Enhancing the Cytotoxicity and Apoptotic Efficacy of Parasporin-2-Derived Variants (Mpp46Aa1) on Cancer Cell Lines

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Construction of the Library of Mutants from PS2Aa1
2.2. Protein Extraction
2.3. Cytotoxicity of PS2Aa1 and Variants in Colon Cancer and Leukemia Cells
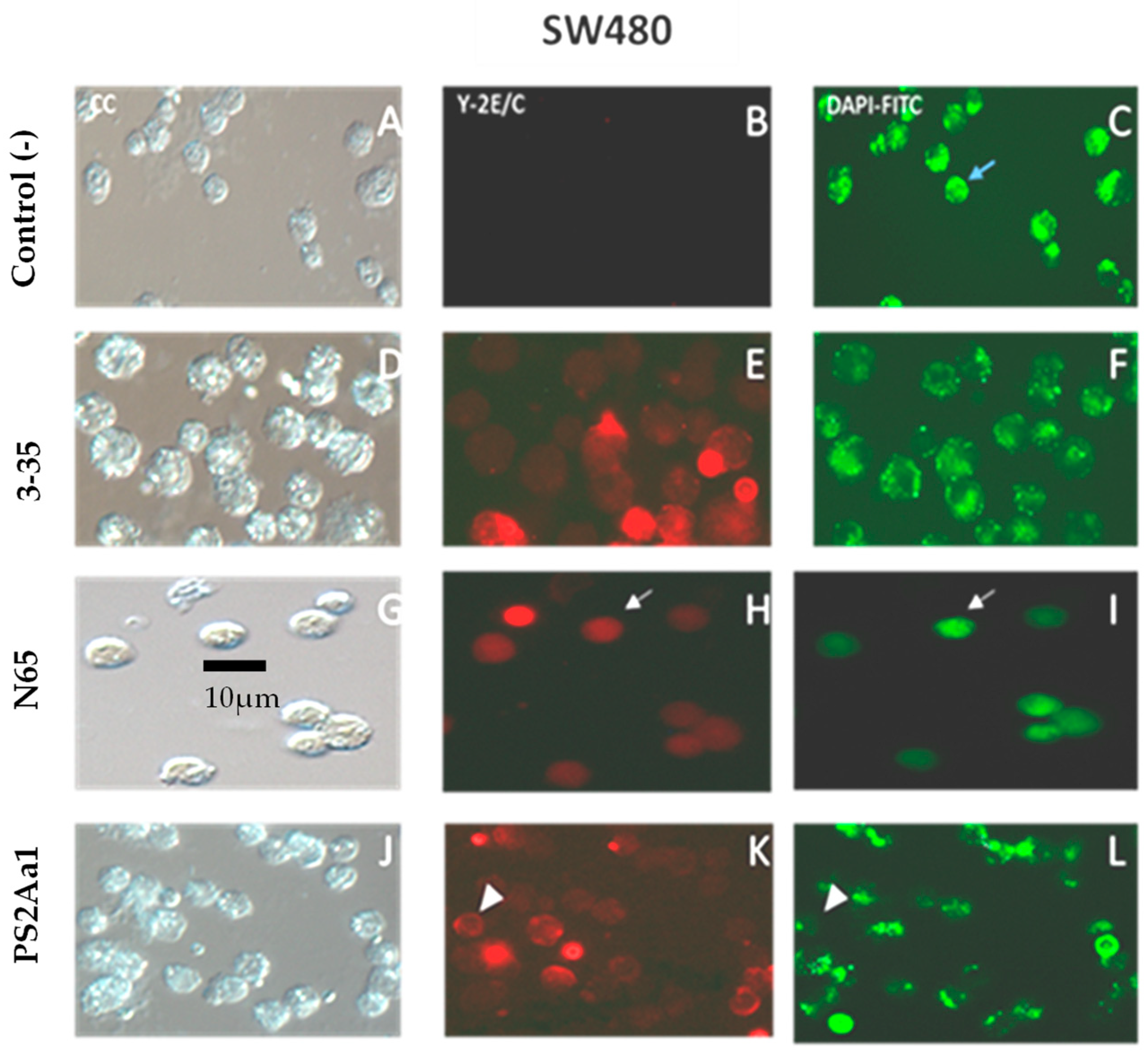
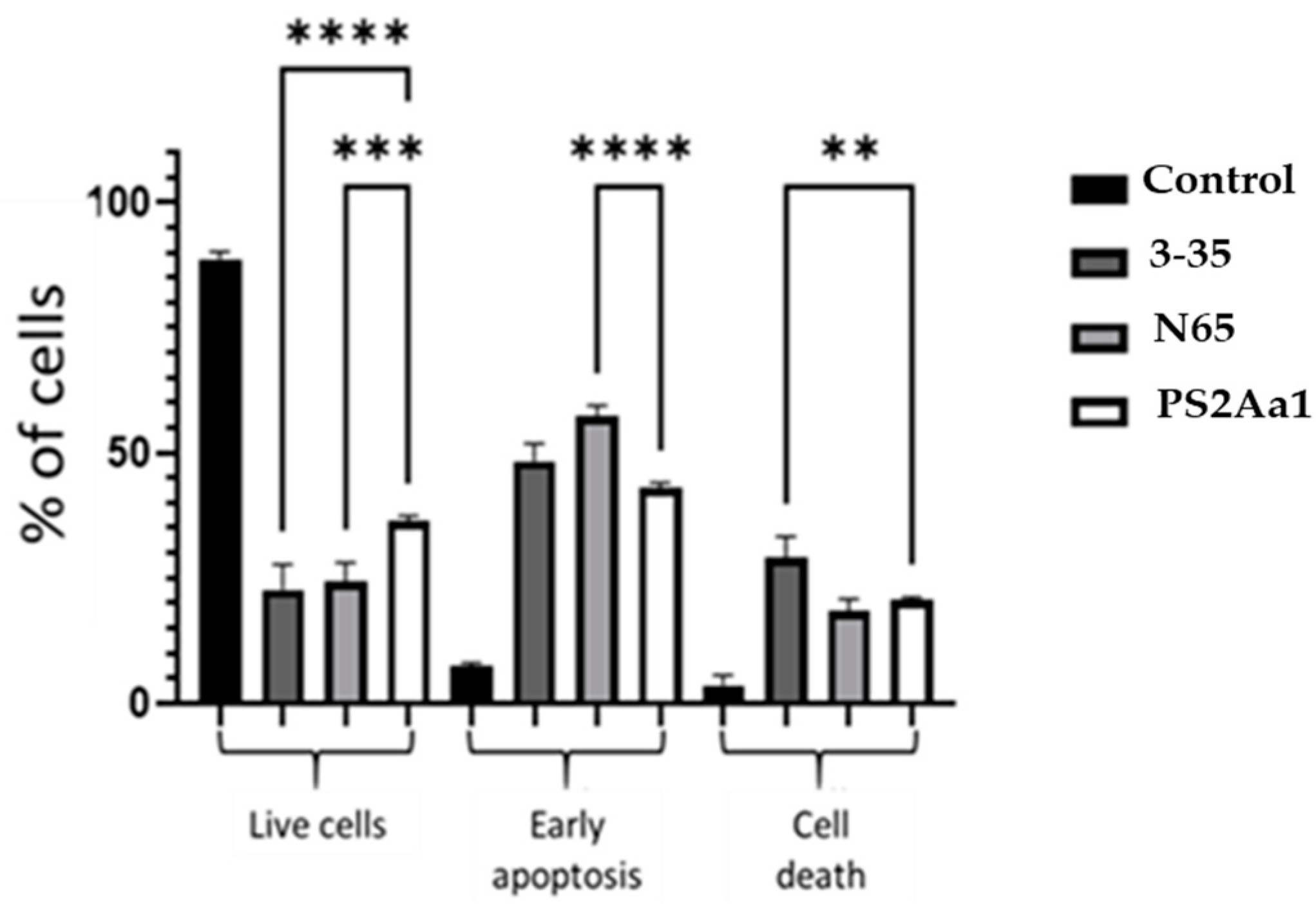
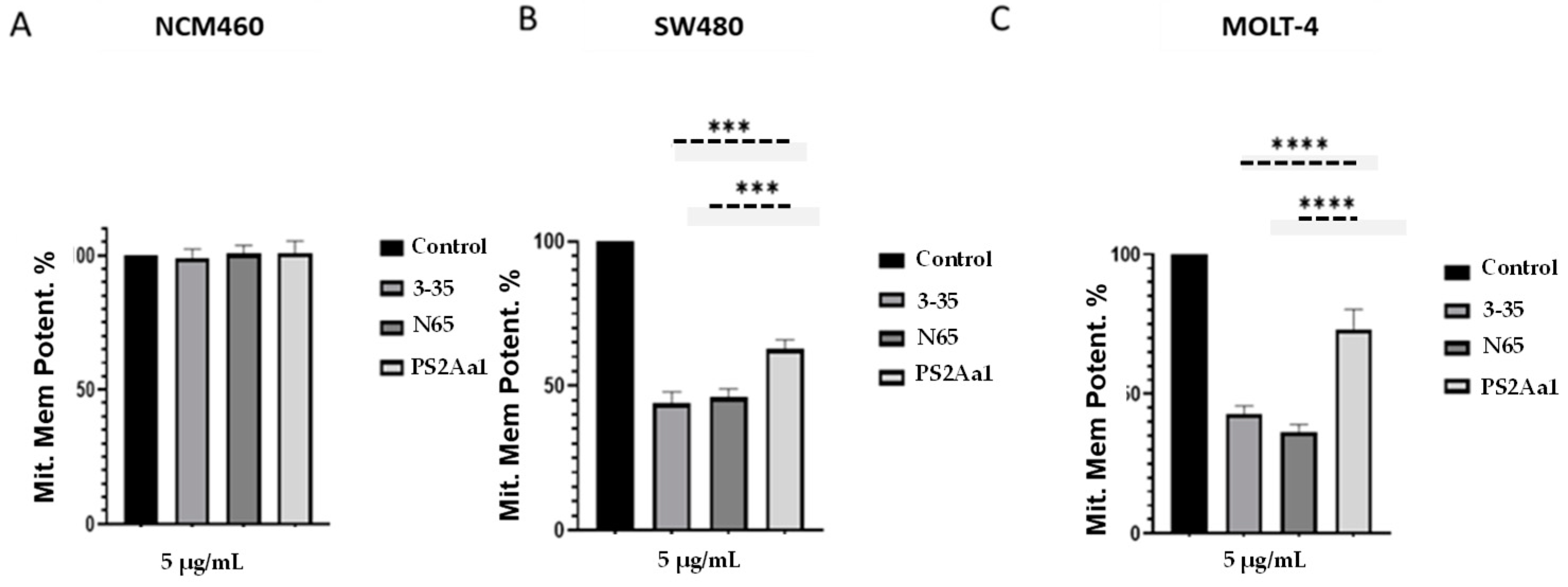
2.4. PS2Aa1 Variants Induce Cell Death via Apoptotic Mechanisms in Colon Cancer and Leukemia Cells
2.5. Molecular Dynamics of PS2Aa1 (Mpp46Aa1) Interaction with the Possible Main Receptor, GPI–CD59
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Bacterial Strains and Culture Condition
5.2. Cell Culture Conditions
5.3. Generation of Parasporin Mutants from Site-Directed Mutagenesis
5.4. Extraction and Activation of Parasporins
5.5. Cytotoxicity Assay and Selectivity Calculation
5.6. Apoptosis Detection via Annexin V-Cy3/6-DFDA Assay
5.7. Caspases 3/7 and 9 Assay
5.8. Determination of Mitochondrial Permeability Potential
5.9. Molecular Dynamic Analysis
5.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.Y.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]
- Chubicka, T.; Girija, D.; Deepa, K.; Salini, S.; Meera, N.; Raghavamenon, A.C.; Divya, M.K.; Babu, T.D. A Parasporin from Bacillus Thuringiensis Native to Peninsular India Induces Apoptosis in Cancer Cells through Intrinsic Pathway. J. Biosci. 2018, 43, 407–416. [Google Scholar] [CrossRef]
- Palma, L.; Munoz, D.; Berry, C.; Murillo, J.; Caballero, P. Bacillus Thuringiensis Toxins: An Overview of Their Biocidal Activity. Toxins 2014, 6, 3296–3325. [Google Scholar] [CrossRef]
- Okassov, A.; Nersesyan, A.; Kitada, S.; Ilin, A. Parasporins as New Natural Anticancer Agents: A Review. J. BUON 2015, 20, 5–16. [Google Scholar]
- Borin, D.B.; Castrejón-Arroyo, K.; Cruz-Nolasco, A.; Peña-Rico, M.; Rorato, M.S.; Santos, R.C.V.; de Baco, L.S.; Pérez-Picaso, L.; Camacho, L.; Navarro-Mtz, A.K. Parasporin A13-2 of Bacillus Thuringiensis Isolates from the Papaloapan Region (Mexico) Induce a Cytotoxic Effect by Late Apoptosis against Breast Cancer Cells. Toxins 2021, 13, 476. [Google Scholar] [CrossRef]
