1. Introduction
Cyanobacterial blooms occur globally, affecting humans and animals through contamination of water and foodstuffs with harmful toxins [
1]. Toxin classes most frequently encountered are the cyclic microcystins (MCs) [
2,
3] the cyclic pentapeptides nodularins (NODs), cylindrospermopsin and the neurotoxins including anatoxins and saxitoxins [
2,
4,
5,
6]. MCs and cylindrospermopsin are usually primarily described as hepatotoxins, but there is also evidence linking these to neurotoxicity, cardiotoxicity, immunotoxicity and developmental toxicity in a wide range of species [
7,
8]. MCs are produced primarily by freshwater cyanobacteria genera such as
Microcystis,
Planktothrix,
Anabaena/Dolichospermum and
Oscillatoria [
9] with at least 310 analogues reported in blooms, cultures and/or from biotransformation reactions in the cell, environment or during sample processing [
10,
11,
12], with increasing reports of MCs found in the marine environment [
13,
14,
15]. Nodularin-R (NOD-R), along with a low number of structural analogues, is produced by
Nodularia spumigena which thrives in brackish water [
16], thereby resulting in impacts in both freshwater and saline environments [
17,
18,
19]. Human health impacts from cyanotoxins have occurred through drinking from contaminated water supplies, recreational exposure, aerosol exposure under certain meteorological conditions and through eating contaminated aquatic foodstuffs, health food supplements, or even terrestrial foods irrigated with contaminated water [
17,
18,
19,
20,
21,
22,
23,
24,
25,
26]. In recent years, the risk from cyanotoxin poisoning following human consumption of seafood contaminated with cyanotoxins has been highlighted, with shellfish being one of the primary routes of toxins through to higher trophic levels [
6,
13,
14,
27,
28,
29] and in some instances with toxin concentrations reaching dangerously high levels (reviewed by [
6]).
Figure 1 illustrates the general structure of MCs and NODs, with both families containing the Adda moiety (3-amino-9-methoxy-2,6,8-trimethyl-10-phenyldeca-4(
E),6(
E)-dienoic acid [
30]. The toxins act through inhibition of serine/threonine protein phosphatases resulting in cell damage and death, with acute symptoms including gastroenteritis, neurotoxicity, liver damage and potentially fatality [
31,
32]. Structural variations in the MCs include most notably variable L-amino acid residues at positions 2 and 4, as indicated by a two-letter suffix (e.g., MC-LR equating to L (leucine) and R (arginine)), although all other residues have been found to exhibit some variability as highlighted by a prefix to the name (e.g., [D-Asp
3]MC-LR which contains D-aspartic acid at position 3) [
11]. Although health impacts from cyanotoxins are found globally, few regulations exist in global legislation. The World Health Organization (WHO) has provided guideline values of 1 µg/L and 12 µg/L thresholds for lifetime and short-term exposure, respectively, to MC-LR in drinking waters [
32,
33].
Numerous approaches are published for the detection and quantitation of MCs and NODs [
34], but with the majority to date focused primarily on water or cyanobacterial sample matrices [
35,
36]. The most common approaches include sensitive biochemical assays such as enzyme-linked immunosorbent assays (ELISA) [
37] and protein phosphate inhibition assays (PPIA) [
38], as well as instrumental chemical detection methods, primarily high-performance liquid chromatography with photodiode array detection (HPLC-PDA) [
39], or LC coupled to mass spectrometry (LC-MS) [
40,
41,
42]. Broadly speaking the biochemical assays are able to detect a wide range of structural analogues providing a quantitative assessment of total MC/NOD presence. Whilst PPIA methods show similar response for both Adda and non-Adda substituted MCs, the commercial Eurofins Abraxis ELISA is incapable of detecting the non Adda-substituted analogues, with cross-reactivities of <0.25% in comparison with MC-LR [
37,
43]. The low cross-reactivities determined for [ADMAdda
5] and [DMAdda
5]-MCs consequently evidences potential under-estimation of toxicity when conducting ELISA quantitation [
37,
43]. A multihapten ELISA based on polyclonal antibodies was, however, raised against a mixture of five MC analogues, resulting in wider cross-reactivities [
44]. Instrumental detection methods typically target known compounds for which reference standards are available, although high-resolution mass spectrometry has also been applied for non-targeted analysis of other MC analogues unavailable as commercial standards [
25,
45,
46], sometimes in combination with a thiol-derivatization approaches prior to analysis [
47,
48].
