx, F.; Lingwood, C.A. Detergent-resistant globotriaosyl ceramide may define verotoxin/glomeruli-restricted hemolytic uremic syndrome pathology. Kidney Int. 2009, 75, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid Rafts as a Membrane-Organizing Principle. Science 2009, 327, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.G.N. Lipid rafts generate digital-like signal transduction in cell plasma membranes. Biotechnol. J. 2012, 7, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.M.; Brumell, J.H.; Finlay, B. Microbial pathogenesis: Lipid rafts as pathogen portals. Curr. Biol. 2000, 10, R823–R825. [Google Scholar] [CrossRef] [Green Version]
- Heung, L.J.; Luberto, C.; Del Poeta, M. Role of Sphingolipids in Microbial Pathogenesis. Infect. Immun. 2006, 74, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utskarpen, A.; Massol, R.; Van Deurs, B.; Lauvrak, S.U.; Kirchhausen, T.; Sandvig, K. Shiga Toxin Increases Formation of Clathrin-Coated Pits through Syk Kinase. PLoS ONE 2010, 5, e10944. [Google Scholar] [CrossRef] [Green Version]
- Torgersen, M.L.; Lauvrak, S.U.; Sandvig, K. The A-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005, 272, 4103–4113. [Google Scholar] [CrossRef]
- Khine, A.A.; Lingwood, C.A. Capping and receptor-mediated endocytosis of cell-bound verotoxin (shiga-like toxin) 1: Chemical identification of an amino acid in the B subunit necessary for efficient receptor glycolipid binding and cellular internalization. J. Cell. Physiol. 1994, 161, 319–332. [Google Scholar] [CrossRef]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.E.; Fraisier, V.; Florent, J.-C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nat. Cell Biol. 2007, 450, 670–675. [Google Scholar] [CrossRef]
- Renard, H.-F.; Simunovic, M.; Lemière, J.; Boucrot, E.; Garcia-Castillo, M.D.; Arumugam, S.; Chambon, V.; Lamaze, C.; Wunder, C.; Kenworthy, A.; et al. Endophilin-A2 functions in membrane scission in clathrin-independent endocytosis. Nat. Cell Biol. 2015, 517, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L. Shiga Toxin—A Model for Glycolipid-Dependent and Lectin-Driven Endocytosis. Toxins 2017, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Falguières, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga Toxin B-Subunit to Retrograde Transport Route in Association with Detergent-resistant Membranes. Mol. Biol. Cell 2001, 12, 2453–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin triggers endothelial and podocyte injury: The role of complement activation. Pediatr. Nephrol. 2019, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Detzner, J.; Gloerfeld, C.; Pohlentz, G.; Legros, N.; Humpf, H.-U.; Mellmann, A.; Karch, H.; Müthing, J. Structural Insights into Escherichia coli Shiga Toxin (Stx) Glycosphingolipid Receptors of Porcine Renal Epithelial Cells and Inhibition of Stx-Mediated Cellular Injury Using Neoglycolipid-Spiked Glycovesicles. Microorganisms 2019, 7, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kociurzynski, R.; Makshakova, O.N.; Knecht, V.; Römer, W. Multiscale Molecular Dynamics Studies Reveal Different Modes of Receptor Clustering by Gb3-Binding Lectins. J. Chem. Theory Comput. 2021, 17, 2488–2501. [Google Scholar] [CrossRef]
- Sandvig, K.; Bergan, J.; Dyve, A.-B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon 2010, 56, 1181–1185. [Google Scholar] [CrossRef]
- Hazes, B.; Read, R. Accumulating Evidence Suggests That Several AB-Toxins Subvert the Endoplasmic Reticulum-Associated Protein Degradation Pathway To Enter Target Cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Saleh, M.T.; Ferguson, J.; Boggs, J.M.; Gariépy, J. Insertion and Orientation of a Synthetic Peptide Representing the C-Terminus of the A1Domain of Shiga Toxin into Phospholipid Membranes. Biochemistry 1996, 35, 9325–9334. [Google Scholar] [CrossRef]
- Nowakowska-Gołacka, J.; Sominka, H.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. Int. J. Mol. Sci. 2019, 20, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The Mammalian Endoplasmic Reticulum-Associated Degradation System. Cold Spring Harb. Perspect. Biol. 2012, 5, a013185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigotti, M.; Carnicelli, D.; Alvergna, P.; Mazzaracchio, R.; Sperti, S.; Montanaro, L. The RNA-N-glycosidase activity of Shiga-like toxin I: Kinetic parameters of the native and activated toxin. Toxicon 1997, 35, 1431–1437. [Google Scholar] [CrossRef]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar] [PubMed] [Green Version]
- Fujinaga, Y.; Wolf, A.A.; Rodighiero, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to ER. Mol. Biol. Cell. 2003, 14, 4783–4793. [Google Scholar] [CrossRef] [PubMed]
- Lencer, W.I.; Saslowsky, D. Raft trafficking of AB5 subunit bacterial toxins. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1746, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Lopata, A.; Kniss, A.; Löhr, F.; Rogov, V.V.; Dötsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369. [Google Scholar] [CrossRef]
- Shi, J.; Hu, X.; Guo, Y.; Wang, L.; Ji, J.; Li, J.; Zhang, Z.-R. A technique for delineating the unfolding requirements for substrate entry into retrotranslocons during endoplasmic reticulum–associated degradation. J. Biol. Chem. 2019, 294, 20084–20096. [Google Scholar] [CrossRef]
- Wiertz, E.J.H.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec6l-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nat. Cell Biol. 1996, 384, 432–438. [Google Scholar] [CrossRef]
- Schäfer, A.; Wolf, D.H. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009, 28, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.-L.; Römisch, K. Proteasome 19S RP Binding to the Sec61 Channel Plays a Key Role in ERAD. PLoS ONE 2015, 10, e0117260. [Google Scholar] [CrossRef] [PubMed]
- Römisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef]
- Bernardi, K.M.; Forster, M.L.; Lencer, W.I.; Tsai, B. Derlin-1 Facilitates the Retro-Translocation of Cholera Toxin. Mol. Biol. Cell 2008, 19, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, Y.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Nagata, K.; Mori, K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006, 172, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballar, P.; Pabuççuoğlu, A.; Kose, F.A. Different p97/VCP complexes function in retrotranslocation step of mammalian Er-associated degradation (ERAD). Int. J. Biochem. Cell Biol. 2011, 43, 613–621. [Google Scholar] [CrossRef]