- Akiba, T.; Okumura, S. Parasporins 1 and 2: Their Structure and Activity. J. Invertebr. Pathol. 2017, 142, 44–49. [Google Scholar] [CrossRef]
- Abe, Y.; Inoue, H.; Ashida, H.; Maeda, Y.; Kinoshita, T.; Kitada, S. Glycan Region of GPI Anchored-Protein Is Required for Cytocidal Oligomerization of an Anticancer Parasporin-2, Cry46Aa1 Protein, from Bacillus Thuringiensis Strain A1547. J. Invertebr. Pathol. 2017, 142, 71–81. [Google Scholar] [CrossRef]
- Akiba, T.; Abe, Y.; Kitada, S.; Kusaka, Y.; Ito, A.; Ichimatsu, T.; Katayama, H.; Akao, T.; Higuchi, K.; Mizuki, E.; et al. Crystal Structure of the Parasporin-2 Bacillus Thuringiensis Toxin That Recognizes Cancer Cells. J. Mol. Biol. 2009, 386, 121–133. [Google Scholar] [CrossRef]
- Brasseur, K.; Auger, P.; Asselin, E.; Parent, S.; Cote, J.C.; Sirois, M. Parasporin-2 from a New Bacillus Thuringiensis 4R2 Strain Induces Caspases Activation and Apoptosis in Human Cancer Cells. PLoS ONE 2015, 10, e0135106. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, B.C.; Yu, Z.; Sun, M. Structural Insights into Bacillus Thuringiensis Cry, Cyt and Parasporin Toxins. Toxins 2014, 6, 2732–2770. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, A.; Kkani, P.; Chandrasekaran, B.; Ponnusamy, S.; Viswanathan, S.; Selvanayagam, P.; Rajaiah, S. Screening and Characterization of a Non-Insecticidal Bacillus Thuringiensis Strain Producing Parasporal Protein with Selective Toxicity against Human Colon Cancer Cell Lines. Ann. Microbiol. 2016, 66, 1167–1178. [Google Scholar] [CrossRef]
- Kitada, S.; Abe, Y.; Maeda, T.; Shimada, H. Parasporin-2 Requires GPI-Anchored Proteins for the Efficient Cytocidal Action to Human Hepatoma Cells. Toxicology 2009, 264, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Soberon, M.; Portugal, L.; Garcia-Gomez, B.I.; Sanchez, J.; Onofre, J.; Gomez, I.; Pacheco, S.; Bravo, A. Cell Lines as Models for the Study of Cry Toxins from Bacillus Thuringiensis. Insect Biochem. Mol. Biol. 2018, 93, 66–78. [Google Scholar] [CrossRef]