Due to the presence of a reactive α,β-unsaturated amide functional group in the Mdha
7 amino acid common to most MCs, they form conjugates with thiol-containing biomolecules such as glutathione (GSH) and cysteine. Whilst LC-MS/MS can be used for direct measurement of these soluble small molecule conjugates [
49,
50,
51,
52], or potentially the parent toxins following deconjugation [
18], most published instrumental methods focus solely on the free (soluble) toxins. However, there is the potential for conjugation with larger biomolecules in seafood tissue that are not readily extractable during sample preparation [
52]. For example, non-extractable MCs are known to bind covalently to protein phosphatases and other thiol-containing proteins in tissues [
53,
54,
55,
56,
57,
58,
59], so may potentially transfer to higher trophic levels. As such, assuming that covalently bound MCs are bioavailable and inhibit protein phosphatases after digestion [
20,
60,
61], detection methods are required which can quantify both soluble and bound (total) toxins, noting the huge variability in proportions of soluble to bound toxins reported to date [
58]. Consequently, whilst ELISA and LC-MS/MS results have been shown to compare well for algal and powdered algal product matrices [
62,
63], for biological tissue samples the ELISA may return higher quantitative results than targeted LC-MS/MS methods. This can be attributed to the MC antibodies used in the assays cross-reacting with the conjugates as well as the potential for MS-related ion suppression, if either matrix-matched calibrants, standard additional quantitation or isotope-dilution recovery correction are not applied [
64,
65].
A common approach to the analysis of total MCs and NODs combined (MCs + NODs) involves the use of a specific chemical reaction called the Lemieux oxidation. This reaction involves the catalyzed oxidative cleavage of an olefin to form two aldehydes or ketones. For MCs + NODs this reaction yields 3-methoxy-2-methyl-4-phenylbutyric acid (MMPB) from the Adda moiety within MCs and NOD-R, regardless of their initial amino acid configuration, which are then detected using mass spectrometric methods such as LC-MS/MS. This approach consequently provides a measure of total Adda-containing MCs + NODs [
66,
67], irrespective of form (soluble, bound or partially degraded [
65]). This MMPB formation method has been used for this purpose in a variety of sample matrices including canine tissues [
65] and shellfish [
58,
68] as well as enzymatic hydrolysis to form soluble microcystin-conjugated peptides [
69], although these irreversible reactions result in the loss of information regarding original MC profiles. Furthermore, the method has been further refined to incorporate oxidative cleavage for MC analogues containing modified Adda moieties such as ADMAdda and DMAdda, resulting in the formation of oxidation products analogous to MMPB [
37].
Use of a single detection method presents risk. Non-specific biochemical methods are associated with matrix effects, are unable to determine toxin profiles, have cross-reactivity towards inactive metabolites and are linked to false positive detection [
70,
71,
72]. Risks of false negatives and the absence of accurate toxicity equivalence factors for many cyanotoxin analogues [
73] can affect the accuracy of targeted instrumental methods for assessing toxicity levels. Furthermore, impracticalities including sample throughput and turnaround time limitations are still associated with the potential for routine high-throughput monitoring using non-targeted mass spectrometry methods and potentially also MMPB analysis. Consequently, there are clear benefits to utilising multiple complementary detection methods to ensure toxicity is not under- or over-estimated, ideally incorporating a measure of both soluble and total MCs.
