Cells Responding to Closely Related Cholesterol-Dependent Cytolysins Release Extracellular Vesicles with a Common Proteomic Content Including Membrane Repair Proteins

 , , , , and
, , , , and 
Abstract
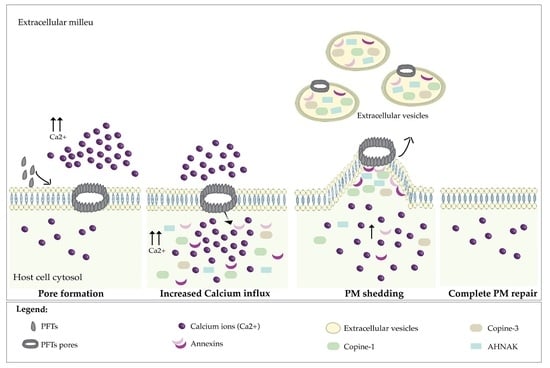
1. Introduction
2. Results and Discussion
2.1. The Concentration of CDCs Determines the Extent of PM Damage and Efficacy of Repair
2.2. Repair of CDC-Induced PM Damage Relies on an Abrupt Shot of Ca2+ Early in Intoxication
2.3. Cells Intoxicated with CDCs Release Large Extracellular Vesicles during PM Damage Repair
2.4. The Release of Large Extracellular Vesicles Contributes to the Removal of CDCs from the PM
2.5. Proteins Present in EVs Released by Intoxicated Cells Are Potential Actors in PM Repair
2.6. Copine-1 and Copine-3 Are Required for Efficient PM Repair
3. Conclusions
4. Materials and Methods
4.1. Cell Lines and Reagents Culture
4.2. Cell Intoxications
4.3. Flow Cytometry Analysis
4.4. Immunofluorescence Microscopy Quantifications
4.5. Isolation of Extracellular Vesicles (EVs)
4.6. Nanoparticle Tracking Assay
4.7. Electron Microscopy and Immunogold Labeling
4.8. Proteomics Sample Preparation and LC-MS/MS Analysis
4.9. Proteomics Data Analysis
4.10. Establishment of CPNE1 and CPNE3 Knockout HeLa Cells by CRISPR/Cas9
4.11. Generation of GFP-Tagged CPNE1 and CPNE3
4.12. Transfection and Immunofluorescence Microscopy
4.13. Immunoblotting Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Draeger, A.; Schoenauer, R.; Atanassoff, A.P.; Wolfmeier, H.; Babiychuk, E.B. Dealing with damage: Plasma membrane repair mechanisms. Biochimie 2014, 107, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, F.; Ros, M.; Bonedeau, E.; Croissant, C.; Frelin, L.; Saltel, F.; Moreau, V.; Bouter, A. Defective membrane repair machinery impairs survival of invasive cancer cells. Sci. Rep. 2020, 10, 21821. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Cabanes, D.; Mesquita, F.S.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 2019, 76, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lieberman, J. Knocking ’em Dead: Pore-Forming Proteins in Immune Defense. Annu. Rev. Immunol. 2020, 38, 455–485. [Google Scholar] [CrossRef] [PubMed]
- Ulhuq, F.R.; Mariano, G. Bacterial pore-forming toxins. Microbiology 2022, 168, 001154. [Google Scholar] [CrossRef]
- Peraro, M.D.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef]
- Cassidy, S.K.; O’Riordan, M.X. More than a pore: The cellular response to cholesterol-dependent cytolysins. Toxins 2013, 5, 618–636. [Google Scholar] [CrossRef]
- Farrand, A.J.; LaChapelle, S.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA 2010, 107, 4341–4346. [Google Scholar] [CrossRef]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 10, 1337–1348. [Google Scholar] [CrossRef]
- Pereira, J.M.; Xu, S.; Leong, J.M.; Sousa, S. The Yin and Yang of Pneumolysin During Pneumococcal Infection. Front. Immunol. 2022, 13, 1717. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Brito, C.; Moya, M.J.M.; Pinheiro, J.C.; Mostowy, S.; Cabanes, D.; Sousa, S. Endoplasmic reticulum chaperone Gp96 controls actomyosin dynamics and protects against pore-forming toxins. EMBO Rep. 2016, 18, 303–318. [Google Scholar] [CrossRef]
- Klenow, M.B.; Heitmann, A.S.B.; Nylandsted, J.; Simonsen, A.C. Timescale of hole closure during plasma membrane repair estimated by calcium imaging and numerical modeling. Sci. Rep. 2021, 11, 4226. [Google Scholar] [CrossRef]
- Zhen, Y.; Radulovic, M.; Vietri, M.; Stenmark, H. Sealing holes in cellular membranes. EMBO J. 2021, 40, e106922. [Google Scholar] [CrossRef]
- Brito, C.; Mesquita, F.S.; Bleck, C.K.E.; Sellers, J.R.; Cabanes, D.; Sousa, S. Perfringolysin O-Induced Plasma Membrane Pores Trigger Actomyosin Remodeling and Endoplasmic Reticulum Redistribution. Toxins 2019, 11, 419. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Brito, C.; Cabanes, D.; Sousa, S. Control of cytoskeletal dynamics during cellular responses to pore forming toxins. Commun. Integr. Biol. 2017, 10, e1349582. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2010, 18, 80–89. [Google Scholar] [CrossRef]