- Suhaili, S.H.; Karimian, H.; Stellato, M.; Lee, T.H.; Aguilar, M.I. Mitochondrial Outer Membrane Permeabilization: A Focus on the Role of Mitochondrial Membrane Structural Organization. Biophys. Rev. 2017, 9, 443. [Google Scholar] [CrossRef]
- Suárez-Barrera, M.O.; Visser, L.; Pinzón-Reyes, E.H.; Rondón Villarreal, P.; Alarcón-Aldana, J.S.; Rueda-Forero, N.J. Site-Directed Mutants of Parasporin PS2Aa1 with Enhanced Cytotoxic Activity in Colorectal Cancer Cell Lines. Molecules 2022, 27, 7262. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Sinha, B.; Domschke, W.; Peters, G.; Schulze-Osthoff, K.; Jänicke, R.U. Alpha-Toxin Is a Mediator of Staphylococcus Aureus-Induced Cell Death and Activates Caspases via the Intrinsic Death Pathway Independently of Death Receptor Signaling. J. Cell Biol. 2001, 155, 637–647. [Google Scholar] [CrossRef]
- Cruz, J.; Suárez-Barrera, M.O.; Rondón-Villarreal, P.; Olarte-Diaz, A.; Guzmán, F.; Visser, L.; Rueda-Forero, N.J. Computational Study, Synthesis and Evaluation of Active Peptides Derived from Parasporin-2 and Spike Protein from Alphacoronavirus against Colorectal Cancer Cells. Biosci. Rep. 2021, 41, BSR20211964. [Google Scholar] [CrossRef]
- Suárez-Barrera, M.O.; Visser, L.; Rondón-Villarreal, P.; Herrera-Pineda, D.F.; Alarcón-Aldana, J.S.; Van den Berg, A.; Orozco, J.; Pinzón-Reyes, E.H.; Moreno, E.; Rueda-Forero, N.J. Genetic Modification Approaches for Parasporins Bacillus Thuringiensis Proteins with Anticancer Activity. Molecules 2021, 26, 7476. [Google Scholar] [CrossRef]
- Mahalakshmi, A.; Shenbagarathai, R. Homology Modeling of Cry10Aa Toxin from B. Thuringiensis Israelensis and B. Thuringiensis Subsp. LDC-9. J. Biomol. Struct. Dyn. 2010, 28, 363–378. [Google Scholar] [CrossRef]