Noting the priority for development of complementary cyanotoxin testing methods in complex matrices [
6], before any testing methods can be implemented into monitoring programs, extensive validation of method performance is required. In addition, routine running of instrumental methods requires the application of a range of quality assurance and quality control procedures. These both require the use of matrix Reference Materials (RMs), so the availability of materials prepared from matrices of direct relevance to the test samples is important to laboratories wishing to implement and maintain cyanotoxin assays. Various RMs have been prepared for cyanotoxins in the past but focused on algal matrix. These include a certified reference material (CRM) for total MCs from the National Institute for Environmental Studies (NIES) in Japan which contained seven microcystin variants [
74], as well as a multi-analyte feasibility study RM from NRC Canada containing seven MC analogues, anatoxins and cylindrospermopsins [
75]. Multiple shellfish RMs have been developed, characterised and even certified in recent years for marine toxins [
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88,
89], but to date no shellfish tissue RM exists for cyanotoxins.
The need for such a RM prompted the culturing of toxic
Microcystis and
Nodularia with subsequent feeding studies in mussels in a controlled laboratory environment to assess the uptake and depuration of toxins within mussel tissues [
26], with the resulting tissue materials utilized for RM preparation and characterization. The aim was to produce a shellfish tissue cyanotoxin RM which was fully characterised and provided proof of concept for future development of a fully certified shellfish RM containing a wide range of toxins from cyanobacterial origin.
4. Materials and Methods
4.1. Chemicals and Standards
For shellfish feeding, Shellfish diet 1800 (approximately 7.4 × 1011 cells/mL) was purchased from ReedMariculture Inc. (Campbell, CA, USA), and dilutions were made in water/seawater (10:0.86, v/v). For LC-MS/MS analysis and toxin quantitation, analytical grade chemicals and HPLC-grade solvents were used throughout the study. Mobile phases were prepared from LC-MS-grade acetonitrile (Fisher Optima, ThermoFisher, Hemel, Hempstead, UK) and water used for LC-MS was obtained in-house. Toxin standards used for preparation of calibration solutions (MC-RR, MC-LA, MC-LY, MC-LF, MC-LW, MC-YR, MC-WR, MC-HilR, MC-HtyR, MC-LR, [D-Asp3]MC-LR and NOD-R) were obtained from Enzo Life Sciences, Exeter, UK (≥95% purity). A certified standard of [Dha7]MC-LR (~99.6% purity) was obtained from Biotoxin Metrology, National Research Council Canada (NRCC; Halifax, NS, Canada), but this was not incorporated into the final mixed calibrant solutions given the lack of chromatographic separation from [D-Asp3]MC-LR. A NRCC CRM for MC-LR (~97.5% purity) was also used for calibration of ELISA and MMPB methods. Reference standards received as solid films were dissolved in water:methanol (1:1 v/v), to form stock solutions. A mixed stock solution was subsequently prepared by combining aliquots of each stock, followed by further dilutions in solvent to create seven-level suite of working calibration standards between 0.33 ng/mL to 327 ng/mL per toxin.
4.2. Culturing of Cyanobacteria and Shellfish Feeding
Cyanobacterial culturing and shellfish feeding was conducted as detailed in [
24]. In brief, two cyanobacterial cultures were grown in modified BG-11 medium with 75 g/L sodium nitrate:
Nodularia spumigena KAC 66 (Kalmar Algae Collection, Kalmar, Sweden) and
Microcystis aeruginosa PCC 7813 (Pasteur Culture Collection of Cyanobacteria, Paris, France).