- Letsiou, E.; Alves, L.G.T.; Fatykhova, D.; Felten, M.; Mitchell, T.J.; Müller-Redetzky, H.; Hocke, A.C.; Witzenrath, M. Microvesicles released from pneumolysin-stimulated lung epithelial cells carry mitochondrial cargo and suppress neutrophil oxidative burst. Sci. Rep. 2021, 11, 9529. [Google Scholar] [CrossRef]
- Köffel, R.; Wolfmeier, H.; Larpin, Y.; Besançon, H.; Schoenauer, R.; Babiychuk, V.S.; Drücker, P.; Pabst, T.; Mitchell, T.J.; Babiychuk, E.B.; et al. Host-Derived Microvesicles Carrying Bacterial Pore-Forming Toxins Deliver Signals to Macrophages: A Novel Mechanism of Shaping Immune Responses. Front. Immunol. 2018, 9, 1688. [Google Scholar] [CrossRef]
- Wolfmeier, H.; Radecke, J.; Schoenauer, R.; Koeffel, R.; Babiychuk, V.S.; Drücker, P.; Hathaway, L.J.; Mitchell, T.J.; Zuber, B.; Draeger, A.; et al. Active release of pneumolysin prepores and pores by mammalian cells undergoing a Streptococcus pneumoniae attack. Biochim. Biophys. Acta 2016, 1860, 2498–2509. [Google Scholar] [CrossRef] [PubMed]
- Larpin, Y.; Besançon, H.; Iacovache, M.; Babiychuk, V.S.; Babiychuk, E.B.; Zuber, B.; Draeger, A.; Köffel, R. Bacterial pore-forming toxin pneumolysin: Cell membrane structure and microvesicle shedding capacity determines differential survival of immune cell types. FASEB J. 2020, 34, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.J.; Sani, M.-A.; Parker, M.W.; Separovic, F. Cholesterol-Dependent Cytolysins: Membrane and Protein Structural Requirements for Pore Formation. Chem. Rev. 2019, 119, 7721–7736. [Google Scholar] [CrossRef] [PubMed]
- Pathak-Sharma, S.; Zhang, X.; Lam, J.G.T.; Weisleder, N.; Seveau, S.M. High-Throughput Microplate-Based Assay to Monitor Plasma Membrane Wounding and Repair. Front. Cell. Infect. Microbiol. 2017, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Wolfmeier, H.; Schoenauer, R.; Atanassoff, A.P.; Neill, D.R.; Kadioglu, A.; Draeger, A.; Babiychuk, E.B. Ca2+-dependent repair of pneumolysin pores: A new paradigm for host cellular defense against bacterial pore-forming toxins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 2045–2054. [Google Scholar] [CrossRef]
- Billington, S.J.; Jost, B.H.; Songer, J.G. Thiol-activated cytolysins: Structure, function and role in pathogenesis. FEMS Microbiol. Lett. 2000, 182, 197–205. [Google Scholar] [CrossRef]
- Atanassoff, A.P.; Wolfmeier, H.; Schoenauer, R.; Hostettler, A.; Ring, A.; Draeger, A.; Babiychuk, E.B. Microvesicle Shedding and Lysosomal Repair Fulfill Divergent Cellular Needs during the Repair of Streptolysin O-Induced Plasmalemmal Damage. PLoS ONE 2014, 9, e89743. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef]
- Maurer, J.; Hupp, S.; Pillich, H.; Mitchell, T.J.; Chakraborty, T.; Iliev, A.I. Missing elimination via membrane vesicle shedding contributes to the diminished calcium sensitivity of listeriolysin O. Sci. Rep. 2018, 8, 15846. [Google Scholar] [CrossRef]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Schoenauer, R.; Larpin, Y.; Babiychuk, E.B.; Drücker, P.; Babiychuk, V.S.; Avota, E.; Schneider-Schaulies, S.; Schumacher, F.; Kleuser, B.; Köffel, R.; et al. Down-regulation of acid sphingomyelinase and neutral sphingomyelinase-2 inversely determines the cellular resistance to plasmalemmal injury by pore-forming toxins. FASEB J. 2018, 33, 275–285. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Mi, H.; Thomas, P. PANTHER Pathway: An Ontology-Based Pathway Database Coupled with Data Analysis Tool. In Protein Networks and Pathway Analysis; Nikolsky, Y., Bryant, J., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 123–140. [Google Scholar]
- Panther Classification System. Available online: http://pantherdb.org/ (accessed on 9 November 2022).