- Walters, F.S.; Stacy, C.M.; Lee, M.K.; Palekar, N.; Chen, J.S. An Engineered Chymotrypsin/Cathepsin G Site in Domain I Renders Bacillus Thuringiensis Cry3A Active against Western Corn Rootworm Larvae. Appl. Environ. Microbiol. 2008, 74, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C.; Gayen, S.; Basu, A.; Ghosh, K.S.; Dasgupta, S.; Maiti, M.K.; Sen, S.K. Prediction-Based Protein Engineering of Domain I of Cry2A Entomocidal Toxin of Bacillus Thuringiensis for the Enhancement of Toxicity against Lepidopteran Insects. Protein Eng. Des. Sel. 2007, 20, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Gowdhami, B.; Manojkumar, Y.; Vimala, R.T.V.; Ramya, V.; Karthiyayini, B.; Kadalmani, B.; Akbarsha, M.A. Cytotoxic Cobalt (III) Schiff Base Complexes: In Vitro Anti-Proliferative, Oxidative Stress and Gene Expression Studies in Human Breast and Lung Cancer Cells. BioMetals 2022, 35, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation and Disease. Immunity 2019, 50, 1352. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of In-Vitro Bioassay Methods: Application in Herbal Drug Research. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar] [CrossRef]
- Okumura, S.; Saitoh, H.; Ishikawa, T.; Inouye, K.; Mizuki, E. Mode of Action of Parasporin-4, a Cytocidal Protein from Bacillus Thuringiensis. Biochim. Biophys. Acta 2011, 1808, 1476–1482. [Google Scholar] [CrossRef]
- Kennedy, C.L.; Lyras, D.; Cordner, L.M.; Melton-Witt, J.; Emmins, J.J.; Tweten, R.K.; Rood, J.I. Pore-Forming Activity of Alpha-Toxin Is Essential for Clostridium Septicum-Mediated Myonecrosis. Infect. Immun. 2009, 77, 943. [Google Scholar] [CrossRef]
- Pal, S.; Ray, S.D.; Homechaudhuri, S. Evaluation of In Vivo Non-Specific Immunity and Oxidative Stress in Labeo Rohita (Hamilton, 1822) Infected with Aeromonas Hydrophila as Biomarker for Early Diagnosis. Int. J. Fish. Aquat. Stud. 2015, 3, 116–124. [Google Scholar]
- Patel, B.; Kumar, P.; Banerjee, R.; Basu, M.; Pal, A.; Samanta, M.; Das, S. Lactobacillus Acidophilus Attenuates Aeromonas Hydrophila Induced Cytotoxicity in Catla Thymus Macrophages by Modulating Oxidative Stress and Inflammation. Mol. Immunol. 2016, 75, 69–83. [Google Scholar] [CrossRef]
