Assessing the Validity of Normalizing Aflatoxin B1-Lysine Albumin Adduct Biomarker Measurements to Total Serum Albumin Concentration across Multiple Human Population Studies

 , , , , and
, , , , and
Abstract
:1. Introduction
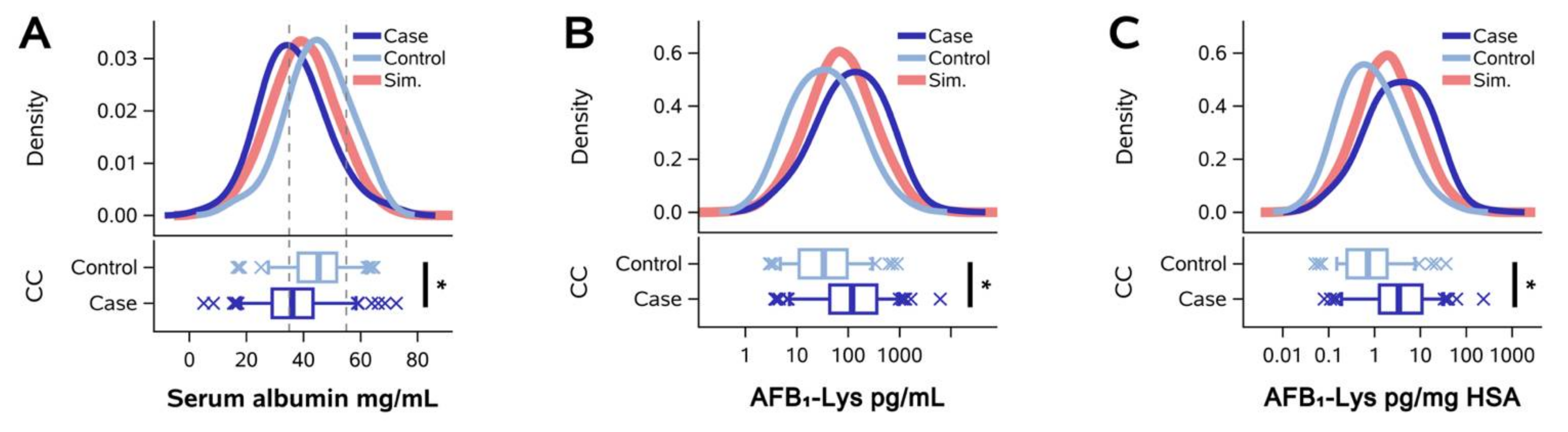
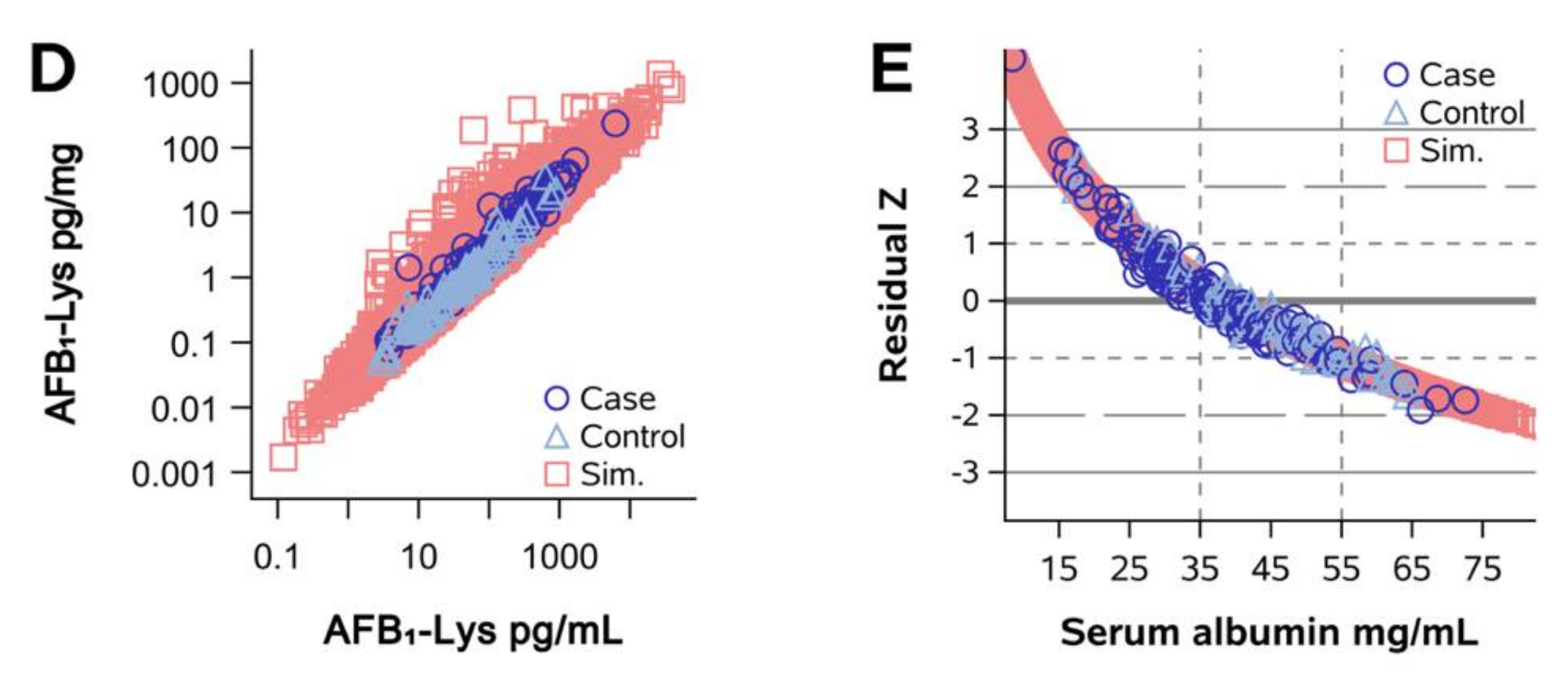
2. Results
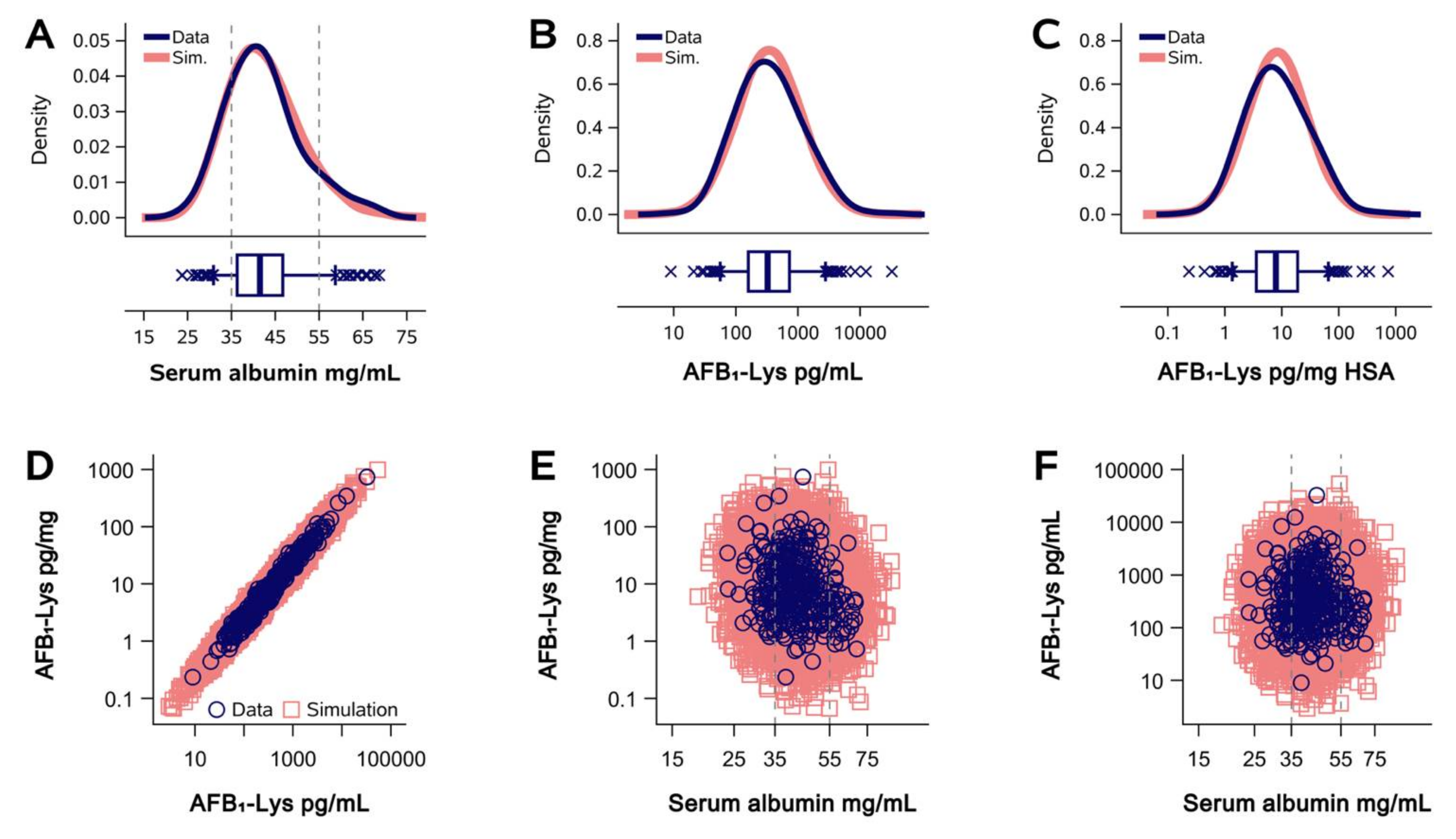
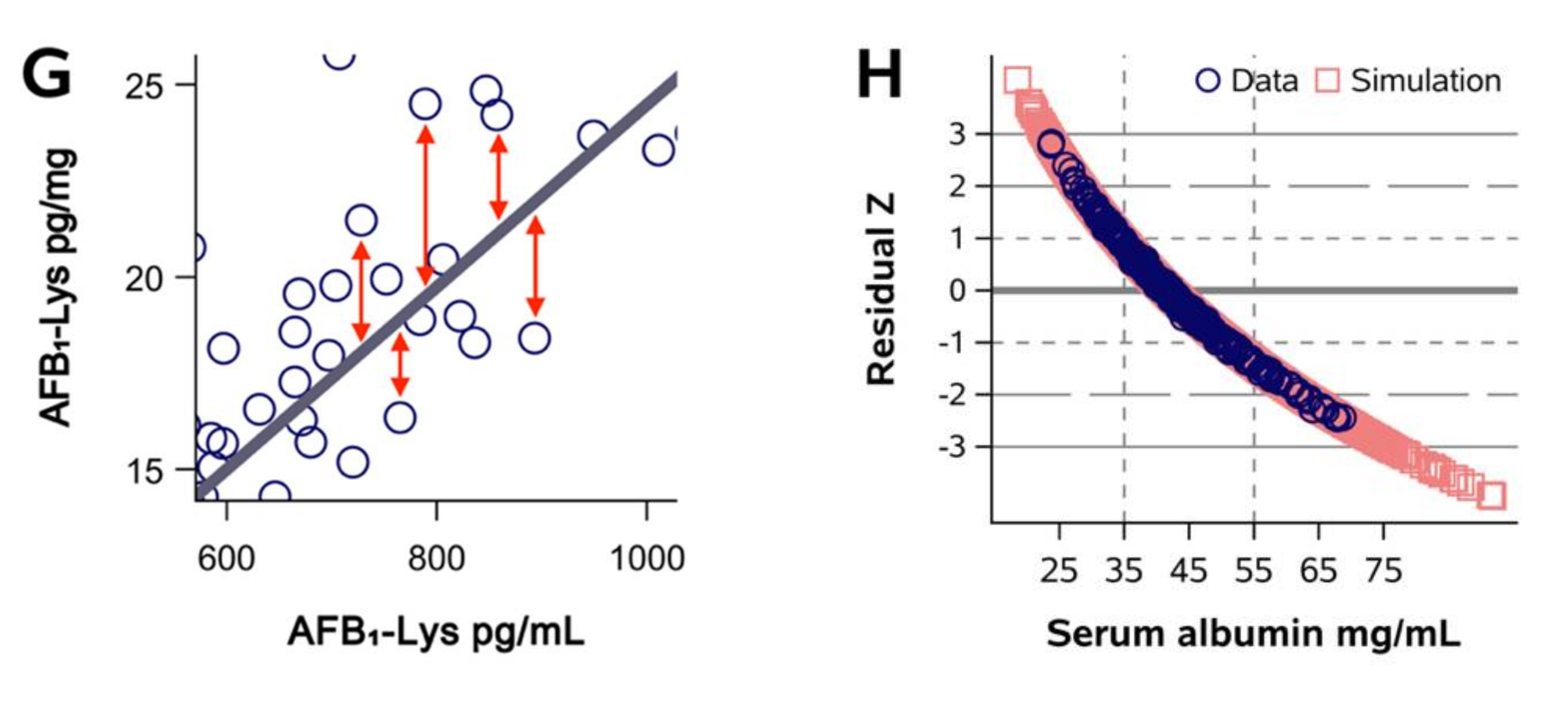
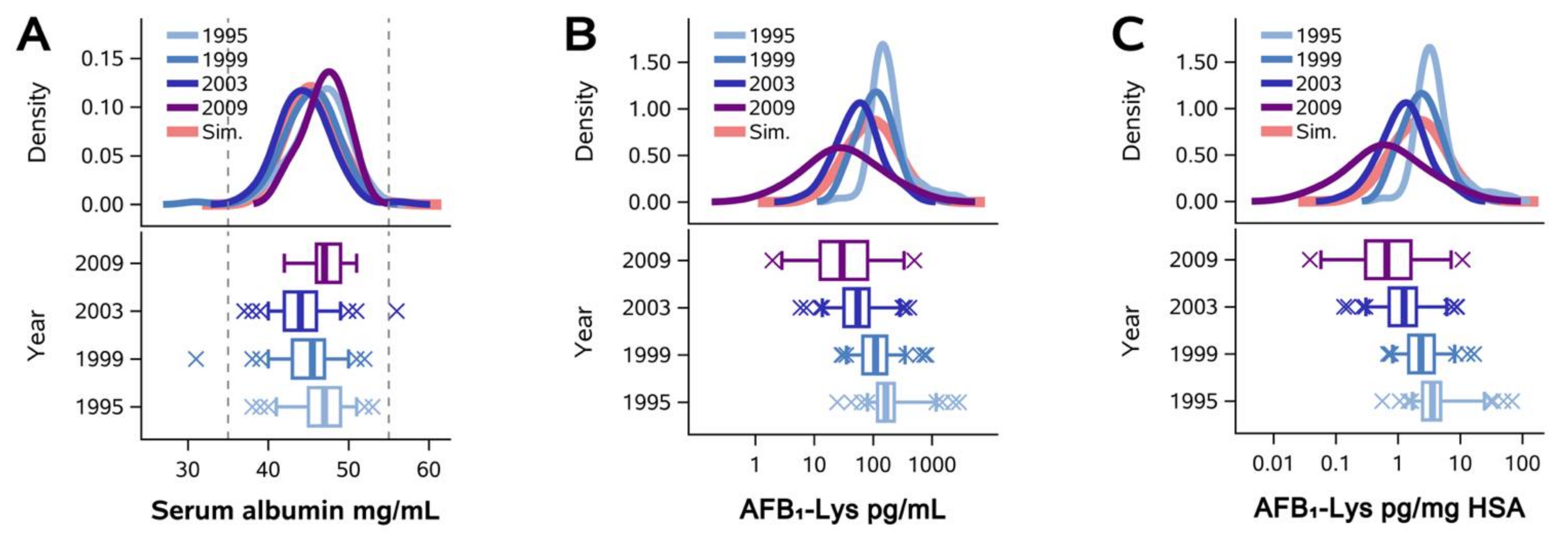
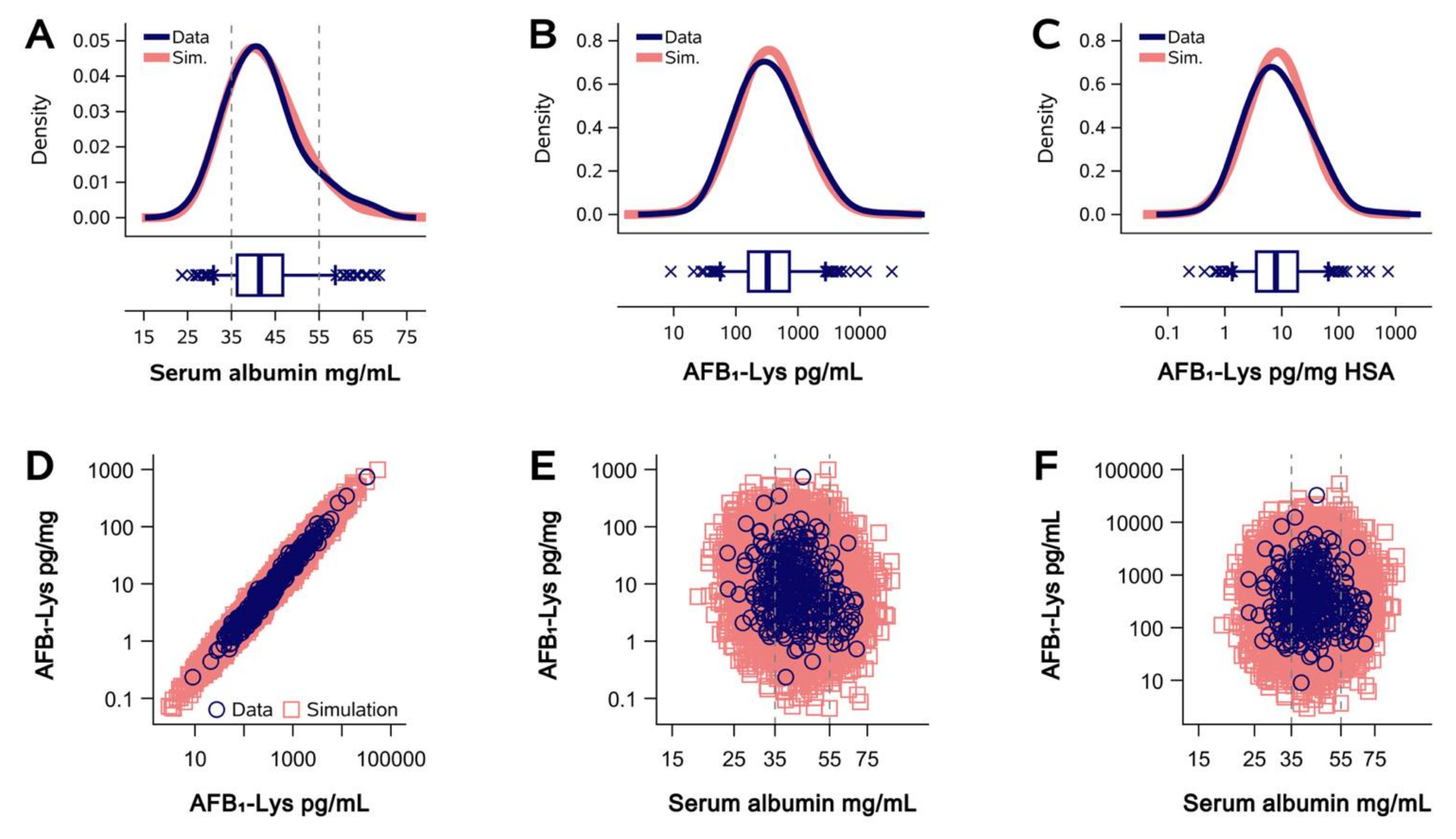
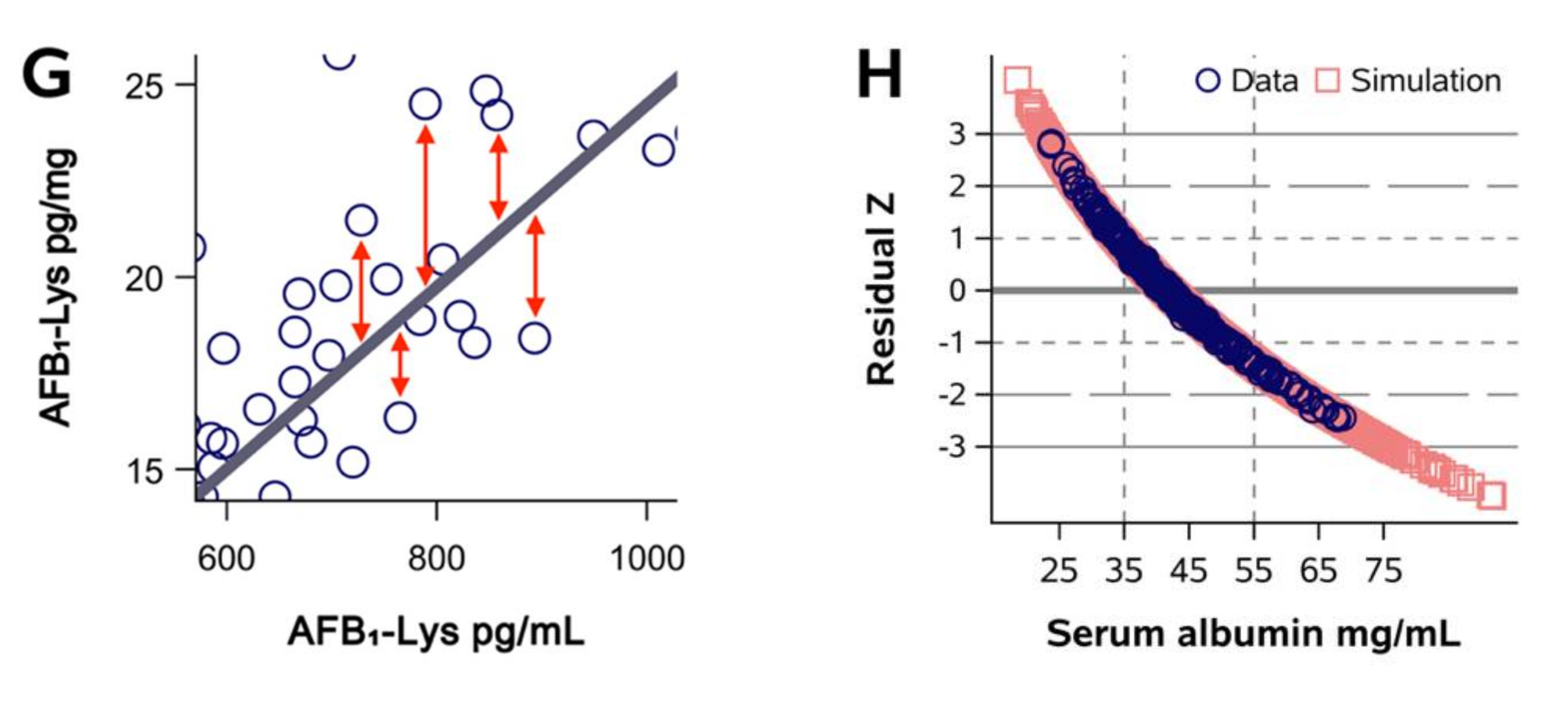
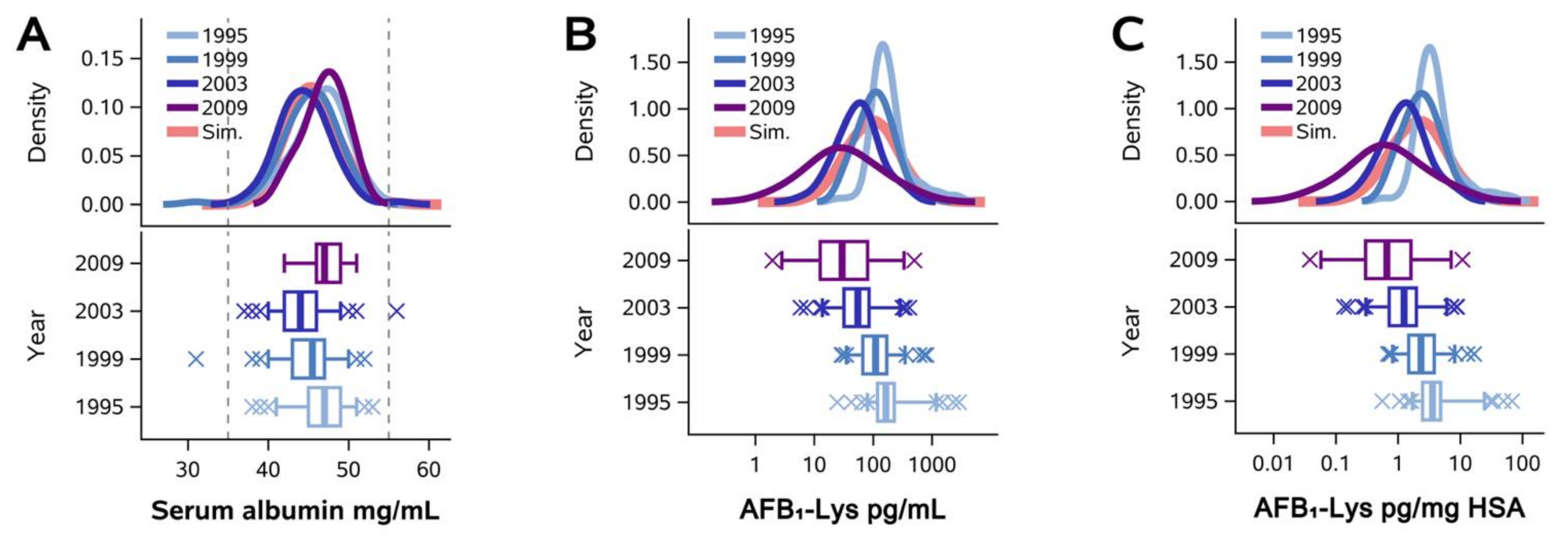
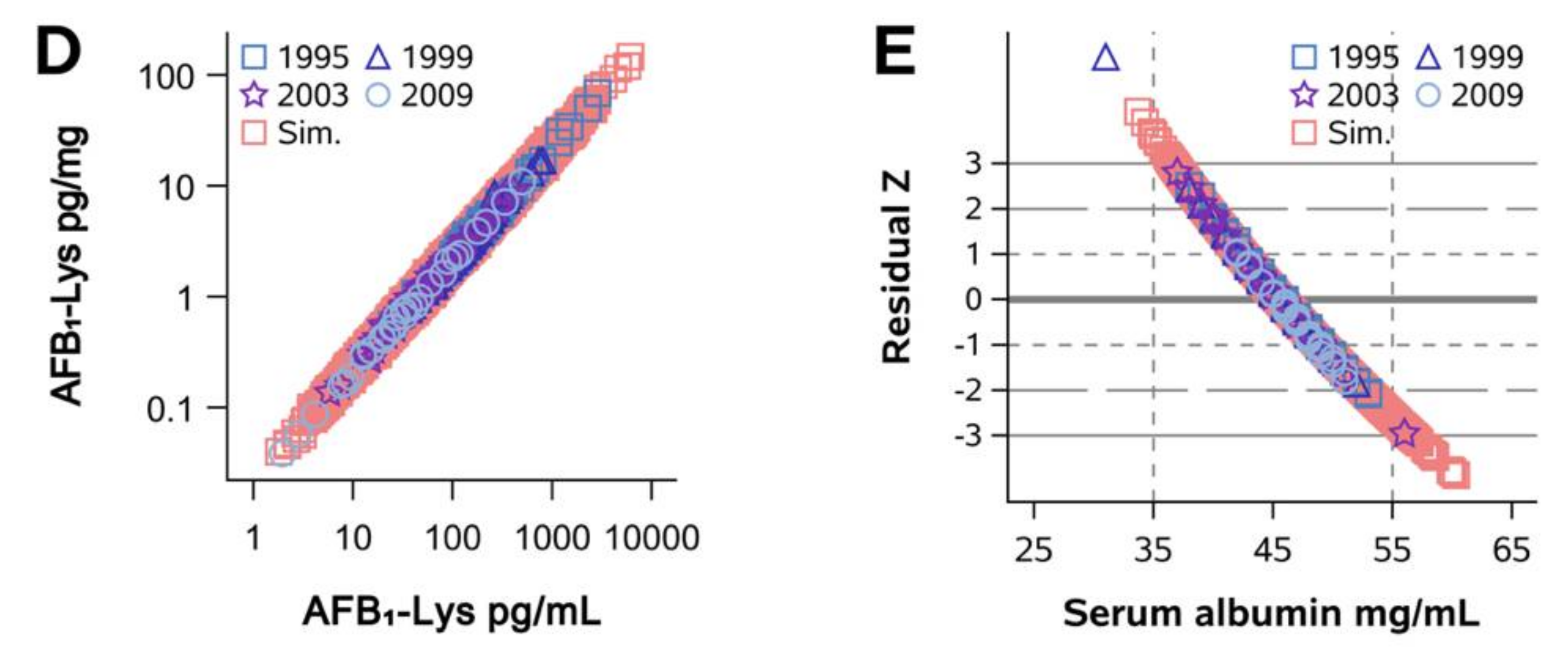
2.1. Normalization of AFB1-Lys Adduct Concentrations to Total HSA Corrects Estimates of AFB1 Internal Dose No Better than Chance in Guatemalan and Chinese Adults
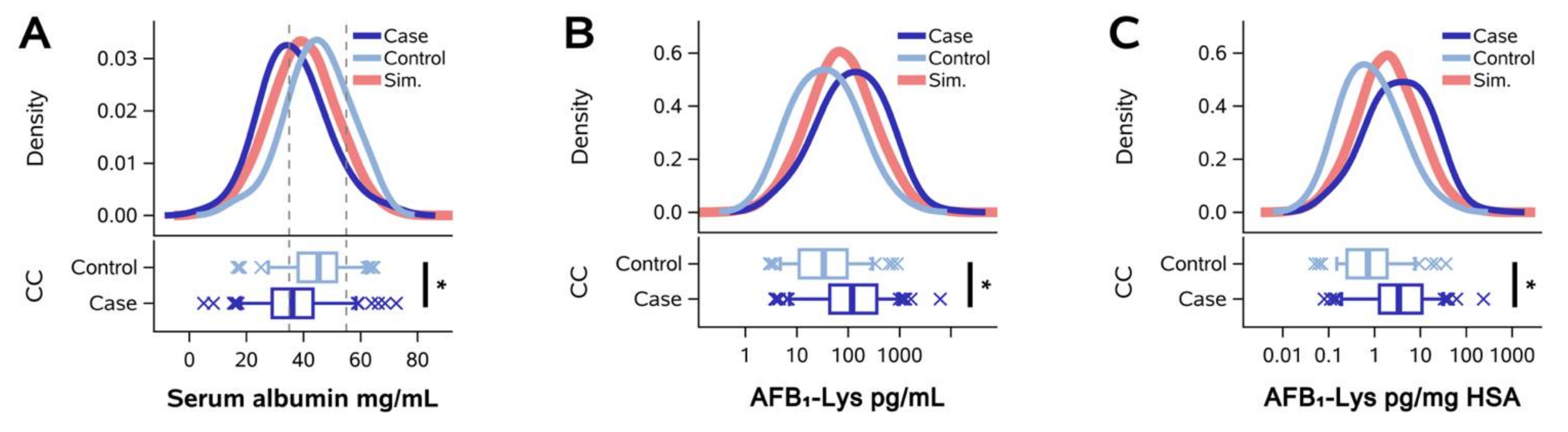
2.2. Normalization of AFB1-Lys Adduct Concentrations to Total HSA Corrects Estimates of AFB1 Internal Dose No Better than Chance in Adults with Gallbladder Cancer
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Guatemala Study
5.2. Qidong Community Longitudinal Study
5.3. Gallbladder Cancer Case-Control Studies
5.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
- 1 μg daily AFB1 dose, or 3.20 nmol/day (AFB1 mol. weight, 312.3 g/mol);
- Hepatic HSA synthesis rate of 150 mg/kg/day [31], or 2.26 μmol/kg/day (HSA mol. weight, 66,472 g/mol)
References
- Council for Agricultural Science and Technology. Mycotoxins: Risks in Plant, Animal, and Human Systems; Council for Agricultural Science and Technology: Ames, IA, USA, 2003; ISBN 0194-4088. [Google Scholar]