- Garza, R.M.; Sato, B.K.; Hampton, R.Y. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J. Biol. Chem. 2009, 284, 14710–14722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, E.M.; Kreft, S.G.; Greenblatt, W.H.; Swanson, R.; Hochstrasser, M. Aberrant substrate engagement of the ER translocon triggers degradation by the Hrd1 ubiquitin ligase. J. Cell Biol. 2012, 197, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonça, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E.; et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, E582–E591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasic, V.; Denkert, N.; Schmidt, C.; Riedel, D.; Stein, A.; Meinecke, M. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 2020, 22, 274–281. [Google Scholar] [CrossRef]
- Li, S.; Spooner, R.A.; Hampton, R.Y.; Lord, J.M.; Roberts, L.M. Cytosolic Entry of Shiga-Like Toxin A Chain from the Yeast Endoplasmic Reticulum Requires Catalytically Active Hrd1p. PLoS ONE 2012, 7, e41119. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Roberts, L.M.; Lencer, W.I. Entry of Protein Toxins into Mammalian Cells by Crossing the Endoplasmic Reticulum Membrane: Co-opting Basic Mechanisms of Endoplasmic Reticulum-Associated Degradation. Curr. Top. Microbiol. Immunol. 2005, 300, 149–168. [Google Scholar] [CrossRef]
- Teter, K.; Allyn, R.L.; Jobling, M.; Holmes, R.K. Transfer of the Cholera Toxin A1 Polypeptide from the Endoplasmic Reticulum to the Cytosol Is a Rapid Process Facilitated by the Endoplasmic Reticulum-Associated Degradation Pathway. Infect. Immun. 2002, 70, 6166–6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, G.; Mikoryak, C.; Hayslett, T.; Bhat, A.; Draper, R.K. Cholera Toxin Up-Regulates Endoplasmic Reticulum Proteins That Correlate with Sensitivity to the Toxin. Exp. Biol. Med. 2008, 233, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Taga, S.; Carlier, K.; Mishal, Z.; Capoulade, C.; Mangeney, M.; Lecluse, Y.; Coulaud, D.; Tetaud, C.; Pritchard, L.L.; Tursz, T.; et al. Intracellular signaling events in CD77-mediated apoptosis of Burkitt’s lymphoma cells. Blood 1997, 90, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Uchida, H.; Kiyokawa, N.; Mori, T.; Sato, N.; Horie, H.; Takeda, T.; Fujimoto, J. Verotoxins induce apoptosis in human renal tubular epithelium derived cells. Kidney Int. 1998, 53, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Karpman, D.; Håkansson, A.; Perez, M.-T.R.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of Renal Cortical Cells in the Hemolytic-Uremic Syndrome: In Vivo and In Vitro Studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Koide, N.; Sugiyama, T.; Mori, I.; Yokochi, T. A Novel Caspase Dependent Pathway Is Involved in Apoptosis of Human Endothelial Cells by Shiga Toxins. Microbiol. Immunol. 2002, 46, 697–700. [Google Scholar] [CrossRef]
- Ching, J.C.Y.; Jones, N.L.; Ceponis, P.J.M.; Karmali, M.A.; Sherman, P.M. Escherichia coli Shiga-Like Toxins Induce Apoptosis and Cleavage of Poly(ADP-Ribose) Polymerase via In Vitro Activation of Caspases. Infect. Immun. 2002, 70, 4669–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, P.; Smith, S.J.; A Giembycz, M.; Rotondo, D.; Plevin, R. Verotoxin activates mitogen-activated protein kinase in human peripheral blood monocytes: Role in apoptosis and proinflammatory cytokine release. Br. J. Pharmacol. 2003, 140, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Fujii, J.; Wood, K.; Matsuda, F.; Carneiro-Filho, B.A.; Schlegel, K.H.; Yutsudo, T.; Binnington-Boyd, B.; Lingwood, C.A.; Obata, F.; Kim, K.S.; et al. Shiga Toxin 2 Causes Apoptosis in Human Brain Microvascular Endothelial Cells via C/EBP Homologous Protein. Infect. Immun. 2008, 76, 3679–3689. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef]
- Debernardi, J.; Hollville, E.; Lipinski, M.; Wiels, J.; Robert, A. Differential role of FL-BID and t-BID during verotoxin-1-induced apoptosis in Burkitt’s lymphoma cells. Oncogene 2018, 37, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Mangeney, M.; A Lingwood, C.; Taga, S.; Caillou, B.; Tursz, T.; Wiels, J. Apoptosis induced in Burkitt’s lymphoma cells via Gb3/CD77, a glycolipid antigen. Cancer Res. 1993, 53, 5314–5319. [Google Scholar]
- Nakagawa, I.; Nakata, M.; Kawabata, S.; Hamada, S. Regulated expression of the Shiga toxin B gene induces apoptosis in mammalian fibroblastic cells. Mol. Microbiol. 2002, 33, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovbasnjuk, O.; Mourtazina, R.; Baibakov, B.; Wang, T.; Elowsky, C.; Choti, M.A.; Kane, A.; Donowitz, M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 19087–19092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tétaud, C.; Falguières, T.; Carlier, K.; Lécluse, Y.; Garibal, J.; Coulaud, D.; Busson, P.; Steffensen, R.; Clausen, H.; Johannes, L.; et al. Two distinct Gb3/CD77 signaling pathways leading to apoptosis are triggered by anti-Gb3/CD77 mAb and verotoxin-1. J. Biol. Chem. 2003, 278, 45200–45208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercatelli, D.; Bortolotti, M.; Giorgi, F.M. Transcriptional network inference and master regulator analysis of the response to ribosome-inactivating proteins in leukemia cells. Toxicology 2020, 441, 152531. [Google Scholar] [CrossRef]
- Mori, T.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Sato, N.; Ohmi, K.; Nakajima, H.; Takeda, T.; et al. Globotriaosyl ceramide (CD77/Gb3) in the glycolipid-enriched membrane domain participates in B-cell receptor-mediated apoptosis by regulating lyn kinase activity in human B cells. Exp. Hematol. 2000, 28, 1260–1268. [Google Scholar] [CrossRef]
- Fujii, J.; Matsui, T.; Heatherly, D.P.; Schlegel, K.H.; Lobo, P.I.; Yutsudo, T.; Ciraolo, G.M.; Morris, R.E.; Obrig, T. Rapid Apoptosis Induced by Shiga Toxin in HeLa Cells. Infect. Immun. 2003, 71, 2724–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Peng, L.; Shen, M.; Xia, Y.; Li, Z.; He, N. Shiga-like toxin I exerts specific and potent anti-tumour efficacy against gastric cancer cell proliferation when driven by tumour-preferential Frizzled-7 promoter. Cell Prolif. 2019, 52, e12607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, D.; Johansson, A.; Grankvist, K.; Andersson, U.; Henriksson, R.; Bergström, P.; Brännström, T.; Behnam-Motlagh, P. Verotoxin-1 induction of apoptosis in Gb3-expressing human glioma cell lines. Cancer Biol. Ther. 2006, 5, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Johansson, D.; Kosovac, E.; Moharer, J.; Ljuslinder, I.; Brännström, T.; Johansson, A.; Behnam-Motlagh, P. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer 2009, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debernardi, J.; Pioche-Durieu, C.; Le Cam, E.; Wiels, J.; Robert, A. Verotoxin-1-Induced ER Stress Triggers Apoptotic or Survival Pathways in Burkitt Lymphoma Cells. Toxins 2020, 12, 316. [Google Scholar] [CrossRef] [PubMed]