- Krishnan, V.; Domanska, B.; Elhigazi, A.; Afolabi, F.; West, M.J.; Crickmore, N. The Human Cancer Cell Active Toxin Cry41Aa from Bacillus Thuringiensis Acts like Its Insecticidal Counterparts. Biochem. J. 2017, 474, 1591–1602. [Google Scholar] [CrossRef]
- Degiacomi, M.T.; Iacovache, I.; Pernot, L.; Chami, M.; Kudryashev, M.; Stahlberg, H.; van der Goot, F.G.; Dal Peraro, M. Molecular Assembly of the Aerolysin Pore Reveals a Swirling Membrane-Insertion Mechanism. Nat. Chem. Biol. 2013, 9, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Shimada, H.; Kitada, S. Raft-Targeting and Oligomerization of Parasporin-2, a Bacillus Thuringiensis Crystal Protein with Anti-Tumour Activity. J. Biochem. 2008, 143, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Goto, H.; Fujii, H.; Naruto, T.; Takeuchi, M.; Tanoshima, R.; Kato, H.; Yanagimachi, M.; Kajiwara, R.; Yokota, S. Flow Cytometric Chemosensitivity Assay Using JC-1, a Sensor of Mitochondrial Transmembrane Potential, in Acute Leukemia. Cancer Chemother. Pharmacol. 2013, 72, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Kühnel, J.M.; Perrot, J.Y.; Faussat, A.M.; Marie, J.P.; Schwaller, M.A. Functional Assay of Multidrug Resistant Cells Using JC-1, a Carbocyanine Fluorescent Probe. Leukemia 1997, 11, 1147–1155. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single-Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Klähn, M.; Zacharias, M. Transformations in Plasma Membranes of Cancerous Cells and Resulting Consequences for Cation Insertion Studied with Molecular Dynamics. Phys. Chem. Chem. Phys. 2013, 15, 14427–14441. [Google Scholar] [CrossRef]

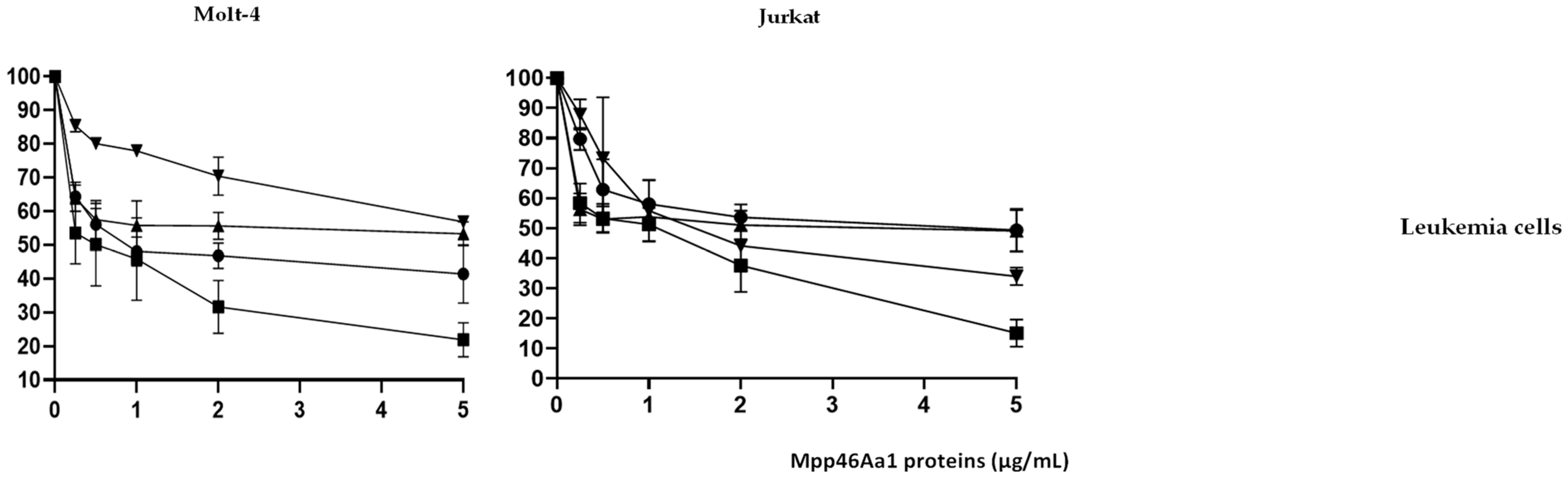
 ) and variants 3–35 (
) and variants 3–35 ( ), N65 (
), N65 ( ), and 0–15 (
), and 0–15 ( ) at different concentrations (from 0.5 to 5 µg/mL).
) and variants 3–35 (), N65 (), and 0–15 () at different concentrations (from 0.5 to 5 µg/mL).
) at different concentrations (from 0.5 to 5 µg/mL).