- Kensler, T.W.; Roebuck, B.D.; Wogan, G.N.; Groopman, J.D. Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci. 2011, 120, S28–S48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Zhao, L.; Zhang, N.-Y.; Karrow, N.A.; Krumm, C.S.; Qi, D.-S.; Sun, L.-H. Aflatoxin B1 metabolism: Regulation by phase I and II metabolizing enzymes and chemoprotective agents. Mutat. Res. Rev. Mutat. Res. 2018, 778, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W.; Groopman, J.D. Aflatoxins. In Encyclopedia of Cancer, 3rd ed.; Boffetta, P., Hainaut, P.L., Eds.; Elsevier: Cambridge, MA, USA, 2018. [Google Scholar]
- McCoy, L.F.; Scholl, P.F.; Schleicher, R.L.; Groopman, J.D.; Powers, C.D.; Pfeiffer, C.M. Analysis of aflatoxin B1-lysine adduct in serum using isotope-dilution liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Jiang, Y.-Z.; Sabbioni, G.; Chapot, B.; Montesano, R. Evaluation of methods for quantitation of aflatoxin-albumin adducts and their application to human exposure assessment. Cancer Res. 1990, 50, 245–251. [Google Scholar]
- Sabbioni, G.; Ambs, S.; Wogan, G.N.; Groopman, J.D. The aflatoxin-lysine adduct quantified by high-performance liquid chromatography from human serum albumin samples. Carcinogenesis 1990, 11, 2063–2066. [Google Scholar] [CrossRef]
- Smith, J.W.; Kroker-Lobos, M.F.; Lazo, M.; Rivera-Andrade, A.; Egner, P.A.; Wedemeyer, H.; Torres, O.; Freedman, N.D.; McGlynn, K.A.; Guallar, E.; et al. Aflatoxin and viral hepatitis exposures in Guatemala: Molecular biomarkers reveal a unique profile of risk factors in a region of high liver cancer incidence. PLoS ONE 2017, 12, e0189255. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.S.; Hernández, E.; Escobar, K.; Villagrán, C.I.; Kroker-Lobos, M.F.; Rivera-Andrade, A.; Smith, J.W.; Egner, P.A.; Lazo, M.; Freedman, N.D.; et al. Aflatoxin B 1 exposure and liver cirrhosis in Guatemala: A case–control study. BMJ Open Gastroenterol. 2020, 7, e000380. [Google Scholar] [CrossRef]