- Koerdt, S.N.; Ashraf, A.P.; Gerke, V. Annexins and plasma membrane repair. Curr. Top. Membr. 2019, 84, 43–65. [Google Scholar]
- Demonbreun, A.R.; Bogdanovic, E.; Vaught, L.A.; Reiser, N.L.; Fallon, K.S.; Long, A.M.; Oosterbaan, C.C.; Hadhazy, M.; Page, P.G.; Joseph, P.R.B.; et al. A conserved annexin A6–mediated membrane repair mechanism in muscle, heart, and nerve. JCI Insight 2022, 7, e158107. [Google Scholar] [CrossRef]
- Rezvanpour, A.; Santamaria-Kisiel, L.; Shaw, G.S. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J. Biol. Chem. 2011, 286, 40174–40183. [Google Scholar] [CrossRef]
- Cowland, J.B.; Carter, D.; Bjerregaard, M.D.; Johnsen, A.H.; Borregaard, N.; Lollike, K. Tissue expression of copines and isolation of copines I and III from the cytosol of human neutrophils. J. Leukoc. Biol. 2003, 74, 379–388. [Google Scholar] [CrossRef]
- Creutz, C.E.; Tomsig, J.L.; Snyder, S.L.; Gautier, M.-C.; Skouri, F.; Beisson, J.; Cohen, J. The Copines, a Novel Class of C2 Domain-containing, Calciumdependent, Phospholipid-binding Proteins Conserved from Paramecium to Humans. J. Biol. Chem. 1998, 273, 1393–1402. [Google Scholar] [CrossRef]
- Tang, H.; Pang, P.; Qin, Z.; Zhao, Z.; Wu, Q.; Song, S.; Li, F. The CPNE Family and Their Role in Cancers. Front. Genet. 2021, 12, 689097. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT Machinery Is Required for Plasma Membrane Repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca2+ operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.S.; Yeung, F.; Stoddard, P.B.; Li, D.; Creutz, C.E.; Mayo, M.W. Copine-I represses NF-κB transcription by endoproteolysis of p65. Oncogene 2008, 27, 3516–3526. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-C.; Zhang, F.-L.; Geng, Q.; Yu, T.; Cui, Y.-Q.; Liu, X.-H.; Li, J.; Yan, M.-X.; Liu, L.; He, X.; et al. Quantitative Proteomic Analysis Identifies CPNE3 as a Novel Metastasis-promoting Gene in NSCLC. J. Proteome Res. 2013, 12, 3423–3433. [Google Scholar] [CrossRef] [PubMed]
- Ghislat, G.; Knecht, E. New Ca2+-dependent regulators of autophagosome maturation. Commun. Integr. Biol. 2012, 5, 308–311. [Google Scholar] [CrossRef]
- Ilacqua, A.N.; Price, J.E.; Graham, B.N.; Buccilli, M.J.; McKellar, D.R.; Damer, C.K. Cyclic AMP signaling in Dictyostelium promotes the translocation of the copine family of calcium-binding proteins to the plasma membrane. BMC Cell Biol. 2018, 19, 13. [Google Scholar] [CrossRef]
- Haase, H.; Pagel, I.; Khalina, Y.; Zacharzowsky, U.; Person, V.; Lutsch, G.; Petzhold, D.; Kott, M.; Schaper, J.; Morano, I. The carboxyl-terminal ahnak domain induces actin bundling and stabilizes muscle contraction. FASEB J. 2004, 18, 839–841. [Google Scholar] [CrossRef]
- Benaud, C.; Gentil, B.J.; Assard, N.; Court, M.; Garin, J.; Delphin, C.; Baudier, J. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol. 2004, 164, 133–144. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Podini, P.; Meldolesi, J. Enlargeosome Traffic: Exocytosis Triggered by Various Signals Is Followed by Endocytosis, Membrane Shedding or Both. Traffic 2007, 8, 742–757. [Google Scholar] [CrossRef]