) and variants 3–35 (), N65 (), and 0–15 () at different concentrations (from 0.5 to 5 µg/mL).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
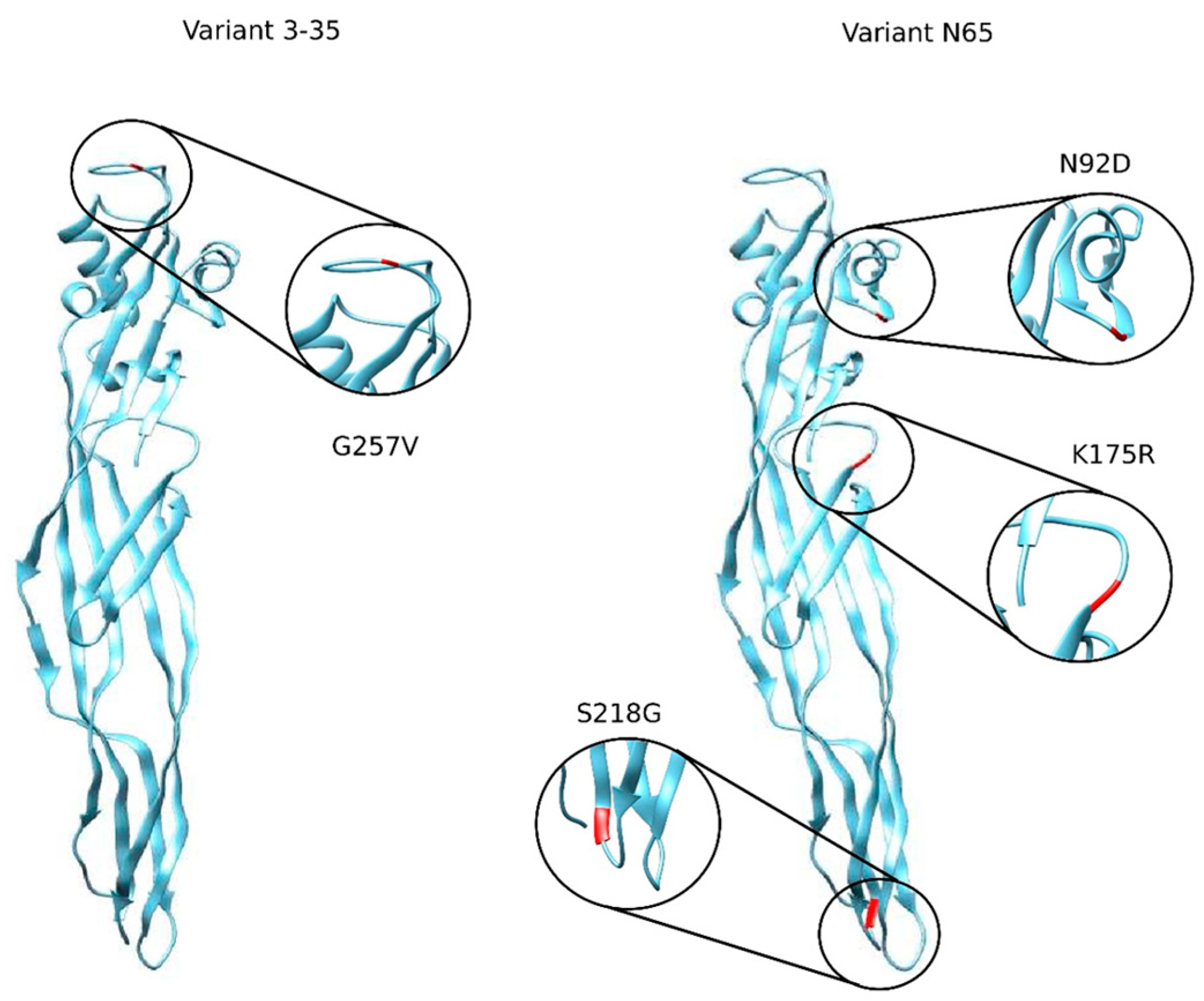
| Nomenclature | Type/Location of the Mutation | Notation of the Mutation |
|---|---|---|
| PS2Aa1 | PS2Aa1 (PDB 2ZTB) | - |
| 0–15 | SUS/256 | G256A |
| 3–35 | SUS/257 | G257V |
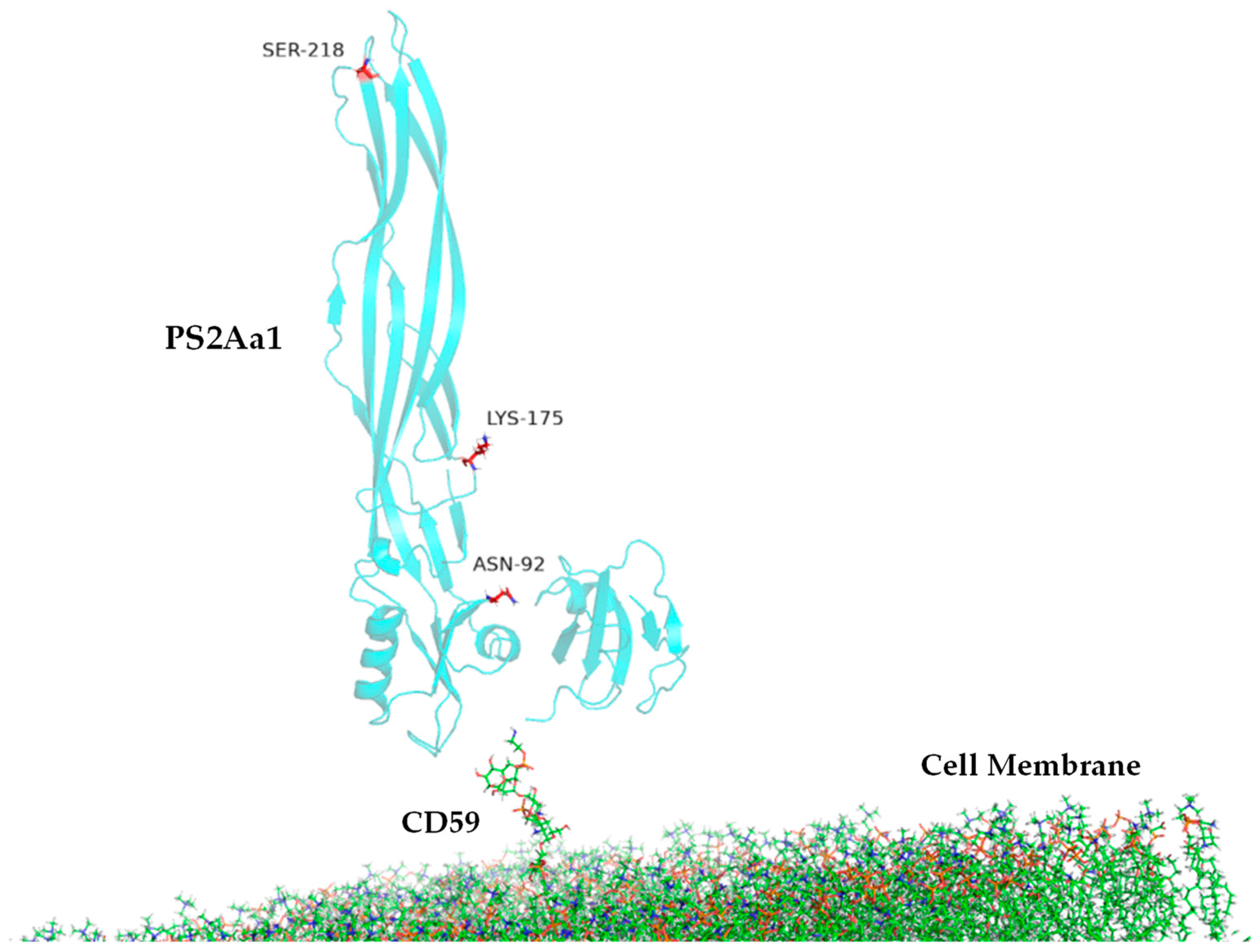
| N65 | SUS/92,175,218 | N92D, K175R, S218G |
| Parasporin/Cell Line | IC50 of Parasporins in µg/mL (95% CI) | ||||||
|---|---|---|---|---|---|---|---|
| SW480 | SW620 | CaCO-2 | MOLT-4 | Jurkat | CHOK1 | NCM460 | |
| 0–15 | 2.9 (2.2–3.4) | 2 (1.6–2.5) | 73.32 (11.6–122.4) | 1.7 (0.8–3.6) | 1.2 (0.6–2.6) | 20.2 (20–20.3) | 77.7 (71.6–8) |
| 3–35 | 0.9 (0.6–1.1) | 1.2 (0.9–1.5) | 0.88 (0.81–0.9) | 1.0 (0.6–1.8) | 1.9 (1.2–3.2) | 39.9 (39.8–4) | 78.8 (70.5–89.6) |
| N65 | 1.2 (1–1.3) | 1.0 (0.9–1.2) | 0.51 (0.427–0.6) | 0.6 (0.4–0.9) | 0.7 (0.5–1) | 40 (39.8–40.2) | 84.2 (79.2–9) |
| PS2Aa1 | 2.1 (1.5–2.8) | 2.3 (1.7–2.9) | 2.57 (1.84–4.1) | 4.7 (3.6–6.5) | 1.6 (1.2–2.2) | 40.1 (40–40.3) | 67.7 (63.6–72.1) |