- Kroker-Lobos, M.F.; Alvarez, C.S.; Rivera-Andrade, A.; Smith, J.W.; Egner, P.A.; Torres, O.; Lazo, M.; Freedman, N.D.; Guallar, E.; Graubard, B.I.; et al. Association between aflatoxin-albumin adduct levels and tortilla consumption in Guatemalan adults. Toxicol. Rep. 2019, 6, 465–471. [Google Scholar] [CrossRef]
- Nogueira, L.; Foerster, C.; Groopman, J.D.; Egner, P.A.; Koshiol, J.; Ferreccio, C. Association of aflatoxin with gallbladder cancer in Chile. JAMA 2015, 313, 26–28. [Google Scholar] [CrossRef] [Green Version]
- Koshiol, J.; Gao, Y.T.; Dean, M.; Egner, P.A.; Nepal, C.; Jones, K.; Wang, B.; Rashid, A.; Luo, W.; Van Dyke, A.L.; et al. Association of aflatoxin and gallbladder cancer. Gastroenterology 2017, 153, 488–494. [Google Scholar] [CrossRef]
- Groopman, J.D.; Egner, P.A.; Schulze, K.J.; Wu, L.S.-F.; Merrill, R.; Mehra, S.; Shamim, A.A.; Ali, H.; Shaikh, S.; Gernand, A.; et al. Aflatoxin exposure during the first 1000 days of life in rural South Asia assessed by aflatoxin B1-lysine albumin biomarkers. Food Chem. Toxicol. 2014, 74, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-G.; Egner, P.A.; Ng, D.; Jacobson, L.P.; Muñoz, A.; Zhu, Y.-R.; Qian, G.-S.; Wu, F.; Yuan, J.-M.; Groopman, J.D.; et al. Reduced aflatoxin exposure presages decline in liver cancer mortality in an endemic region of China. Cancer Prev. Res. 2013, 6, 1038–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, C.P.; Hudson, G.J.; Sabbioni, G.; Chapot, B.; Hall, A.J.; Wogan, G.N.; Whittle, H.; Montesano, R.; Groopman, J.D. Dietary intake of aflatoxins and the level of albumin-bound aflatoxin in peripheral blood in The Gambia, West Africa. Cancer Epidemiol. Biomarkers Prev. 1992, 1, 229–234. [Google Scholar] [PubMed]