N. spumigena media was supplemented with 20% (
w/
v) Instant Ocean artificial seawater (Aquarium Systems Inc., Sarrebourg, France). Cultures were maintained at 20–23 °C, under continuous illumination (10–15 µmol/m
2/s) and sparged with sterile air at 2.3 L/min until four days before harvesting. Cells were collected and maintained in 25 L carboys at 17 ± 1 °C with mild aeration and a light cycle of 17 h illumination (24 µmol/m
2/s) and 7 h darkness for four weeks prior to shellfish feeding. Live mussels
Mytilus edulis (sex undetermined) were obtained from the Shetland Islands (N Scotland, UK), acclimatized to laboratory tank conditions for a week and cleaned of barnacles and other debris prior to the experiment. Two seawater tanks (each 300 L, 122 cm long, 102 cm wide, 77 cm high and containing approximately 150 L seawater) equipped with ultraviolet sterilizers (class 1 IP64, twin UV 24 W, 240 V, 50 Hz, Tropical Marine Centre, Rickmansworth, Greater London, UK) were maintained at 16 ± 1 °C. One of the tanks was used for the exposure of 420
M. edulis to
M. aeruginosa (3.9 × 10
6 cells/L final concentration) and
N. spumigena (3.1 × 10
6 cells/L final concentration) for a total period of three days of daily feeding. Mussels were separated into three separate sub-compartments (A, D and E). Live cyanobacterial cells were homogenised and fed to live mussels. The second control tank contained 420 mussels separated into three sub-compartments B, C and F and fed shellfish diet only (
Figure 6). After checking toxin concentrations within the tissues of a sub-sample, the exposure was terminated and the mussels removed for processing.
4.3. Reference Material Preparation
Just over 2.0 kg of toxin-containing mussel tissue from the exposed tank sub-compartments A, D and E was available for processing after the exposure study had completed and a previous study on MC/NOD uptake in mussels was completed [
26]. Mussels were shucked to remove shells and whole tissues incorporating all the visceral mass and edible muscle combined were homogenised thoroughly in batches using high speed Waring industrial blenders. Each batch of homogenised tissue was combined into a large polypropylene container, with approximately 450 mL deionized water added to further aid homogenization, resulting in an overall water content of ~80%. Re-homogenization was conducted using a high-speed hand blender and an aliquot of tissue was taken for analysis to confirm the presence of a suitable number of cyanotoxin analytes at appropriate concentration levels. After testing, a final homogenization step was conducted and a magnetic stirrer added to the vessel, enabling continuous stirring during reference material (RM) aliquoting. Prior to dispensing the samples, 7.0 mL polypropylene Bijou vials were labelled with unique LRM identification numbers. Aliquots (4.5 g) were weighed into the tubes, to provide enough material for two 2.0 g sub-samples to be taken from each vial. As soon as each vial was dispensed, a second operator capped the tube. A total of 520 vials of RM were dispensed and capped, before placing all upright in a −20 °C freezer in order to freeze the contents in the bottom of the vials. After 24 h, the vials were transferred to long-term storage at −80 °C.