| Parasporin/Cell Lines | SW480 | SW620 | CaCo-2 | MOLT-4 | Jurkat |
|---|---|---|---|---|---|
| 0–15 | 27.0 | 38.2 | 1.1 | 45.7 | 63.7 |
| N65 | 67.4 | 77.1 | 165.1 | 131.3 | 107.9 |
| 3–35 | 97.8 | 72.5 | 89.5 | 78.7 | 43.4 |
| PS2Aa1 | 31.9 | 29.5 | 26.3 | 14.2 | 42.3 |
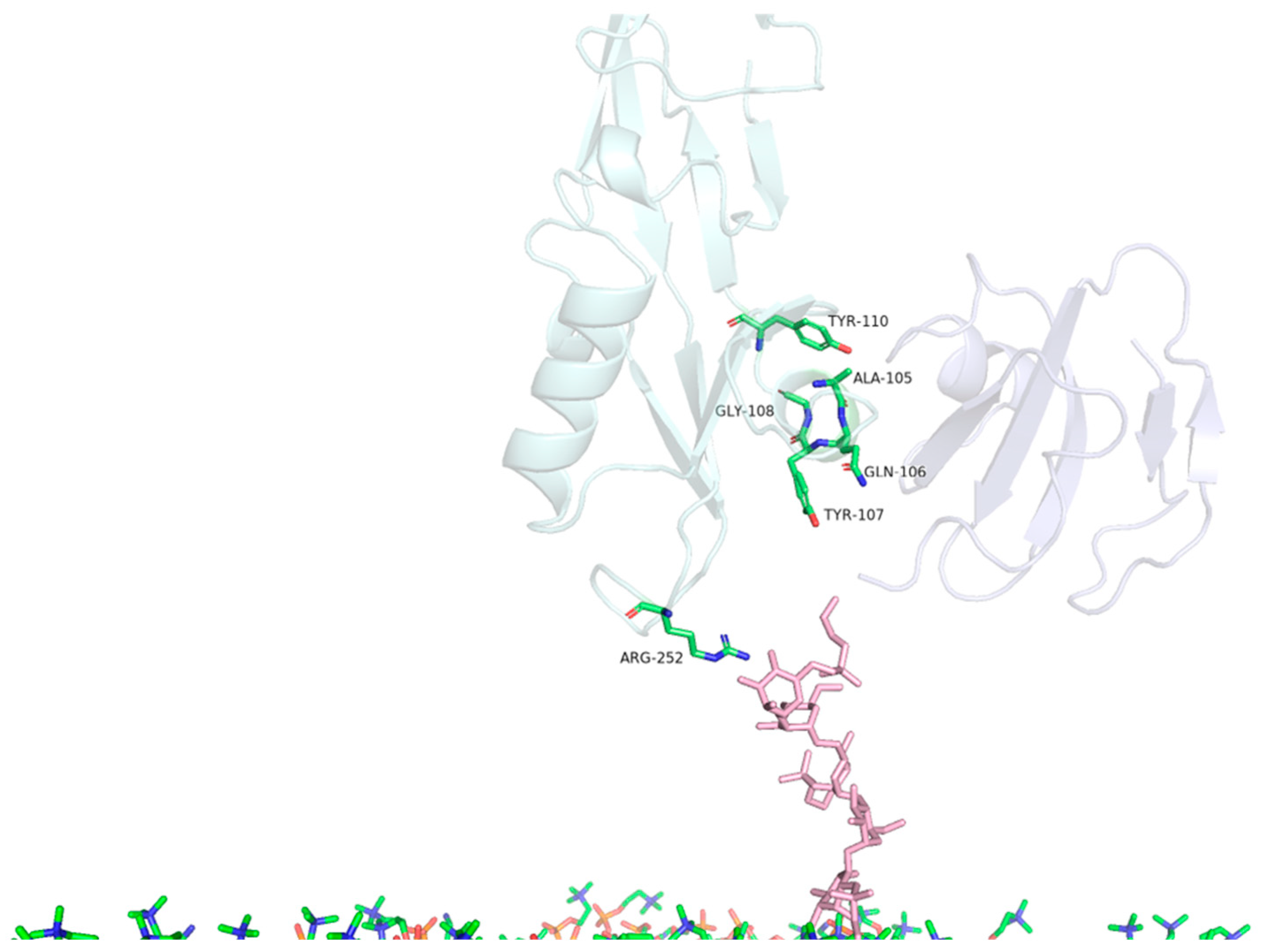
| MD | Contact Residues (>70%) | |
|---|---|---|
| PS2Aa1 Residues—CD59 | PS2Aa1 Residues—GPI | |
| Replicate 1 | ALA105, GLN106, TYR107, GLY108, TYR110 | ARG252 |
| Replicate 2 | TYR57, ASN92, PRO101, ALA105, GLN106, TYR107, GLY108, TYR110 | ARG252 |
| Replicate 3 | TYR57, ASN92, PRO101, ALA105, GLN106, TYR107, GLY108, TYR110 | ARG252 |
| Comparison | PS2Aa1 Residues—CD59 |
|---|---|
| ALA105 | 5.6 Å |
| GLN106 | 5.7 Å |
| TYR107 | 3.6 Å |
| GLY108 | 4.8 Å |
| TYR110 | 3.8 Å |
| TYR57 | 5.4 Å |
| ASN92 | 5.6 Å |
| PRO101 | 3.6 Å |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alarcón-Aldana, J.S.; Visser, L.; Rueda-Forero, N.J.; Pinzón-Reyes, E.H.; Rondón-Villarreal, P.; Suárez-Barrera, M.O. Enhancing the Cytotoxicity and Apoptotic Efficacy of Parasporin-2-Derived Variants (Mpp46Aa1) on Cancer Cell Lines. Toxins 2024, 16, 415. https://doi.org/10.3390/toxins16100415