- Peng, T.; Li, L.Q.; Peng, M.H.; Liu, Z.M.; Liu, T.W.; Yan, L.N.; Shen, H.M.; Wang, L.; Wang, Q.; Wang, K.B.; et al. Is correction for protein concentration appropriate for protein adduct dosimetry? Hypothesis and clues from an aflatoxin B1-exposed population. Cancer Sci. 2007, 98, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, J.; Wang, G.; Groopman, J.D.; Kensler, T.W. Qidong: A crucible for studies on liver cancer etiology and prevention. Cancer Biol. Med. 2019, 16, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Goussous, N.; Maqsood, H.; Patel, K.; Ferdosi, H.; Muhammad, N.; Sill, A.M.; Kowdley, G.C.; Cunningham, S.C. Clues to predict incidental gallbladder cancer. Hepatobil. Pancreat. Dis. Int. 2018, 17, 149–154. [Google Scholar] [CrossRef]
- Koshenkov, V.P.; Koru-Sengul, T.; Franceschi, D.; DiPasco, P.J.; Rodgers, S.E. Predictors of incidental gallbladder cancer in patients undergoing cholecystectomy for benign gallbladder disease. J. Surg. Oncol. 2013, 107, 118–123. [Google Scholar] [CrossRef]
- Pitt, S.C.; Jin, L.X.; Hall, B.L.; Strasberg, S.M.; Pitt, H.A. Incidental gallbladder cancer at cholecystectomy. Ann. Surg. 2014, 260, 128–133. [Google Scholar] [CrossRef]
- Duly, E.B.; Grimason, S.; Grimason, P.; Barnes, G.; Trinick, T.R. Measurement of serum albumin by capillary zone electrophoresis, bromocresol green, bromocresol purple, and immuoassay methods. J. Clin. Pathol. 2003, 56, 780–782. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Salvagno, G.L.; Montagnana, M.; Brocco, G.; Guidi, G.C. Influence of hemolysis on routine clinical chemistry testing. Clin. Chem. Lab. Med. 2006, 44, 311–316. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M.; Di Somma, S.; Cervellin, G. Hemolyzed specimens: A major challenge for emergency departments and clinical laboratories. Crit. Rev. Clin. Lab. Sci. 2011, 48, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W.; Matchado, A.J.; Wu, L.S.-F.; Arnold, C.D.; Burke, S.M.; Maleta, K.M.; Ashorn, P.; Stewart, C.P.; Shaikh, S.; Ali, H.; et al. Longitudinal assessment of prenatal, perinatal, and early-life aflatoxin B1 exposure in 828 mother-child dyads from Bangladesh and Malawi. Curr. Dev. Nutr. 2022, 6, nzab153. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Ballmer, P.E.; Walshe, D.; McNurlan, M.A.; Watson, H.; Brunt, P.W.; Garlic, P.J. Albumin synthesis rates in cirrhosis: Correlation with Child-Turcotte classification. Hepatology 1993, 18, 292–297. [Google Scholar] [CrossRef]
- Ballmer, P.E.; Reichen, J.; Mcnurlan, M.A.; Sterchi, A.-B.; Anderson, S.E.; Garlick, P.J. Albumin but not fibronogen synthesis correlates with galactose elimination capacity in patients with cirrhosis of the liver. Hepatology 1996, 24, 53–59. [Google Scholar] [CrossRef]
- Branch, R.A.; James, J.A.; Read, A.E. The clearance of antipyrine and indocyanine green in normal subjects and in patients with chronic lever disease. Clin. Pharmacol. Ther. 1976, 20, 81–89. [Google Scholar] [CrossRef]
- Hepner, G.W.; Vesell, E.S. Assessment of aminopyrine metabolism in man by breath analysis after oral administration of 14C-aminopyrine. Effects of phenobarbital, disulfiram and portal cirrhosis. N. Engl. J. Med. 1974, 291, 1384–1388. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostatis: A new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int. J. Gen. Med. 2016, 6, 229–255. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, P.B.; Ranek, L.; Statland, B.E.; Tygstrup, N. Clearance of antipyrine-dependence of quantitative liver function. Eur. J. Clin. Investig. 1974, 4, 129–134. [Google Scholar] [CrossRef]