4.4. Targeted Toxin Analysis
Vials of mussels were opened carefully and 2.00 ± 0.01 g sub-samples were weighed accurately into 50 mL polypropylene centrifuge tubes. Extraction and analysis by LC-MS/MS was conducted using the validated and ISO17025:2005 accredited method of [
90]. Briefly, each homogenised tissue was extracted with a single dispersive extraction using 8.0 mL of methanol:water (80:20,
v/
v) and a 2 min vortex mixing time prior to centrifugation (4500 g; 10 min) and filtration of the resulting supernatant (0.2 µm syringe filter). Targeted LC-MS/MS analysis of cyanotoxins was conducted as detailed in [
90]. A Waters (Manchester, UK) Acquity UHPLC system coupled to a Waters (Manchester, UK) Xevo TQ triple quadrupole mass spectrometer (MS/MS) was used with a 1.7 µm, 2.1 × 50 mm Waters Acquity UPLC BEH C18 column in conjunction with a Waters BEH C18 guard cartridge. The column was held at +60 °C, and a 5 µL injection volume utilized, together with mobile phase flow rate of 0.6 mL/min. Mobile phase A1 consisted of water containing 0.025% (
v/
v) of formic acid, mobile phase B1 comprised acetonitrile (MeCN) with 0.025% (
v/
v) formic acid. The UHPLC gradient started at 98% A1, dropping to 75% A1 at 0.5 min holding until 1.5 min, dropping further to 60% A1 at 3.0 min, decreasing further to 50% A1 at 4 min, before a sharp drop to 5% A1 at 4.1 min, holding until 4.5 min before increasing back to 98% A1 for column equilibration at 5 min for a further 0.5 min. The MS/MS source parameters were exactly as specified in [
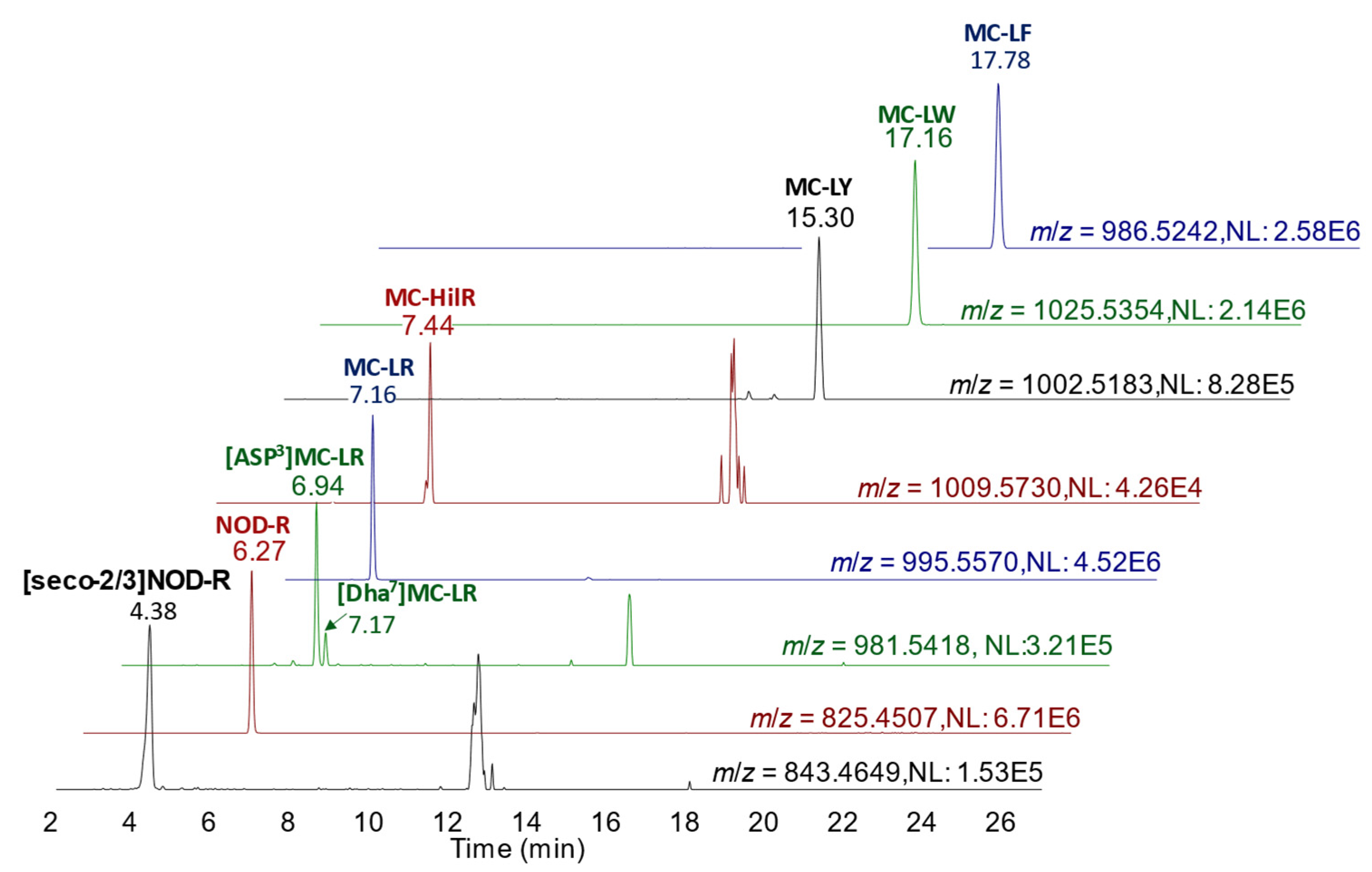
90], with 150 °C source temperature, 600 °C desolvation temperature, 600 L/h desolvation gas flow, 0.15 mL/min collision gas flow and capillary voltage at 1.0 kV. Quantitation of MCs was performed against external calibration standards with results calculated in terms of µg/kg wet weight of shellfish tissue. Throughout each LC-MS/MS sequence used for characterization, homogeneity and stability testing, quality control (QC) measures were applied following ISO17025-accredited protocols. This included the assessment of instrument blanks, method sensitivity, calibration linearity, positive control response, method procedural blanks and method precision. All QC checks passed stipulated thresholds. For minor toxin analogue analysis, the same chromatographic conditions as above were used. However, LC effluent was directed into the ESI source of a Waters Xevo TQ-S triple quadrupole mass spectrometer operating in SRM scan mode using positive ionization. SRM transitions are shown in the chromatograms of
Figure 2 and were obtained using a collision energy of 70 eV as per [
90].
