Shining a Light on Venom-Peptide Receptors: Venom Peptides as Targeted Agents for In Vivo Molecular Imaging †



Abstract
:1. Introduction
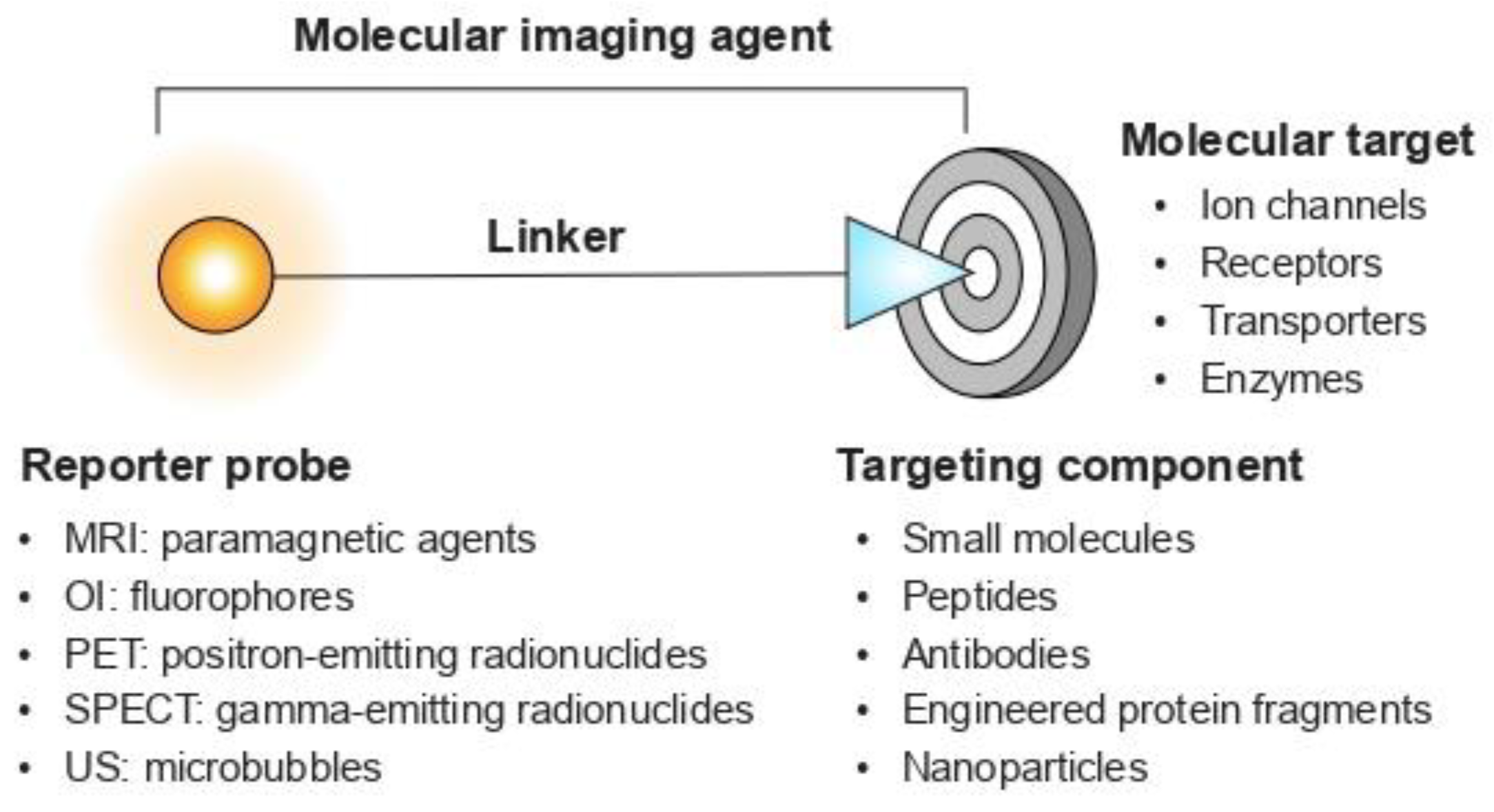
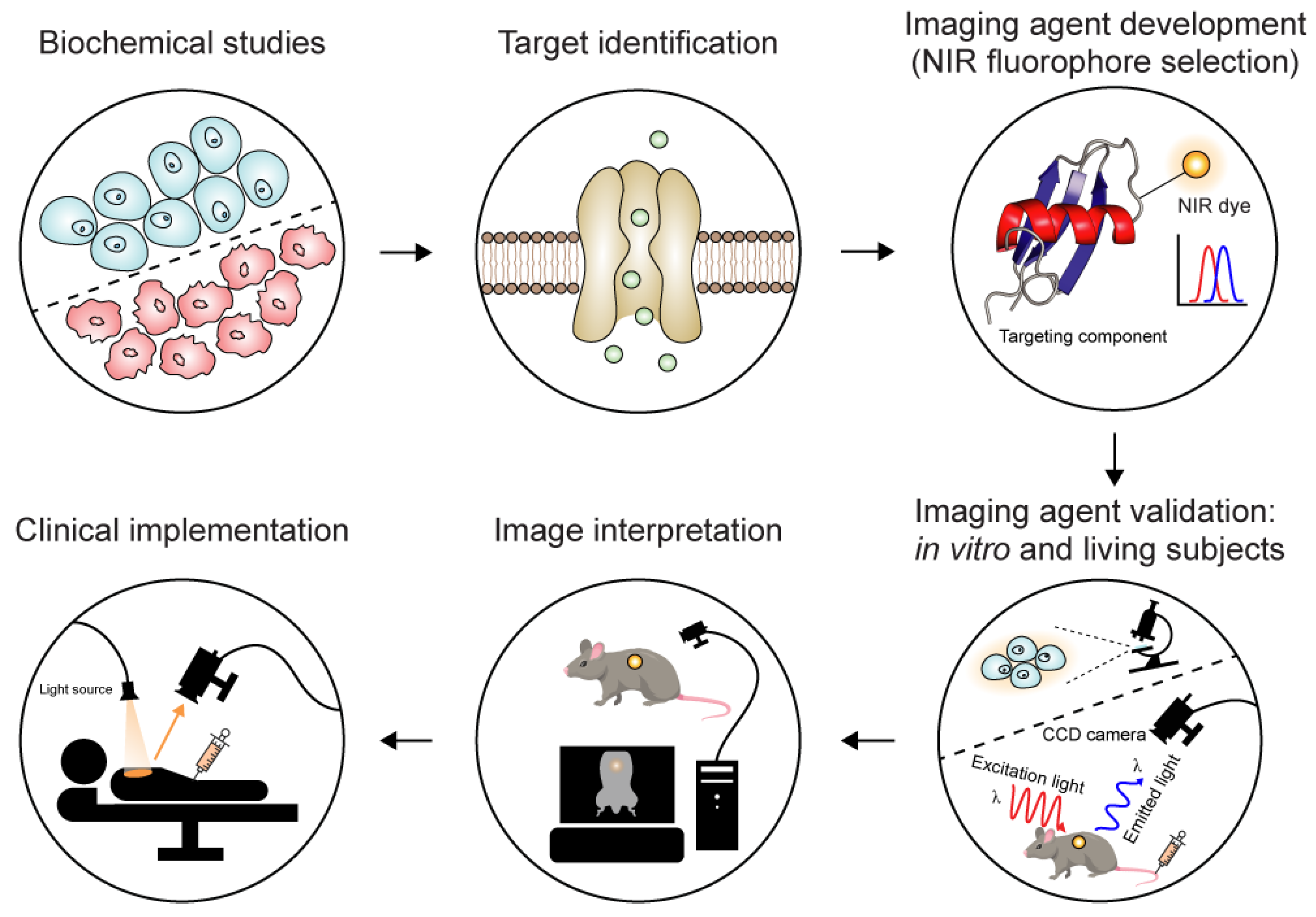
1.1. A Quick Look at Molecular Imaging Agents
1.2. Animal Venoms: A Rich Source of Imaging Agents
1.3. Near-Infrared Fluorescent Imaging
2. Chlorotoxin: An Intraoperative Agent for Imaging Brain Tumours
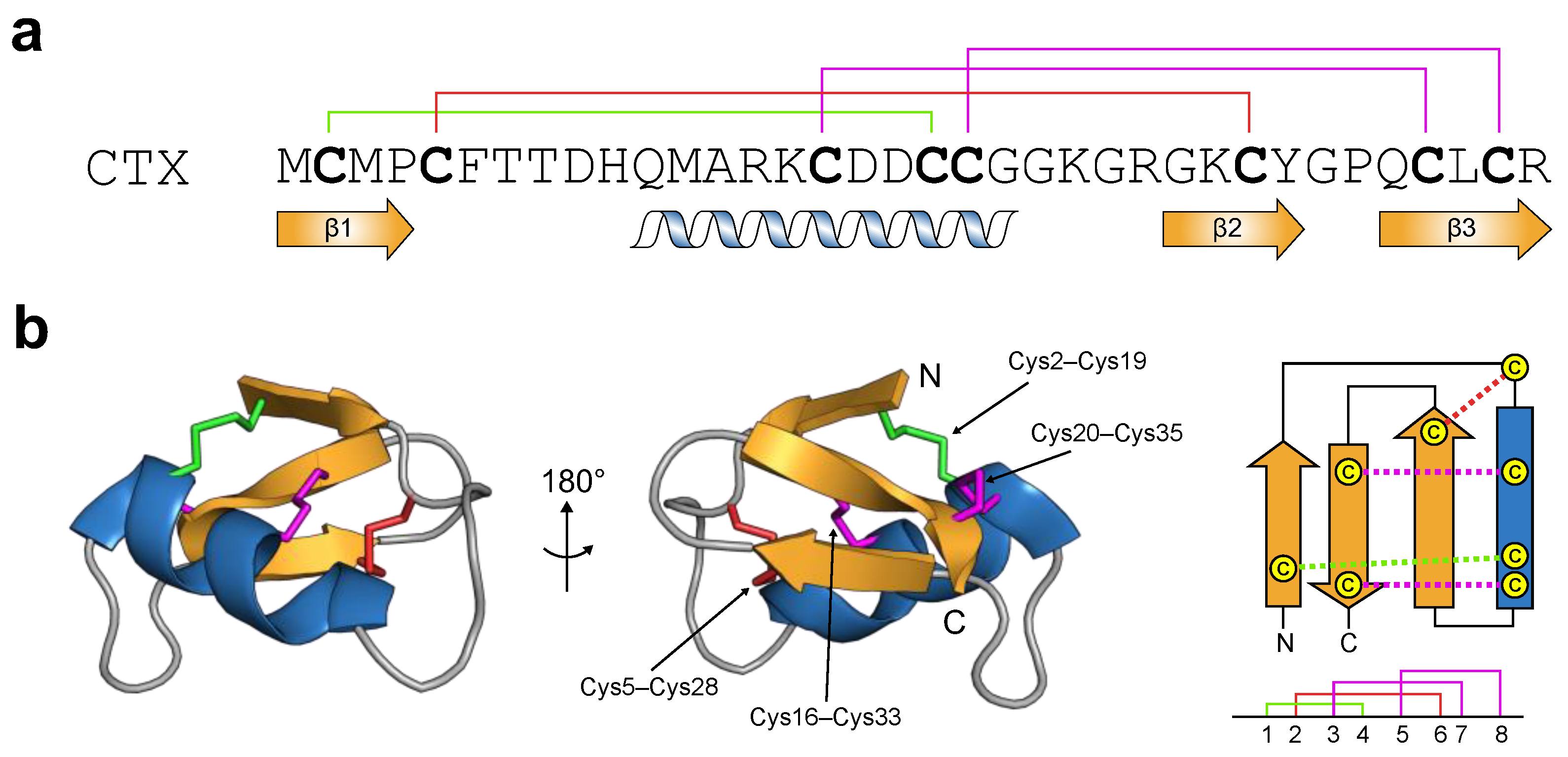
2.1. Chlorotoxin: From Primary to Tertiary Structure
2.2. Chlorotoxin: A Biological Chameleon
2.3. Chlorotoxin: Imaging Applications
3. Peripheral Nerve Injury
3.1. Intraoperative Preservation of Peripheral Nerve Tissues
3.2. NaV1.7: Contribution of Peripheral Nerves
3.3. Spider-Venom Peptides as Nerve-Targeted Agents
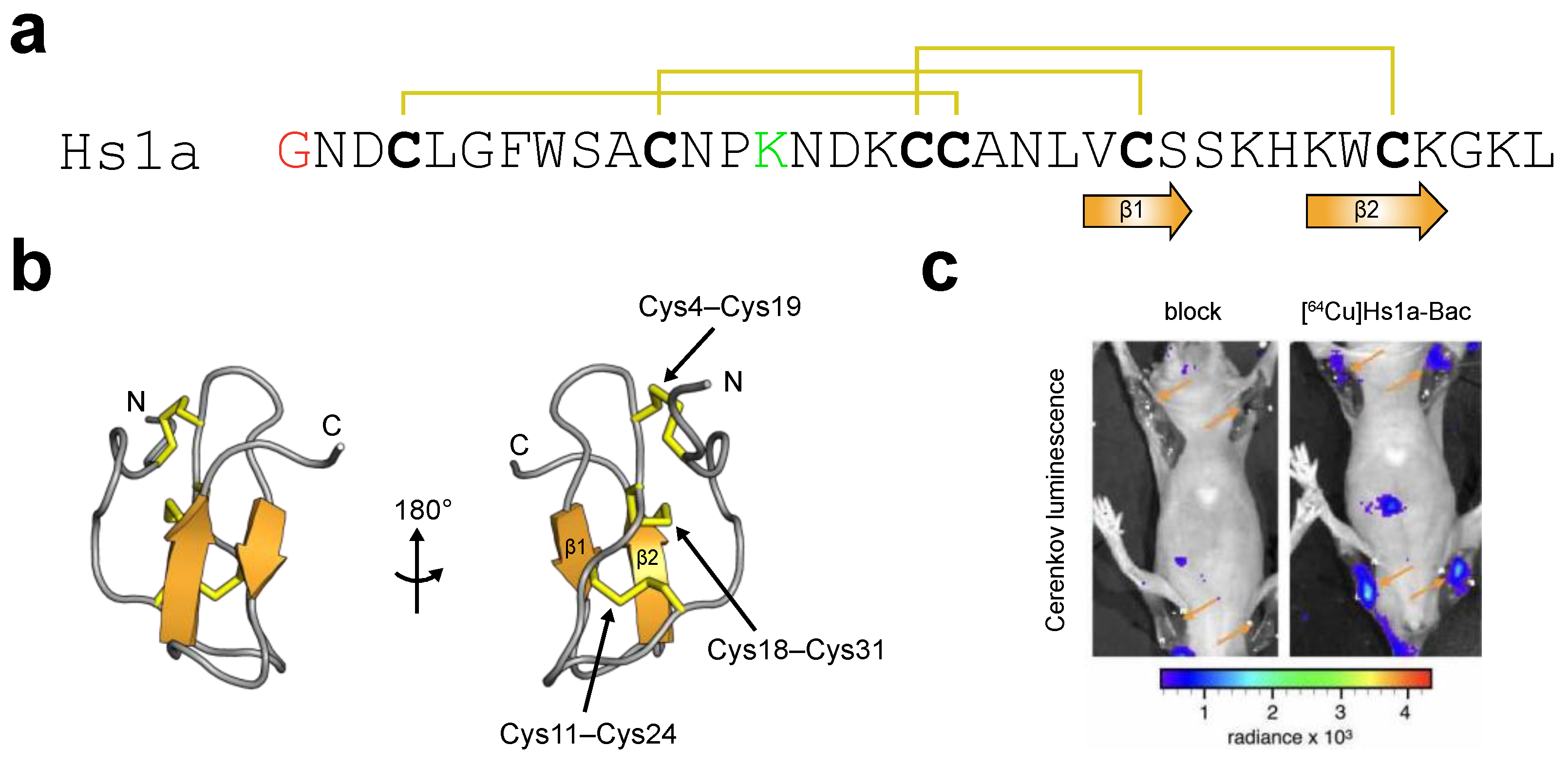
3.3.1. μ-theraphotoxin-Hs1a
3.3.2. μ-theraphotoxin-Tsp1a
3.4. Development of Nerve-Targeted Imaging Agents
4. Potential Applications of Molecular Imaging
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Massoud, T.F.; Gambhir, S.S. Molecular imaging in living subjects: Seeing fundamental biological processes in a new light. Genes Dev. 2003, 17, 545–580. [Google Scholar] [CrossRef]