- Larson, G.; Samuelsson, B.E. Blood Group Type Glycosphingolipids of Human Cord Blood Erythrocytes1. J. Biochem. 1980, 88, 647–657. [Google Scholar] [CrossRef]
- Kundu, S.K.; Evans, A.; Rizvi, J.; Glidden, H.; Marcus, D.M. A new pkphenotype in the p blood group system. Eur. J. Immunogenet. 1980, 7, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Wiels, J.; Fellous, M.; Tursz, T. Monoclonal antibody against a Burkitt lymphoma-associated antigen. Proc. Natl. Acad. Sci. USA 1981, 78, 6485–6488. [Google Scholar] [CrossRef] [Green Version]
- Fellous, M.; Wiels, J.; Cartron, J.P.; Tursz, T. A monoclonal antibody, specific for Burkitt’s lymphoma, is also a blood group Pk antibody. Dev. Boil. Stand. 1984, 57, 293–298. [Google Scholar]
- Balana, A.; Wiels, J.; Tetaud, C.; Tursz, T.; Mishal, Z. Induction of cell differentiation in burkitt lymphoma lines. BLA: A glycolipid marker of B-cell differentiation. Int. J. Cancer 1985, 36, 453–460. [Google Scholar] [CrossRef]
- Mangeney, M.; Richard, Y.; Coulaud, D.; Tursz, T.; Wiels, J. CD77: An antigen of germinal center B cells entering apoptosis. Eur. J. Immunol. 1991, 21, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, G.; Zeuthen, J. Distribution of the Burkitt’s-lymphoma-associated antigen (BLA) in normal human tissue and malignant lymphoma as defined by immunohistological staining with monoclonal antibody 38.13. J. Cancer Res. Clin. Oncol. 1987, 113, 78–86. [Google Scholar] [CrossRef]
- Bien, T.; Perl, M.; Machmüller, A.C.; Nitsche, U.; Conrad, A.; Johannes, L.; Müthing, J.; Soltwisch, J.; Janssen, K.-P.; Dreisewerd, K. MALDI-2 Mass Spectrometry and Immunohistochemistry Imaging of Gb3Cer, Gb4Cer, and Further Glycosphingolipids in Human Colorectal Cancer Tissue. Anal. Chem. 2020, 92, 7096–7105. [Google Scholar] [CrossRef]
- Zhang, T.; Van Die, I.; Tefsen, B.; Van Vliet, S.J.; Laan, L.C.; Zhang, J.; Dijke, P.T.; Wuhrer, M.; Belo, A.I. Differential O- and Glycosphingolipid Glycosylation in Human Pancreatic Adenocarcinoma Cells With Opposite Morphology and Metastatic Behavior. Front. Oncol. 2020, 10, 732. [Google Scholar] [CrossRef]
- Stimmer, L.; Dehay, S.; Nemati, F.; Massonnet, G.; Richon, S.; Decaudin, D.; Klijanienko, J.; Johannes, L. Human breast cancer and lymph node metastases express Gb3 and can be targeted by STxB-vectorized chemotherapeutic compounds. BMC Cancer 2014, 14, 916. [Google Scholar] [CrossRef] [PubMed]
- Arab, S.; Russel, E.; Chapman, W.B.; Rosen, B.; A Lingwood, C. Expression of the verotoxin receptor glycolipid, globotriaosylceramide, in ovarian hyperplasias. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1997, 9, 553–563. [Google Scholar]
- Farkas-Himsley, H.; Hill, R.P.; Rosen, B.; Arab, S.; Lingwood, C.A. The bacterial colicin active against tumor cells in vitro and in vivo is verotoxin 1. Proc. Natl. Acad. Sci. USA 1995, 92, 6996–7000. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Klock, J.; Macher, B. Neutral glycospingolipids in hairy cell leukemia. Biochemistry 1981, 20, 6505–6508. [Google Scholar] [CrossRef]
- Furukawa, K.; Yokoyama, K.; Sato, T.; Wiels, J.; Hirayama, Y.; Ohta, M.; Furukawa, K. Expression of the Gb3/CD77 synthase gene in megakaryoblastic leukemia cells: Implication in the sensitivity to verotoxins. J. Biol. Chem. 2002, 277, 11247–11254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacasse, E.C.; Bray, M.; Patterson, B.; Lim, W.M.; Perampalam, S.; Radvanyi, L.G.; Keating, A.; Stewart, A.K.; Buckstein, R.; Sandhu, J.S.; et al. Shiga-like toxin-1 receptor on human breast cancer, lymphoma, and myeloma and absence from CD34(+) hematopoietic stem cells: Implications for ex vivo tumor purging and autologous stem cell transplantation. Blood 1999, 94, 2901–2910. [Google Scholar]
- Arbus, G.S.; Grisaru, S.; Segal, O.; Dosch, M.; Pop, M.; Lala, P.; Nutikka, A.; A Lingwood, C. Verotoxin targets lymphoma infiltrates of patients with post-transplant lymphoproliferative disease. Leuk. Res. 2000, 24, 857–864. [Google Scholar] [CrossRef]
- Ishitoya, S.; Kurazono, H.; Nishiyama, H.; Nakamura, E.; Kamoto, T.; Habuchi, T.; Terai, A.; Ogawa, O.; Yamamoto, S. Verotoxin Induces Rapid Elimination of Human Renal Tumor Xenografts in SCID Mice. J. Urol. 2004, 171, 1309–1313. [Google Scholar] [CrossRef]
- Falguières, T.; Maak, M.; von Weyhern, C.; Sarr, M.; Sastre, X.; Poupon, M.-F.; Robine, S.; Johannes, L.; Janssen, K.-P. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol. Cancer Ther. 2008, 7, 2498–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, P.E.; Maak, M.; Nitsche, U.; Perl, M.; Novotny, A.; Slotta-Huspenina, J.; Dransart, E.; Holtorf, A.; Johannes, L.; Janssen, K.-P. Gastric Adenocarcinomas Express the Glycosphingolipid Gb3/CD77: Targeting of Gastric Cancer Cells with Shiga Toxin B-Subunit. Mol. Cancer Ther. 2016, 15, 1008–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, C.; Fukushi, Y.; Satoh, M.; Saitoh, S.; Orikasa, S.; Nudelman, E.; Straud, M.; Hakomori, S.-I. Changes in glycolipid expression in human testicular tumor. Int. J. Cancer 1990, 45, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Souady, J.; Hülsewig, M.; Drmić-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S.; et al. Shiga Toxin Receptor Gb3Cer/CD77: Tumor-Association and Promising Therapeutic Target in Pancreas and Colon Cancer. PLoS ONE 2009, 4, e6813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maak, M.; Nitsche, U.; Keller, L.; Wolf, P.; Sarr, M.; Thiebaud, M.; Rosenberg, R.; Langer, R.; Kleeff, J.; Friess, H.; et al. Tumor-Specific Targeting of Pancreatic Cancer with Shiga Toxin B-Subunit. Mol. Cancer Ther. 2011, 10, 1918–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storck, W.; Meisen, I.; Gianmoena, K.; Pläger, I.; Kouzel, I.U.; Bielaszewska, M.; Haier, J.; Mormann, M.; Humpf, H.-U.; Karch, H.; et al. Shiga toxin glycosphingolipid receptor expression and toxin susceptibility of human pancreatic ductal adenocarcinomas of differing origin and differentiation. Biol. Chem. 2012, 393, 785–799. [Google Scholar] [CrossRef]
- Li, S.C.; Kundu, S.K.; Degasperi, R.; Li, Y.T. Accumulation of globotriaosylceramide in a case of leiomyosarcoma. Biochem. J. 1986, 240, 925–927. [Google Scholar] [CrossRef]
- Arab, S.; Murakami, M.; Dirks, P.; Boyd, B.; Hubbard, S.L.; Lingwood, C.A.; Rutka, J.T. Verotoxins inhibit the growth of and induce apoptosis in human astrocytoma cells. J. Neuro Oncol. 1998, 40, 137–150. [Google Scholar] [CrossRef]
- Gariépy, J. The use of Shiga-like toxin 1 in cancer therapy. Crit. Rev. Oncol. 2001, 39, 99–106. [Google Scholar] [CrossRef]