- Borgonovo, B.; Cocucci, E.; Racchetti, G.; Podini, P.; Bachi, A.; Meldolesi, J. Regulated exocytosis: A novel, widely expressed system. Nat. Cell Biol. 2002, 4, 955–963. [Google Scholar] [CrossRef]
- Lalioti, V.S.; Ilari, A.; O’Connell, D.J.; Poser, E.; Sandoval, I.V.; Colotti, G. Sorcin Links Calcium Signaling to Vesicle Trafficking, Regulates Polo-Like Kinase 1 and Is Necessary for Mitosis. PLoS ONE 2014, 9, e85438. [Google Scholar] [CrossRef]
- Los, F.C.; Kao, C.-Y.; Smitham, J.; McDonald, K.L.; Ha, C.; Peixoto, C.A.; Aroian, R.V. RAB-5- and RAB-11-Dependent Vesicle-Trafficking Pathways Are Required for Plasma Membrane Repair after Attack by Bacterial Pore-Forming Toxin. Cell Host Microbe 2011, 9, 147–157. [Google Scholar] [CrossRef]
- Hamon, M.A.; Batsché, E.; Régnault, B.; Tham, T.N.; Seveau, S.; Muchardt, C.; Cossart, P. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. USA 2007, 104, 13467–13472. [Google Scholar] [CrossRef]
- Hamon, M.; Cossart, P. Histone Modifications and Chromatin Remodeling during Bacterial Infections. Cell Host Microbe 2008, 4, 100–109. [Google Scholar] [CrossRef]
- Parseghian, M.H.; Luhrs, K.A. Beyond the walls of the nucleus: The role of histones in cellular signaling and innate immunity. Biochem. Cell Biol. 2006, 84, 589–604. [Google Scholar] [CrossRef]
- Sork, H.; Corso, G.; Krjutskov, K.; Johansson, H.J.; Nordin, J.; Wiklander, O.P.B.; Lee, Y.X.F.; Westholm, J.O.; Lehtiö, J.; Wood, M.J.A.; et al. Heterogeneity and interplay of the extracellular vesicle small RNA transcriptome and proteome. Sci. Rep. 2018, 8, 10813. [Google Scholar] [CrossRef]
- Yang, Y.; Boza-Serrano, A.; Dunning, C.J.R.; Clausen, B.H.; Lambertsen, K.L.; Deierborg, T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflam. 2018, 15, 168. [Google Scholar] [CrossRef]
- Glomski, I.J.; Gedde, M.M.; Tsang, A.W.; Swanson, J.; Portnoy, D.A. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 2002, 156, 1029–1038. [Google Scholar] [CrossRef]
- Thery, C.; Clayton, A.; Amigorena, S.; Raposo, G. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. In Current Protocols in Cell Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Volume 30, pp. 3.22.1–3.22.29. [Google Scholar]
- Nato, F.; Reich, K.; Lhopital, S.; Rouyre, S.; Geoffroy, C.; Mazie, J.C.; Cossart, P. Production and characterization of neutralizing and nonneutralizing monoclonal antibodies against listeriolysin O. Infect. Immun. 1991, 59, 4641–4646. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- UniPro. Available online: http://www.uniprot.org (accessed on 15 September 2022).
- Hamouda, N.N.; Haute, C.V.D.; Vanhoutte, R.; Sannerud, R.; Azfar, M.; Mayer, R.; Calabuig, Á.C.; Swinnen, J.V.; Agostinis, P.; Baekelandt, V.; et al. ATP13A3 is a major component of the enigmatic mammalian polyamine transport system. J. Biol. Chem. 2021, 296, 100182. [Google Scholar] [CrossRef] [PubMed]
- Benchling. Available online: www.benchling.com (accessed on 9 November 2022).