- Groopman, J.D.; Hall, A.J.; Montesano, R.; Wild, C.P.; Whittle, H.; Hudson, G.J.; Wogan, G.N. Molecular dosimetry of aflatoxin-N7-guanine in human urine obtained in The Gambia, west Africa. Cancer Epidemiol. Biomarkers Prev. 1992, 1, 221–227. [Google Scholar]
- Yiengst, M.J.; Shock, N.W. Blood and plasma volume in adult males. J. Appl. Physiol. 1962, 17, 195–198. [Google Scholar] [CrossRef]
- Crispell, K.R.; Porter, B.; Nieset, R.T. Studies of plasma volume using human serum albumin tagged with radioactive iodine-181. J. Clin. Investig. 1950, 29, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.N.; Fadem, S.Z. Reassessment of albumin as a nutritional marker in kidney disease. J. Am. Soc. Nephrol. 2010, 21, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.V.; Guenter, P.; Jensen, G.; Malone, A.; Schofield, M. Consensus statement of the Academy of Nutrition and Dietetics/American Society for Parenteral and Enteral Nutrition: Indicators recommended for the identification and documentation of pediatric malnutrition (undernutrition). J. Acad. Nutr. Diet. 2014, 114, 1988–2000. [Google Scholar] [CrossRef]
- Wogan, G.N.; Edwards, G.S.; Shank, R.C. Excretion and tissue distribution of radioactivity from aflatoxin B1-14C in rats. Cancer Res. 1967, 27, 1729–1736. [Google Scholar]
- Groopman, J.D.; Fowler, K.W.; Busby, W.F.J.; Wogan, G.N. Nuclear distribution of aflatoxin B2 and its interaction with histones in rat liver in vivo. Carcinogenesis 1980, 2, 1371–1373. [Google Scholar] [CrossRef]