- Salhia, B.; Rutka, J.T.; Lingwood, C.; Nutikka, A.; Van Furth, W.R. The Treatment of Malignant Meningioma with Verotoxin. Neoplasia 2002, 4, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Couture, O.; Dransart, E.; Dehay, S.; Nemati, F.; Decaudin, D.; Johannes, L.; Tanter, M. Tumor Delivery of Ultrasound Contrast Agents Using Shiga Toxin B Subunit. Mol. Imaging 2011, 10, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Arab, S.; Rutka, J.; Lingwood, C. Verotoxin induces apoptosis and the complete, rapid, long-term elimination of human astrocytoma xenografts in nude mice. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1999, 11, 33–39. [Google Scholar]
- Viel, T.; Dransart, E.; Nemati, F.; Henry, E.; Thézé, B.; Decaudin, D.; Lewandowski, D.; Boisgard, R.; Johannes, L.; Tavitian, B. In Vivo Tumor Targeting by the B-Subunit of Shiga Toxin. Mol. Imaging 2008, 7, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Heath-Engel, H.M.; Lingwood, C.A. Verotoxin sensitivity of ECV304 cells in vitro and in vivo in a xenograft tumour model: VT1 as a tumour neovascular marker. Angiogenesis 2003, 6, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Desselle, A.; Chaumette, T.; Gaugler, M.-H.; Cochonneau, D.; Fleurence, J.; Dubois, N.; Hulin, P.; Aubry, J.; Birklé, S.; Paris, F. Anti-Gb3 Monoclonal Antibody Inhibits Angiogenesis and Tumor Development. PLoS ONE 2012, 7, e45423. [Google Scholar] [CrossRef] [Green Version]
- Birklé, S.; Desselle, A.; Chaumette, T.; Gaugler, M.-H.; Cochonneau, D.; Fleurence, J.; Dubois, N.; Hulin, P.; Aubry, J.; Paris, F. Inhibition of tumor angiogenesis by globotriaosylceramide immunotargeting. Oncoimmunology 2013, 2, e23700. [Google Scholar] [CrossRef] [Green Version]
- Junqua, S.; Larsen, A.K.; Wils, P.; Mishal, Z.; Wiels, J.; Le Pecq, J.-B. Decreased accessibility of globotriaosylceramide associated with decreased tumorigenicity in Burkitt’s lymphoma variants induced by immunoselection. Cancer Res. 1989, 49, 6480–6486. [Google Scholar]
- Sibold, J.; Ahadi, S.; Werz, D.B.; Steinem, C. Chemically synthesized Gb3 glycosphingolipids: Tools to access their function in lipid membranes. Eur. Biophys. J. 2021, 50, 109–126. [Google Scholar] [CrossRef]
- Kiarash, A.; Boyd, B.; Lingwood, C. Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11146. [Google Scholar] [CrossRef]
- Arab, S.; Lingwood, C.A. Influence of phospholipid chain length on verotoxin/globotriaosyl ceramide binding in model membranes: Comparison of a surface bilayer film and liposomes. Glycoconj. J. 1996, 13, 159–166. [Google Scholar] [CrossRef]
- Chark, D.; Nutikka, A.; Trusevych, N.; Kuzmina, J.; Lingwood, C. Differential carbohydrate epitope recognition of globotriaosyl ceramide by verotoxins and a monoclonal antibody. Role in human renal glomerular binding. JBIC J. Biol. Inorg. Chem. 2004, 271, 405–417. [Google Scholar] [CrossRef]
- Kim, M.; Binnington, B.; Sakac, D.; Fernandes, K.R.; Shi, S.P.; Lingwood, C.A.; Branch, D.R. Comparison of detection methods for cell surface globotriaosylceramide. J. Immunol. Methods 2011, 371, 48–60. [Google Scholar] [CrossRef]
- Greenshields, K.N.; Halstead, S.K.; Zitman, F.M.; Rinaldi, S.; Brennan, K.M.; O’Leary, C.; Chamberlain, L.H.; Easton, A.; Roxburgh, J.; Pediani, J.; et al. The neuropathic potential of anti-GM1 autoantibodies is regulated by the local glycolipid environment in mice. J. Clin. Investig. 2009, 119, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Nyholm, P.-G.; Pascher, I. Steric presentation and recognition of the saccharide chains of glycolipids at the cell surface: Favoured conformations of the saccharide-lipid linkage calculated using molecular mechanics (MM3). Int. J. Biol. Macromol. 1993, 15, 43–51. [Google Scholar] [CrossRef]
- Hooper, N.M. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae (Review). Mol. Membr. Biol. 1999, 16, 145–156. [Google Scholar] [CrossRef]
- Yahi, N.; Aulas, A.; Fantini, J. How cholesterol constrains glycolipid conformation for optimal recognition of Alzheimer’s beta amyloid peptide (Abeta1-40). PLoS ONE 2010, 5, e9079. [Google Scholar] [CrossRef]
- Lingwood, D.; Binnington, B.; Róg, T.; Vattulainen, I.; Grzybek, M.; Coskun, Ü.; ALingwood, C.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar] [CrossRef]
- Mahfoud, R.; Manis, A.; Binnington, B.; Ackerley, C.; Lingwood, C.A. A Major Fraction of Glycosphingolipids in Model and Cellular Cholesterol-containing Membranes Is Undetectable by Their Binding Proteins. J. Biol. Chem. 2010, 285, 36049–36059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Christian, A.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef]
- Matencio, A.; Navarro-Orcajada, S.; González-Ramón, A.; García-Carmona, F.; López-Nicolás, J.M. Recent advances in the treatment of Niemann pick disease type C: A mini-review. Int. J. Pharm. 2020, 584, 119440. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Binnington, B.; Ngan, B.; Chadwick, K.; Fleshner, N.; A Lingwood, C. Cholesterol masks membrane glycosphingolipid tumor-associated antigens to reduce their immunodetection in human cancer biopsies. Glycobiology 2013, 23, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.W. Human teratocarcinoma stem cells: Glycolipid antigen expression and modulation during differentiation. J. Cell. Biochem. 1987, 35, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Bosnakovski, D.; Figueiredo, C.A.; Visser, J.W.; Perlingeiro, R.C.R. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood 2006, 109, 1743–1751. [Google Scholar] [CrossRef]
- Muramatsu, T.; Muramatsu, H. Carbohydrate antigens expressed on stem cells and early embryonic cells. Glycoconj. J. 2004, 21, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-L.; Hung, J.-T.; Cheung, S.K.C.; Lee, H.-Y.; Chu, K.-C.; Li, S.-T.; Lin, Y.-C.; Ren, C.-T.; Cheng, T.-J.R.; Hsu, T.-L.; et al. Carbohydrate-based vaccines with a glycolipid adjuvant for breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 2517–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.-Y.; Khoo, K.-H.; Li, S.-C.; Li, Y.-T.; et al. Ceramide Glycosylation by Glucosylceramide Synthase Selectively Maintains the Properties of Breast Cancer Stem Cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.-J.; Ding, Y.; Levery, S.B.; Lobaton, M.; Handa, K.; Hakomori, S.-I. Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 4968–4973. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Lala, P.; Ito, S.; Lingwood, C.A. Transfection of MDCK cells with the MDR1 gene results in a major increase in globotriaosyl ceramide and cell sensitivity to verocytotoxin: Role of P-gp in glycolipid biosynthesis. J. Biol. Chem. 2000, 275, 6246–6251. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.; Lingwood, C. Apparent cooperativity in multivalent verotoxin globotriaosyl ceramide binding: Kinetic and saturation binding experiments with radiolabelled verotoxin [125I]-VT1. Biochim. Biophys. Acta 2000, 1501, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Gallegos, K.M.; Conrady, D.G.; Karve, S.S.; Gunasekera, T.S.; Herr, A.B.; Weiss, A.A. Shiga Toxin Binding to Glycolipids and Glycans. PLoS ONE 2012, 7, e30368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Alaoui, A.; Schmidt, F.; Amessou, M.; Sarr, M.; Decaudin, D.; Florent, J.C.; Johannes, L. Shiga toxin-mediated retrograde delivery of a topoisomerase I inhibitor prodrug. Angew. Chem. Int. Ed. Engl. 2007, 46, 6469–6472. [Google Scholar] [CrossRef] [PubMed]