- Willmann, J.K.; van Bruggen, N.; Dinkelborg, L.M.; Gambhir, S.S. Molecular imaging in drug development. Nat. Rev. Drug Discov. 2008, 7, 591–607. [Google Scholar] [CrossRef]
- Ashcroft, F.M. From molecule to malady. Nature 2006, 440, 440–447. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Hübner, C.A.; Fuhrmann, J.C. Ion channels: Function unravelled by dysfunction. Nat. Cell Biol. 2004, 6, 1039–1047. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The concise guide to pharmacology 2019/20: Ion channels. Br. J. Pharmacol. 2019, 176, S142–S228. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Bagal, S.K.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- James, M.L.; Gambhir, S.S. A molecular imaging primer: Modalities, imaging agents, and applications. Physiol. Rev. 2012, 92, 897–965. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.W.; Qiu, S.Q.; Zhang, G.J. Molecular and functional imaging in cancer-targeted therapy: Current applications and future directions. Sig. Transduct. Target. Ther. 2023, 8, 89. [Google Scholar] [CrossRef]
- Lee, S.; Xie, J.; Chen, X. Peptide-based probes for targeted molecular imaging. Biochemistry 2010, 49, 1364–1376. [Google Scholar] [CrossRef]
- Wu, A.M. Antibodies and antimatter: The resurgence of immuno-PET. J. Nucl. Med. 2009, 50, 2. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.B.; Wilkinson, T.; Schofield, D.J. Therapeutic monoclonal antibodies to complex membrane protein targets: Antigen generation and antibody discovery strategies. BioDrugs 2018, 32, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, C.J.; Colussi, P.; Clark, T.G. Ion channels as therapeutic antibody targets. mAbs 2019, 11, 265–296. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug. Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Holford, M.; Daly, M.; King, G.F.; Norton, R.S. Venoms to the rescue. Science 2018, 361, 842–844. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef]
- Robinson, S.D.; Undheim, E.A.B.; Ueberheide, B.; King, G.F. Venom peptides as therapeutics: Advances, challenges and the future of venom-peptide discovery. Expert Rev. Proteom. 2017, 14, 931–939. [Google Scholar] [CrossRef]