Appendix A tabulates all SRM transitions. Full method performance characteristics for the method are detailed in [
90]. Limits of Reporting (LOR) range from 0.3 to 1.5 µg/kg wet weight per toxin in mussel tissue, with the majority of calibration regressions >0.99 over a linear range reaching the equivalent of 2.5 mg/kg wet weight. No significant matrix effects were evident, so all calibrations were subsequently prepared in solvent rather than mussel extract matrix.
4.5. Toxin Profile and Homogeneity Testing
Fourteen RM samples were chosen for homogeneity testing. These were selected immediately after aliquoting and initial freezing, to provide a representative cross section of samples aliquoted over the entire production batch. In addition to the first (vial 1) and last (vial 520) aliquots dispensed, remaining test samples were randomly chosen across the production batch. Prior to homogeneity testing, samples were allowed to slowly thaw to room temperature, before the contents of each sample were mixed and two separate 2.00 ± 0.01 aliquots weighed out from each vial into 50 mL polypropylene centrifuge tubes, giving a total of 28 samples for extraction. All samples were extracted in MeOH:water (80:20 v/v) and filtered prior to LC-MS/MS analysis.
4.6. Stability Testing
Material stability was assessed over 181 days using a reverse-isochronous experimental design. Triplicate RM vials were subjected to storage under both frozen (−20 °C) and refrigerated (+4 °C) conditions, incorporating eight time points (specifically 0, 28, 53, 86, 108, 126, 150 and 181 days). The triplicate samples were stored at −80 °C for variable periods of time before being placing in storage at the two elevated temperature conditions until the end of the study. At the end time point, all samples were removed from storage, allowed to equilibrate to room temperature before being extracted, filtered and analysed by LC-MS/MS. During analysis, triplicates were spread across the entire instrumental sequence, to account for any instrumental drift if present. Quantitative results determined at t = 0 were used to calculate mean concentrations, with three-times the standard deviation of the mean used to assigned acceptability limits for toxin concentrations at later time points.
4.7. Untargeted Toxin Analysis
Sample extraction was carried out as in
Section 4.4. LC-HRMS analyses were performed on an Agilent 1200 LC system (Agilent, Santa Clara, CA, USA) coupled to a Q Exactive HF Orbitrap mass spectrometer with a HESI-II heated electrospray ionization interface (ThermoFisher Scientific, Waltham, MA, USA). Liquid chromatography parameters included a 5 µL injection on a SymmetryShield 3.5 µm C18 column (100 × 2.1 mm; Waters, Milford, MA, USA) held at 40 °C with mobile phases A and B of H
2O and CH
3CN, respectively, both of which contained 0.1%
v/
v formic acid. The elution gradient (0.3 mL/min) included a linear increase from 20–100% B over 21 min, a hold at 100% B (6 min), a decrease to 20% B over 0.1 min and equilibration at 20% B for 2.9 min.