- Amessou, M.; Carrez, D.; Patin, D.; Sarr, M.; Grierson, D.S.; Croisy, A.; Tedesco, A.C.; Maillard, P.; Johannes, L. Retrograde delivery of photosensitizer (TPPp-O-beta-GluOH)3 selectively potentiates its photodynamic activity. Bioconjug. Chem. 2008, 19, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Batisse, C.; Dransart, E.; Sarkouh, R.A.; Brulle, L.; Bai, S.K.; Godefroy, S.; Johannes, L.; Schmidt, F. A new delivery system for auristatin in STxB-drug conjugate therapy. Eur. J. Med. Chem. 2015, 95, 483–491. [Google Scholar] [CrossRef]
- Tam, P.; Mahfoud, R.; Nutikka, A.; Khine, A.A.; Binnington, B.; Paroutis, P.; Lingwood, C. Differential Intracellular Trafficking and Binding of Verotoxin 1 and Verotoxin 2 to Globotriaosylceramide-containing Lipid Assemblies. J. Cell Physiol. 2008, 216, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Hagnerelle, X.; Plisson, C.; Lambert, O.; Marco, S.; Rigaud, J.L.; Johannes, L.; Lévy, D. Two-dimensional structures of the Shiga toxin B-subunit and of a chimera bound to the glycolipid receptor Gb3. J. Struct. Biol. 2002, 139, 113–121. [Google Scholar] [CrossRef]
- Haicheur, N.; Bismuth, E.; Bosset, S.; Adotevi, O.; Warnier, G.; Lacabanne, V.; Regnault, A.; Desaymard, C.; Amigorena, S.; Ricciardi-Castagnoli, P.; et al. The B Subunit of Shiga Toxin Fused to a Tumor Antigen Elicits CTL and Targets Dendritic Cells to Allow MHC Class I-Restricted Presentation of Peptides Derived from Exogenous Antigens. J. Immunol. 2000, 165, 3301–3308. [Google Scholar] [CrossRef] [Green Version]
- El Alaoui, A.; Schmidt, F.; Sarr, M.; Decaudin, D.; Florent, J.C.; Johannes, L. Synthesis and properties of a mitochondrial peripheral benzodiazepine receptor conjugate. Chem. Med. Chem. 2008, 3, 1687–1695. [Google Scholar] [CrossRef]
- Patel, C.; Saad, H.; Shenkman, M.; Lederkremer, G.Z. Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD. Cells 2020, 9, 2138. [Google Scholar] [CrossRef]
- Luginbuehl, V.; Meier, N.; Kovar, K.; Rohrer, J. Intracellular drug delivery: Potential usefulness of engineered Shiga toxin subunit B for targeted cancer therapy. Biotechnol. Adv. 2018, 36, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ryou, J.-H.; Sohn, Y.-K.; Hwang, D.-E.; Kim, H.-S. Shiga-like toxin-based high-efficiency and receptor-specific intracellular delivery system for a protein. Biochem. Biophys. Res. Commun. 2015, 464, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Ryou, J.; Sohn, Y.; Hwang, D.; Park, W.; Kim, N.; Heo, W.; Kim, M.-Y.; Kim, H.-S. Engineering of bacterial exotoxins for highly efficient and receptor-specific intracellular delivery of diverse cargos. Biotechnol. Bioeng. 2016, 113, 1639–1646. [Google Scholar] [CrossRef]
- Plavec, T.; Zahirović, A.; Zadravec, P.; Sabotič, J.; Berlec, A. Lectin-Mediated Binding of Engineered Lactococcus lactis to Cancer Cells. Microorganisms 2021, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Mohseni Moghadam, Z.; Halabian, R.; Sedighian, H.; Behzadi, E.; Amani, J.; Imani Fooladi, A.A. Designing and Analyzing the Structure of DT-STXB Fusion Protein as an Anti-tumor Agent: An in Silico Approach. Iran J. Pathol. 2019, 14, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Daniel, T.O. Shiga toxin-endothelial cell interactions. In Recent Advances in Verocytotoxin-Producing Eshcerichia Coli Infections; Karmali, M.A., Goglio, A.G., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 317–324. [Google Scholar]
- Robinson, L.A.; Hurley, R.M.; Lingwood, C.; Matsell, D.G. Escherichia coli verotoxin binding to human paediatric glomerular mesangial cells. Pediatr. Nephrol. 1995, 9, 700–704. [Google Scholar] [CrossRef]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Bitzan, M. Treatment options for HUS secondary to Escherichia coli O157:H7. Kidney Int. 2009, 75, S62–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene varients:frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, H.; Friedrich, A.W.; Gerber, A.; Zimmerhackl, L.B.; Schmidt, M.A.; Bielaszewska, M. New Aspects in the Pathogenesis of Enteropathic Hemolytic Uremic Syndrome. Semin. Thromb. Hemost. 2006, 32, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the Baboon Responses to Shiga-Like Toxin. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Siegler, R.L.; Pysher, T.J.; Tesh, V.L.; Taylor, F.B. Response to Single and Divided Doses of Shiga Toxin-1 in a Primate Model of Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2001, 12, 1458–1467. [Google Scholar] [CrossRef]
- Siegler, R.L.; Obrig, T.G.; Pysher, T.J.; Tesh, V.L.; Denkers, N.D.; Taylor, F.B. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr. Nephrol. 2003, 18, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.; Lingwood, C. Synergistic effect of verotoxin and interferon-alpha on erythropoiesis. Cell. Mol. Boil. 2003, 49, 1363–1369. [Google Scholar]
- Betz, J.; Dorn, I.; Kouzel, I.U.; Bauwens, A.; Meisen, I.; Kemper, B.; Bielaszewska, M.; Mormann, M.; Weymann, L.; Sibrowski, W.; et al. Shiga toxin of enterohaemorrhagic Escherichia coli directly injures developing human erythrocytes. Cell. Microbiol. 2016, 18, 1339–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, M.; Alves-Rosa, F.; Rubel, C.; Fernandez, G.C.; Fernández-Alonso, G.; Alberto, F.; Rivas, M.; Isturiz, M. Pretreatment of mice with lipopolysaccharide (LPS) or IL-1beta exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin. Exp. Immunol. 2000, 119, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; van Haaften, W.C.E.; Tesh, V.L. Regulation of Proinflammatory Cytokine Expression by Shiga Toxin 1 and/or Lipopolysaccharides in the Human Monocytic Cell Line THP-1. Infect. Immun. 2004, 72, 2618–2627. [Google Scholar] [CrossRef] [Green Version]