- Zhang, J.; Orang’o, O.; Tonui, P.; Tong, Y.; Maina, T.; Kiptoo, S.; Muthoka, K.; Groopman, J.D.; Smith, J.W.; Madeen, E.; et al. Detection and concentration of plasma aflatoxin is associated with detection of oncogenic human papillomavirus in Kenyan women. Open Forum Infect. Dis. 2019, 6, ofz354. [Google Scholar] [CrossRef]
- Rivera-Andrade, A.; Kroker-Lobos, M.F.; Lazo, M.; Freedman, N.D.; Smith, J.W.; Torres, O.; McGlynn, K.A.; Groopman, J.D.; Guallar, E.; Ramirez-Zea, M. High prevalence of non-alcoholic fatty liver disease and metabolic risk factors in Guatemala: A population-based study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 191–200. [Google Scholar] [CrossRef]
- Harrison, R.L. Introduction to Monte Carlo Simulation. Proc. AIP Conf. Proc. 2010, 1204, 17–21. [Google Scholar]
- Brinkley, J. Introduction to Data Simulation. In Proceedings of the Southeast SAS Users Group Meeting, St. Pete Beach, FL, USA, 14–17 October 2018; p. 135. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 Normalized (pg/mg HSA) | 1 Raw (pg/mL Serum) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 1 | |||||||
| Parameter | Estimate (SE) | p | R2 | Estimate (SE) | p | R2 | Estimate (SE) | p | R2 |
| Intercept | 3.086 (0.556) | <0.0001 | 0.124 | −1.595 (0.038) | <0.0001 | 0.976 | 3.086 (0.556) | <0.0001 | 0.083 |
| 2 Raw AFB1-lys | - | - | 0.999 (0.009) | <0.0001 | - | - | |||
| 2 HSA | −0.951 (0.336) | 0.005 | - | - | 0.049 (0.336) | 0.883 | |||
| 3 Male | −0.140 (0.057) | 0.016 | −0.003 (0.010) | 0.761 | −0.140 (0.057) | 0.016 | |||
| 4 Urban | −0.194 (0.063) | 0.002 | −0.051 (0.010) | <0.0001 | −0.194 (0.063) | 0.002 | |||
| 5 Education | −0.004 (0.008) | 0.668 | −0.001 (0.001) | 0.602 | −0.004 (0.008) | 0.668 | |||