LC-HRMS source conditions included a capillary temperature of 350 °C, sheath and auxiliary gas flow rates of 25 and 8 units, respectively a spray voltage of +3.7 kV and an S-Lens RF level of 100. Comprehensive HRMS/MS data were first acquired using a combined full scan (FS) and data independent acquisitions (DIA) scan mode. All HRMS/MS data were processed manually in Thermo Xcalibu 4.1 software.
Full scan data were collected from m/z 500–1200 using the 30,000 resolution setting, an AGC target of 1 × 106 and a max IT of 120 ms and MS/MS data was collected using the 15,000 resolution setting, an AGC target of 2 × 105 and a stepped collision energy of 30, 60 and 80 V. For DIA, the max C-trap ion trapping time (max IT) was set to ‘auto’ and 62 m/z wide precursor isolation windows centered at m/z 530, 590, 650, 710, 770, 830, 890, 950, 1010, 1070, 1130, 1190, 1250, 1310, and 1370 were used.
Targeted MS/MS spectra of suspected MCs were then acquired using the Parallel Reaction Monitoring scan mode with a using a resolution setting of 15,000, AGC target of 5 × 105 and, a Maximum IT fill time of 3 sec and an isolation window of 0.5 m/z. Stepped CE for MCs containing one arginine residue were 60, 65 and 70 V and those for MCs containing no arginine residues were 25, 35 and 40 V.
4.8. ELISA
4.8.1. Multihapten ELISA
Mussel tissues were extracted by adding either 8.0 mL or 10.0 mL MeOH:water (80:20
v/
v) to 2.0 g tissue homogenate, vortex mixing and then centrifuging for 10 min (2250 g; 4 °C). The concentration of MCs in each shellfish extract supernatant was determined by indirect competitive ELISA (NVI, Oslo, Norway) as described by Samdal et al. [
42]. The assay was optimized, as reported previously, with only minor adjustments to 0.5 µg/mL of the plate-coating antigen, 1:7000 of the antiserum 80289-5b, and 1:12,000 of the donkey antisheep IgG (H + L)−horseradish peroxidase conjugate (antisheep−HRP from Agrisera antibodies (Vännäs, Sweden)). These concentrations were determined by checkerboard titrations followed by optimization of the standard curve. The MC-LR standard (NRC CRM-MC-LR) in methanol (500 ng/mL) was diluted in Phosphate Buffered Saline with Tween (PBST) to give a methanol concentration of 10%, and then in a threefold dilution series in sample buffer resulting in standard concentrations of 50, 16.7, 5.56, 0.62, 0.20, 0.069, 0.023, 0.0076, and 0.0025 ng/mL. Serial dilutions of standards and samples were analyzed in duplicate on the plate. All incubations were performed at ~20 °C. Absorbances were measured at 450 nm using a SpectraMax i3x plate reader (Molecular Devices, Sunnyvale, CA, USA). Assay standard curves were calculated using 4-parameter logistic treatment of the data using SoftMax Pro version 6.5.1. (Molecular Devices, Sunnyvale, CA, USA). The assay working range was defined as the linear region at 20–80% of maximum absorbance (A
max). The CYNTX RMs were quantitated at 400 and 800 times dilution to remove potential matrix effects, as evidenced by negative control shellfish samples.