- Keusch, G.T.; Acheson, D.W.K.; Aaldering, L.; Erban, J.; Jacewicz, M.S. Comparison of the effects of Shiga-like toxin 1 on cytokine-and butyrate pretreated human umbilical and saphenous vein endothelial cells. J. Infect. Dis. 1996, 173, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Louise, C.B.; Tran, M.C.; Obrig, T.G. Sensitization of human umbilical vein endothelial cells to Shiga toxin: Involvement of protein kinase C and NF-kappaB. Infect. Immun. 1997, 65, 3337–3344. [Google Scholar] [CrossRef] [Green Version]
- Molostvov, G.; Morris, A.; Rose, P.; Basu, S. Interaction of cytokines and growth factor in the regulation of verotoxin-induced apoptosis in cultured human endothelial cells. Br. J. Haematol. 2001, 113, 891–897. [Google Scholar] [CrossRef]
- Stone, M.K.; Kolling, G.L.; Lindner, M.H.; Obrig, T.G. p38 Mitogen-Activated Protein Kinase Mediates Lipopolysaccharide and Tumor Necrosis Factor Alpha Induction of Shiga Toxin 2 Sensitivity in Human Umbilical Vein Endothelial Cells. Infect. Immun. 2007, 76, 1115–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stricklett, P.K.; Hughes, A.K.; Ergonul, Z.; Kohan, D.E. Molecular Basis for Up-Regulation by Inflammatory Cytokines of Shiga Toxin 1 Cytotoxicity and Globotriaosylceramide Expression. J. Infect. Dis. 2002, 186, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Clayton, F.; Pysher, T.J.; Lou, R.; Kohan, D.E.; Denkers, N.D.; Tesh, V.L.; Taylor , F.B., Jr.; Siegler, R.L. Lipopolysaccharide Upregulates Renal Shiga Toxin Receptors in a Primate Model of Hemolytic Uremic Syndrome. Am. J. Nephrol. 2005, 25, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Warnier, M.; Römer, W.; Geelen, J.; Lesieur, J.; Amessou, M.; Heuvel, L.V.D.; Monnens, L.; Johannes, L. Trafficking of Shiga toxin/Shiga-like toxin-1 in human glomerular microvascular endothelial cells and human mesangial cells. Kidney Int. 2006, 70, 2085–2091. [Google Scholar] [CrossRef] [Green Version]
- Hariya, Y.; Shirakawa, S.; Yonekura, N.; Yokosawa, N.; Kohama, G.-I.; Fujii, N. Augmentation of Verotoxin-Induced Cytotoxicity/Apoptosis by Interferon Is Repressed in Cells Persistently Infected with Mumps Virus. J. Interf. Cytokine Res. 1999, 19, 479–485. [Google Scholar] [CrossRef]
- Siegler, R.L.; Pysher, T.J.; Lou, R.; Tesh, V.L.; Taylor , F.B., Jr. Response to Shiga Toxin-1, with and without Lipopolysaccharide, in a Primate Model of Hemolytic Uremic Syndrome. Am. J. Nephrol. 2001, 21, 420–425. [Google Scholar] [CrossRef]
- van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; van den Heuvel, L.P.; van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood 1996, 88, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.H.; Armstrong, C.S.; Sakiri, R.; Tesh, V.L. Shiga Toxin-Induced Tumor Necrosis Factor Alpha Expression: Requirement for Toxin Enzymatic Activity and Monocyte Protein Kinase C and Protein Tyrosine Kinases. Infect. Immun. 2000, 68, 5183–5189. [Google Scholar] [CrossRef] [Green Version]
- Ohara, T.; Kojio, S.; Taneike, I.; Nakagawa, S.; Gondaira, F.; Tamura, Y.; Gejyo, F.; Zhang, H.-M.; Yamamoto, T. Effects of Azithromycin on Shiga Toxin Production by Escherichia coli and Subsequent Host Inflammatory Response. Antimicrob. Agents Chemother. 2002, 46, 3478–3483. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Park, M.J.; Ye, S.-K.; Kim, C.-W.; Kim, Y.-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Percheron, L.; Gramada, R.; Tellier, S.; Salomon, R.; Harambat, J.; Llanas, B.; Fila, M.; Allain-Launay, E.; Lapeyraque, A.-L.; Leroy, V.; et al. Eculizumab treatment in severe pediatric STEC-HUS: A multicenter retrospective study. Pediatr. Nephrol. 2018, 33, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Takahashi, M.; Tamada, M.; Senda, M.; Hibino, M.; Shimizu, E.; Okuta, A.; Miyazawa, A.; Senda, T.; Nishikawa, K. Identification of a peptide motif that potently inhibits two functionally distinct subunits of Shiga toxin. Commun. Biol. 2021, 4, 538. [Google Scholar] [CrossRef]
- Janssen, K.-P.; Vignjevic, D.M.; Boisgard, R.; Falguières, T.; Bousquet, G.; Decaudin, D.; Dollé, F.; Louvard, D.; Tavitian, B.; Robine, S.; et al. In vivo Tumor Targeting Using a Novel Intestinal Pathogen-Based Delivery Approach. Cancer Res. 2006, 66, 7230–7236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCloskey, N.; Pound, J.D.; Holder, M.J.; Williams, J.M.; Roberts, L.M.; Lord, J.M.; Gordon, J. The extrafollicular-to-follicular transition of human B lymphocytes: Induction of functional globotriaosylceramide (CD77) on high threshold occupancy of CD40. Eur. J. Immunol. 1999, 29, 3236–3244. [Google Scholar] [CrossRef]
- Vingert, B.; Adotevi, O.; Patin, D.; Jung, S.; Shrikant, P.; Freyburger, L.; Eppolito, C.; Sapoznikov, A.; Amessou, M.; Quintin-Colonna, F.; et al. The Shiga toxin B-subunit targets antigen in vivo to dendritic cells and elicits anti-tumor immunity. Eur. J. Immunol. 2006, 36, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Tartour, E.; Van der Bruggen, P.; Vantomme, V.; Joyeux, I.; Goud, B.; Fridman, W.H.; Johannes, L. Major histocompatibility complex class I presentation of exogenous soluble tumor antigen fused to the B-fragment of Shiga toxin. Eur. J. Immunol. 1998, 28, 2726–2737. [Google Scholar] [CrossRef]
- Noakes, K.L.; Teisserenc, H.T.; Lord, J.; Dunbar, P.; Cerundolo, V.; Roberts, L.M. Exploiting retrograde transport of Shiga-like toxin 1 for the delivery of exogenous antigens into the MHC class I presentation pathway. FEBS Lett. 1999, 453, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Chapman, D.C.; Williams, D.B. ER quality control in the biogenesis of MHC class I molecules. Semin. Cell Dev. Biol. 2010, 21, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, O.; Vingert, B.; Freyburger, L.; Shrikant, P.; Lone, Y.-C.; Quintin-Colonna, F.; Haicheur, N.; Amessou, M.; Herbelin, A.; Langlade-Demoyen, P.; et al. B Subunit of Shiga Toxin-Based Vaccines Synergize with α-Galactosylceramide to Break Tolerance against Self Antigen and Elicit Antiviral Immunity. J. Immunol. 2007, 179, 3371–3379. [Google Scholar] [CrossRef]
- Tran, T.; Diniz, M.O.; Dransart, E.; Gey, A.; Merillon, N.; Lone, Y.C.; Godefroy, S.; Sibley, C.; Ferreira, L.C.; Medioni, J.; et al. A Therapeutic Her2/neu Vaccine Targeting Dendritic Cells Preferentially Inhibits the Growth of Low Her2/neu-Expressing Tumor in HLA-A2 Transgenic Mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4133–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.C.; Lord, J.M.; Roberts, L.M.; Tartour, E.; Johannes, L. 1st Class Ticket to Class I: Protein Toxins as Pathfinders for Antigen Presentation. Traffic 2002, 3, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, F.; Terme, M.; Nizard, M.; Badoual, C.; Bureau, M.-F.; Freyburger, L.; Clement, O.; Marcheteau, E.; Gey, A.; Fraisse, G.; et al. Mucosal Imprinting of Vaccine-Induced CD8+ T Cells Is Crucial to Inhibit the Growth of Mucosal Tumors. Sci. Transl. Med. 2013, 5, 172ra20. [Google Scholar] [CrossRef] [Green Version]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef] [PubMed]