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Sušac, L.; Vuong, M.T.; Thomas, C.; von Bülow, S.; O’Brien-Ball, C.; Santos, A.M.; Fernandes, R.A.; Hummer, G.; Tampé, R.; Davis, S.J. Structure of a fully assembled tumor-specific T cell receptor ligated by pMHC. Cell 2022, 185, 3201–3213.e19. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster 1 | |
|---|---|
| Calcium binding proteins Annexin A1 Annexin A2 Annexin A3 Annexin A4 Annexin A5 Annexin A6 Annexin A7 Annexin A11 Calmodulin-like protein 5 Copine-1 Copine-3 Hippocalcin-like protein 1 Translational proteins 40S ribosomal protein S20 40S ribosomal protein S18 60S ribosomal protein L8 60S ribosomal protein L31 60S ribosomal protein L23a Eukaryotic translation initiation factor 4B Transporters ATP synthase subunit G2, mitochondrial Cationic amino acid transporter 2 Monocarboxylate transporter 4 Sodium- and chloride-dependent taurine transporter Solute carrier family 12 member 2 Chaperones DnaJ homolog subfamily A member 1 Peptidyl-prolyl cis-trans isomerase B Proteasome assembly chaperone 1 Protein disulfide-isomerase A3 Cell adhesion molecules Cadherin-2 Cadherin-13 C-X-C chemokine receptor type 4 Desmocollin-1 Podocalyxin Chromatin-binding, or -regulatory proteins Histone H2A.V Histone H2B type 1-L Histone H3.3 Histone H4 Scaffold/adaptor proteins A-kinase anchor protein 12 | Cytoskeletal proteins Keratin, type I cytoskeletal 18 Vimentin PDZ and LIM domain protein 5 Defense/immunity proteins HLA class I histocompatibility antigen, B alpha chain HLA class I histocompatibility antigen, C alpha chain UL16-binding protein 3 Transmembrane signal receptors CD44 antigen Ephrin type-A receptor 2 Tissue factor Protein modifying enzymes Protein kinase C alpha type Multifunctional procollagen lysine hydroxylase and glycosyltransferase LH3 Membrane trafficking proteins Alpha-taxilin Gene-specific transcriptional regulators Glucocorticoid receptor Leucine-rich repeat flightless-interacting protein 1 Metabolite interconversion enzymes 5-nucleotidase Carnitine O-palmitoyltransferase 1, liver isoform Others Proteasomal ubiquitin receptor ADRM1 Glycogen synthase, muscle CD59 glycoprotein Neuroblast differentiation-associated protein AHNAK Neuroplastin Brain acid soluble protein 1 CD99 antigen Nuclease-sensitive element-binding protein 1 Y-box-binding protein 3 Myristoylated alanine-rich-C-kinase substrate Sorcin Ubiquitin-40S ribosomal protein S27a Dermcidin RNA-binding protein 14 78 kDa glucose-regulated protein RNA-binding protein 4 Uncharacterized protein C7orf50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, S.; Pereira, J.M.; Mayer, R.L.; Gonçalves, A.D.A.; Impens, F.; Cabanes, D.; Sousa, S. Cells Responding to Closely Related Cholesterol-Dependent Cytolysins Release Extracellular Vesicles with a Common Proteomic Content Including Membrane Repair Proteins. Toxins 2023, 15, 4. https://doi.org/10.3390/toxins15010004
Alves S, Pereira JM, Mayer RL, Gonçalves ADA, Impens F, Cabanes D, Sousa S. Cells Responding to Closely Related Cholesterol-Dependent Cytolysins Release Extracellular Vesicles with a Common Proteomic Content Including Membrane Repair Proteins. Toxins. 2023; 15(1):4. https://doi.org/10.3390/toxins15010004
Chicago/Turabian StyleAlves, Sara, Joana M. Pereira, Rupert L. Mayer, Alexandre D. A. Gonçalves, Francis Impens, Didier Cabanes, and Sandra Sousa. 2023. "Cells Responding to Closely Related Cholesterol-Dependent Cytolysins Release Extracellular Vesicles with a Common Proteomic Content Including Membrane Repair Proteins" Toxins 15, no. 1: 4. https://doi.org/10.3390/toxins15010004
APA StyleAlves, S., Pereira, J. M., Mayer, R. L., Gonçalves, A. D. A., Impens, F., Cabanes, D., & Sousa, S. (2023). Cells Responding to Closely Related Cholesterol-Dependent Cytolysins Release Extracellular Vesicles with a Common Proteomic Content Including Membrane Repair Proteins. Toxins, 15(1), 4. https://doi.org/10.3390/toxins15010004