- Schmidtko, A.; Lötsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Beneski, D.A.; Catterall, W.A. Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc. Natl. Acad. Sci. USA 1980, 77, 639–643. [Google Scholar] [CrossRef]
- Garcia-Calvo, M.; Knaus, H.G.; McManus, O.B.; Giangiacomo, K.M.; Kaczorowski, G.J.; Garcia, M.L. Purification and reconstitution of the high-conductance, calcium-activated potassium channel from tracheal smooth muscle. J. Biol. Chem. 1994, 269, 676–682. [Google Scholar] [CrossRef]
- Koschak, A.; Bugianesi, R.M.; Mitterdorfer, J.; Kaczorowski, G.J.; Garcia, M.L.; Knaus, H.-G. Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom. J. Biol. Chem. 1998, 273, 2639–2644. [Google Scholar] [CrossRef]
- Gehlert, D.R.; Gackenheimer, S.L. Comparison of the distribution of binding sites for the potassium channel ligands [125I]apamin, [125I]charybdotoxin and [125I]iodoglyburide in the rat brain. Neuroscience 1993, 52, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Kuzmenkov, A.I.; Vassilevski, A.A. Labelled animal toxins as selective molecular markers of ion channels: Applications in neurobiology and beyond. Neurosci. Lett. 2018, 679, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, H. Fluorescent labeling techniques in biomolecules: A flashback. RSC Adv. 2012, 2, 7017–7029. [Google Scholar] [CrossRef]