- Rutjes, N.W.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, D.; Ueno, T.; Takashima, S.; Ohta, K.; Miyawaki, T.; Suzuki, T.; Suzuki, Y. Establishment of a monoclonal antibody directed against Gb3Cer/CD77: A useful immunochemical reagent for a differentiation marker in Burkitt’s lymphoma and germinal centre B cells. Glycoconj. J. 1997, 14, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Mangeney, M.; Rousselet, G.; Taga, S.; Tursz, T.; Wiels, J. The fate of human CD77+ germinal center B lymphocytes after rescue from apoptosis. Mol. Immunol. 1995, 32, 333–339. [Google Scholar] [CrossRef]
- Liu, Y.J.; Malisan, F.; De Bouteiller, O.; Guret, C.; Lebecque, S.; Banchereau, J.; Mills, F.C.; E Max, E.; Martinez-Valdez, H. Within Germinal Centers, Isotype Switching of Immunoglobulin Genes Occurs after the Onset of Somatic Mutation. Immunity 1996, 4, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.; Madrid-Marina, V.; Estrov, Z.; Freedman, M.H.; Lingwood, C.A.; Dosch, H.-M. Expression of glycolipid receptors to Shiga-like toxin on human B lymphocytes: A mechanism for the failure of long-lived antibody response to dysenteric disease. Int. Immunol. 1990, 2, 1–8. [Google Scholar] [CrossRef]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Porcellini, E.; Galassi, E.; Valerii, M.C.; Spisni, E. Human monocytes stimulated by Shiga toxin 1a via globotriaosylceramide release proinflammatory molecules associated with hemolytic uremic syndrome. Int. J. Med. Microbiol. 2018, 308, 940–946. [Google Scholar] [CrossRef]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Barbieri, S.; Rocchi, L.; Arfilli, V.; Scavia, G.; Ricci, F.; Bontadini, A.; et al. Endothelial damage induced by Shiga toxins delivered by neutrophils during transmigration. J. Leukoc. Biol. 2010, 88, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Havira, M.S.; Ta, A.; Kumari, P.; Wang, C.; Russo, A.J.; Ruan, J.; Rathinam, V.A.; Vanaja, S.K. Shiga toxin suppresses noncanonical inflammasome responses to cytosolic LPS. Sci. Immunol. 2020, 5, eabc0217. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Watanabe-Takahashi, M.; Hamabata, T.; Furukawa, K.; Funamoto, S.; Nishikawa, K. A nontoxigenic form of Shiga toxin 2 suppresses the production of amyloid β by altering the intracellular transport of amyloid precursor protein through its receptor-binding B-subunit. Biochem. Biophys. Res. Commun. 2021, 557, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Helm, K.V.; Mulvey, G.L.; Armstrong, G.D. Serum Amyloid P Component Binding to Shiga Toxin 2 Requires Both A Subunit and B Pentamer. Infect. Immun. 2003, 71, 6075–6078. [Google Scholar] [CrossRef] [Green Version]
- Hamazaki, H. Ca(2+)-dependent binding of human serum amyloid P component to Alzheimer’s beta-amyloid peptide. J. Biol. Chem. 1995, 270, 10392–10394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamazaki, H. Amyloid P component promotes aggregation of Alzheimer’s beta-amyloid peptide. Biochem. Biophys. Res. Commun. 1995, 211, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Tennent, G.A.; Lovat, L.B.; Pepys, M.B. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc. Natl. Acad. Sci. USA 1995, 92, 4299–4303. [Google Scholar] [CrossRef] [Green Version]
- Pepys, M.B.; Herbert, J.; Hutchinson, W.L.; Tennent, G.A.; Lachmann, H.J.; Gallimore, J.R.; Lovat, L.B.; Bartfai, T.; Alanine, A.; Hertel, C.; et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nat. Cell Biol. 2002, 417, 254–259. [Google Scholar] [CrossRef]
- Reverter, M.; Rentero, C.; de Muga, S.V.; Alvarez-Guaita, A.; Mulay, V.; Cairns, R.; Wood, P.; Monastyrskaya, K.; Pol, A.; Tebar, F.; et al. Cholesterol transport from late endosomes to the Golgi regulates t-SNARE trafficking, assembly and function. Mol. Biol. Cell. 2011, 22, 4108–4123. [Google Scholar] [CrossRef]
- Lingwood, C. Verotoxin Receptor-Based Pathology and Therapies. Front. Cell. Infect. Microbiol. 2020, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Ridsdale, A.; Denis, M.; Gougeon, P.-Y.; Ngsee, J.K.; Presley, J.F.; Zha, X. Cholesterol Is Required for Efficient Endoplasmic Reticulum-to-Golgi Transport of Secretory Membrane Proteins. Mol. Biol. Cell 2006, 17, 1593–1605. [Google Scholar] [CrossRef] [Green Version]
- Marks, D.L.; Pagano, R.E. Endocytosis and sorting of glycosphingolipids in sphingolipid storage disease. Trends Cell Biol. 2002, 12, 605–613. [Google Scholar] [CrossRef]
- Puri, V.; Jefferson, J.R.; Singh, R.D.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Sphingolipid Storage Induces Accumulation of Intracellular Cholesterol by Stimulating SREBP-1 Cleavage. J. Biol. Chem. 2003, 278, 20961–20970. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, C. Is Cholesterol the Key Factor for Autism? Am. J. Biomed. Sci. Res. 2020, 7, 483–486. [Google Scholar]
- Levy, M.; Garmy, N.; Gazit, E.; Fantini, J. The minimal amyloid-forming fragment of the islet amyloid polypeptide is a glycolipid-binding domain. FEBS J. 2006, 273, 5724–5735. [Google Scholar] [CrossRef]
- Di Scala, C.; Yahi, N.; Lelievre, C.; Garmy, N.; Chahinian, H.; Fantini, J. Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s beta amyloid peptide. ACS Chem. Neurosci. 2013, 4, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer’s beta-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4, 120. [Google Scholar] [CrossRef] [Green Version]
- Tamboli, I.Y.; Prager, K.; Barth, E.; Heneka, M.; Sandhoff, K.; Walter, J. Inhibition of glycosphingolipid biosynthesis reduces secretion of the beta-amyloid precursor protein and amyloid beta-peptide. J. Biol. Chem. 2005, 280, 28110–28117. [Google Scholar] [CrossRef] [Green Version]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [Green Version]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum–Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef]
- Gething, M.-J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, S.; Guo, L.; Fisher, E.A.; Brodsky, J.L. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum–associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 2012, 23, 520–532. [Google Scholar] [CrossRef]