4.8.2. Adda ELISA
Tissues were transferred to glass vials as 100 mg subsets (in duplicate) and extracted using 4 mL MeOH:water (75:25
v/
v) in 100 mM acetic acid via bath sonication (20 min). Samples were cooled (4 °C) followed by centrifugation (1500 g; 15 min). Supernatants were retained and pellets vortex rinsed with extractant followed by centrifugation. The supernatants were pooled, mixed, and used for both the Adda ELISA and subsequent MMPB oxidation. One of each duplicate ‘Soluble MCs + NODs’ extract (50 µL corresponding to 1 mg tissue) were evaporated to dryness (60 °C, N
2) and reconstituted in 1 mL phosphate buffer (10 mM; pH 7) for analysis. Further dilutions were conducted as necessary to achieve data within range of the calibration curve (0.15–5.0 ng/mL of NRC-CRM-MC-LR). A MCs + NODs Adda ELISA (Eurofins-Abraxis, Warminster, Pennsylvania, USA) [
109] was utilized with the protocol refined and QC requirements as previously described [
101]. This included the preparation of a standard curve from dilutions of a certified MC-LR standard over a range between 0.15 ng/mL and 4.00 ng/mL, also incorporating fortified samples at 1.0 ng/mL. The assay is sensitive down to a quantification limit of 150 µg/kg wet weight for MCs + NODs as determined from dilution factors (1000-fold) and kit sensitivity (0.15 ng/mL).
4.9. MMPB for Total MCs
4.9.1. Sample Preparation
Samples for the MMPB method were further homogenized by transferring 0.5 g of material to 7 mL vials containing ceramic beads (2.8 mm). Each subsample received 10 mM phosphate buffer (pH = 7) to achieve sample concentrations of 100 mg/mL. An Omni Bead Ruptor was utilized to homogenize samples (6 m/s; 15 s; 2×; dwell 30 s). Aliquots (0.1 g corresponding to 10 mg) were dispensed via Pasteur pipette into glass vials for oxidations in duplicate. In addition to whole tissues, tissue extracts (
Section 4.8.2) were oxidized as 500 µL aliquots (corresponding to 10 mg tissue in acidified MeOH) for freely extractable soluble MCs + NODs. All subsets were spiked with internal standard (
d3-MMPB). Tissue was oxidized with 2.5 mL of oxidant while extracts were oxidized with 1 mL oxidant, which was composed of 0.2 M K
2CO
3, 0.1 M KMnO
4 and 0.1 M NaIO
4. Oxidation was stopped by the addition of sodium bisulfite (40% wt%). Aliquots were cleaned using solid phase extraction (SPE). Preconditioned Strata X Polymeric SPE cartridges (200 mg for tissues, 100 mg for extracts) were loaded with sample, rinsed 3x with deionized water, and eluted with acetonitrile:water (90:10
v/v). Elutions were evaporated to dryness (60 °C, N
2), reconstituted in MeOH:water (5:95
v/v) (sample concentration of 10 mg/mL), filtered (0.2 µm), and analyzed. QC measures incorporated into the testing batch included application of spiked recovery checks and internal standard returns, together with method procedural blanks. All controls fell within method-specified control limits.
4.9.2. LC-MS/MS Analysis for MMPB (Adda MCs + NODs)
The [M-H]− ion of MMPB (m/z 207) was fragmented and the product ion m/z 131 was monitored. The internal standard d3-MMPB (m/z 210 > 131) was used in conjunction with MMPB response for calibration. A matrix matched calibration curve (using a non-exposed toxin-free mussel control sample) was generated for quantification of total Adda MCs + NODs (0–25,000 ng/g of oxidized MC-LR) and a calibration curve generated in water was used for quantification of soluble MCs + NODs (0–500 ng/mL of oxidized MC-LR). A certified reference standard of MC-LR (NRC CRM -MC-LR) was used for all calibrations.
4.10. Data Analysis
Statistics used for the assessment of method equivalence were performed using RStudio (version 1.3. 1056). For the repeated means ANOVA (‘rstatix’package) and Tukey’s pairwise post hoc (‘eemeans’ package) analysis data was log transformed. The NIST Decision Tree was used to provide a recommendation on how to combine the independent measurement results obtained from the ELISAs, MMPB and LC-MS/MS analyses [
91,
92], with a Hierarchical Laplace-Gauss Fit Model applied for determining the consensus estimate and associated standard uncertainty.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