Alarcón-Aldana JS, Visser L, Rueda-Forero NJ, Pinzón-Reyes EH, Rondón-Villarreal P, Suárez-Barrera MO. Enhancing the Cytotoxicity and Apoptotic Efficacy of Parasporin-2-Derived Variants (Mpp46Aa1) on Cancer Cell Lines. Toxins. 2024; 16(10):415. https://doi.org/10.3390/toxins16100415
Chicago/Turabian StyleAlarcón-Aldana, Juan S., Lydia Visser, Nohora J. Rueda-Forero, Efraín H. Pinzón-Reyes, Paola Rondón-Villarreal, and Miguel O. Suárez-Barrera. 2024. "Enhancing the Cytotoxicity and Apoptotic Efficacy of Parasporin-2-Derived Variants (Mpp46Aa1) on Cancer Cell Lines" Toxins 16, no. 10: 415. https://doi.org/10.3390/toxins16100415
APA StyleAlarcón-Aldana, J. S., Visser, L., Rueda-Forero, N. J., Pinzón-Reyes, E. H., Rondón-Villarreal, P., & Suárez-Barrera, M. O. (2024). Enhancing the Cytotoxicity and Apoptotic Efficacy of Parasporin-2-Derived Variants (Mpp46Aa1) on Cancer Cell Lines. Toxins, 16(10), 415. https://doi.org/10.3390/toxins16100415