- Bozaykut, P.; Ozer, N.K.; Karademir, B. Regulation of protein turnover by heat shock proteins. Free. Radic. Biol. Med. 2014, 77, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Benaroudj, N.; Tarcsa, E.; Cascio, P.; Goldberg, A.L. The unfolding of substrates and ubiquitin-independent protein degradation by proteasomes. Biochimie 2001, 83, 311–318. [Google Scholar] [CrossRef]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef]
- Hebert, D.N.; Bernasconi, R.; Molinari, M. ERAD substrates: Which way out? Semin. Cell Dev. Biol. 2010, 21, 526–532. [Google Scholar] [CrossRef]
- Hoff, H.; Zhang, H.; Sell, C.; Giordano, A.; Romano, G. Protein Degradation Via the Proteosome. Cell Cycle Control Dysregulation Protoc. 2004, 285, 079–092. [Google Scholar] [CrossRef]
- Osaki, Y.; Saito, A.; Imaizumi, K. The degradation of mutant proteins by ERAD and the pathogenesis of diseases. Clin. Calcium 2018, 28, 1684–1689. [Google Scholar]
- Adnan, H.; Zhang, Z.; Park, H.-J.; Tailor, C.; Che, C.; Kamani, M.; Spitalny, G.; Binnington, B.; Lingwood, C. Endoplasmic Reticulum-Targeted Subunit Toxins Provide a New Approach to Rescue Misfolded Mutant Proteins and Revert Cell Models of Genetic Diseases. PLoS ONE 2016, 11, e0166948. [Google Scholar] [CrossRef]
- Castilla, J.; Rísquez, R.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Diaz, Y.; Mellet, C.O.; Fernández, J.M.G.; Castillón, S. Conformationally-locked N-glycosides: Exploiting long-range non-glycone interactions in the design of pharmacological chaperones for Gaucher disease. Eur. J. Med. Chem. 2015, 90, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Spano, V.; Montalbano, A.; Carbone, A.; Scudieri, P.; Galietta, L.J.V.; Barraja, P. An overview on chemical structures as DeltaF508-CFTR correctors. Eur. J. Med. Chem. 2019, 180, 430–448. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Benharouga, M.; Hu, W.; Lukacs, G.L. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 2001, 276, 8942–8950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibodeau, P.H.; Richardson, J.M., III; Wang, W.; Millen, L.; Watson, J.; Mendoza, J.L.; Du, K.; Fischman, S.; Senderowitz, H.; Lukacs, G.L.; et al. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2010, 285, 35825–35835. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Ward, C.L.; Kopito, R.R. Cotranslational Ubiquitination of Cystic Fibrosis Transmembrane Conductance Regulator in Vitro. J. Biol. Chem. 1998, 273, 7189–7192. [Google Scholar] [CrossRef] [Green Version]
- van Barneveld, A.; Stanke, F.; Tamm, S.; Siebert, B.; Brandes, G.; Derichs, N.; Ballmann, M.; Junge, S.; Tümmler, B. Functional analysis of F508del CFTR in native human colon. Biochim. Biophys. Acta 2010, 1802, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Bendikov-Bar, I.; Horowitz, M. Gaucher disease paradigm: From ERAD to co-morbidity. Hum. Mutat. 2012, 33, 1398–1407. [Google Scholar] [CrossRef]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- van Doorninck, J.H.; French, P.J.; Verbeek, E.; Peters, R.H.; Morreau, H.; Bijman, J.; Scholte, B.J. A mouse model for the cystic fibrosis delta F508 mutation. Embo J. 1995, 14, 4403–4411. [Google Scholar] [CrossRef] [Green Version]
- Zeiher, B.G.; Eichwald, E.; Zabner, J.; Smith, J.J.; Puga, A.P.; McCray, P.B.; Capecchi, M.R.; Welsh, M.J.; Thomas, K.R. A mouse model for the delta F508 allele of cystic fibrosis. J. Clin. Investig. 1995, 96, 2051–2064. [Google Scholar] [CrossRef] [Green Version]
- Best, J.A.; Quinton, P.M.; Klemcke, H.G.; Vallet, J.L.; Christenson, R.K. Salivary secretion assay for drug efficacy for cystic fibrosis in mice. Exp. Physiol. 2005, 90, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droebner, K.; Sandner, P. Modification of the salivary secretion assay in F508del mice—The murine equivalent of the human sweat test. J. Cyst. Fibros. 2013, 12, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ciciriello, F.; Anjos, S.M.; Carissimo, A.; Liao, J.; Carlile, G.W.; Balghi, H.; Robert, R.; Luini, A.; Hanrahan, J.W.; et al. Ouabain Mimics Low Temperature Rescue of F508del-CFTR in Cystic Fibrosis Epithelial Cells. Front. Pharmacol. 2012, 3, 176. [Google Scholar] [CrossRef] [Green Version]
- Dhooghe, B.; Bouckaert, C.; Capron, A.; Wallemacq, P.; Leal, T.; Noel, S. Resveratrol increases F508del-CFTR dependent salivary secretion in cystic fibrosis mice. Biol. Open 2015, 4, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Carlile, G.W.; Robert, R.; Goepp, J.; Matthes, E.; Liao, J.; Kus, B.; Macknight, S.D.; Rotin, D.; Hanrahan, J.W.; Thomas, D.Y. Ibuprofen rescues mutant cystic fibrosis transmembrane conductance regulator trafficking. J. Cyst. Fibros. 2015, 14, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Sanders, A.; Hemmelgarn, H.; Melrose, H.L.; Hein, L.; Fuller, M.; Clarke, L.A. Transgenic mice expressing human glucocerebrosidase variants: Utility for the study of Gaucher disease. Blood Cells Mol. Dis. 2013, 51, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; Van Dussen, L.; Hollak, C.E.M.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; Van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.M.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; McDermott, B.K.; Chin, S.J.; Fletcher, J.M.; Fuller, M. Expanding the clinical utility of glucosylsphingosine for Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 558–563. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Chen, Y.; Bellamy, W.P.; Seabra, M.C.; Field, M.C.; Ali, B.R. ER-associated protein degradation is a common mechanism underpinning numerous monogenic diseases including Robinow syndrome. Hum. Mol. Genet. 2005, 14, 2559–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, H.; Gou, Y.; Zook, A.; Lozano, M.M.; Dudley, J.P. ERAD and how viruses exploit it. Front. Microbiol. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchini, L.M.; Lingwood, C.A. A Verotoxin 1 B Subunit-Lambda CRO Chimeric Protein Specifically Binds Both DNA and Globotriaosylceramide (Gb3) to Effect Nuclear Targeting of Exogenous DNA in Gb3 Positive Cells. Exp. Cell Res. 2001, 269, 117–129. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lingwood, C. Therapeutic Uses of Bacterial Subunit Toxins. Toxins 2021, 13, 378. https://doi.org/10.3390/toxins13060378
Lingwood C. Therapeutic Uses of Bacterial Subunit Toxins. Toxins. 2021; 13(6):378. https://doi.org/10.3390/toxins13060378
Chicago/Turabian StyleLingwood, Clifford. 2021. "Therapeutic Uses of Bacterial Subunit Toxins" Toxins 13, no. 6: 378. https://doi.org/10.3390/toxins13060378
APA StyleLingwood, C. (2021). Therapeutic Uses of Bacterial Subunit Toxins. Toxins, 13(6), 378. https://doi.org/10.3390/toxins13060378