| 6 Income | −0.097 (0.033) | 0.003 | 0.002 (0.006) | 0.674 | −0.097 (0.033) | 0.003 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.W.; Ng, D.K.; Alvarez, C.S.; Egner, P.A.; Burke, S.M.; Chen, J.-G.; Kensler, T.W.; Koshiol, J.; Rivera-Andrade, A.; Kroker-Lobos, M.F.; et al. Assessing the Validity of Normalizing Aflatoxin B1-Lysine Albumin Adduct Biomarker Measurements to Total Serum Albumin Concentration across Multiple Human Population Studies. Toxins 2022, 14, 162. https://doi.org/10.3390/toxins14030162
Smith JW, Ng DK, Alvarez CS, Egner PA, Burke SM, Chen J-G, Kensler TW, Koshiol J, Rivera-Andrade A, Kroker-Lobos MF, et al. Assessing the Validity of Normalizing Aflatoxin B1-Lysine Albumin Adduct Biomarker Measurements to Total Serum Albumin Concentration across Multiple Human Population Studies. Toxins. 2022; 14(3):162. https://doi.org/10.3390/toxins14030162
Chicago/Turabian StyleSmith, Joshua W., Derek K. Ng, Christian S. Alvarez, Patricia A. Egner, Sean M. Burke, Jian-Guo Chen, Thomas W. Kensler, Jill Koshiol, Alvaro Rivera-Andrade, María F. Kroker-Lobos, and et al. 2022. "Assessing the Validity of Normalizing Aflatoxin B1-Lysine Albumin Adduct Biomarker Measurements to Total Serum Albumin Concentration across Multiple Human Population Studies" Toxins 14, no. 3: 162. https://doi.org/10.3390/toxins14030162
APA StyleSmith, J. W., Ng, D. K., Alvarez, C. S., Egner, P. A., Burke, S. M., Chen, J.-G., Kensler, T. W., Koshiol, J., Rivera-Andrade, A., Kroker-Lobos, M. F., Ramírez-Zea, M., McGlynn, K. A., & Groopman, J. D. (2022). Assessing the Validity of Normalizing Aflatoxin B1-Lysine Albumin Adduct Biomarker Measurements to Total Serum Albumin Concentration across Multiple Human Population Studies. Toxins, 14(3), 162. https://doi.org/10.3390/toxins14030162