- Pragl, B.; Koschak, A.; Trieb, M.; Obermair, G.; Kaufmann, W.A.; Gerster, U.; Blanc, E.; Hahn, C.; Prinz, H.; Schütz, G.; et al. Synthesis, characterization, and application of cy-dye- and alexa-dye-labeled hongotoxin1 analogues. The first high affinity fluorescence probes for voltage-gated K+ channels. Bioconjug. Chem. 2002, 13, 416–425. [Google Scholar] [CrossRef]
- Huang, M.; Zhu, X.; Yang, Y.; Tan, Y.; Luo, S.; Zhangsun, D. Fluorescently labeled α-conotoxin TxID, a new probe for α3β4 neuronal nicotinic acetylcholine receptors. Mar. Drugs 2022, 20, 511. [Google Scholar] [CrossRef]
- Hone, A.J.; Whiteaker, P.; Christensen, S.; Xiao, Y.; Meyer, E.L.; McIntosh, J.M. A novel fluorescent alpha-conotoxin for the study of α7 nicotinic acetylcholine receptors. J. Neurochem. 2009, 111, 80–89. [Google Scholar] [CrossRef]
- Hone, A.J.; Whiteaker, P.; Mohn, J.L.; Jacob, M.H.; McIntosh, J.M. Alexa Fluor 546-ArIB[V11L;V16A] is a potent ligand for selectively labeling α7 nicotinic acetylcholine receptors. J. Neurochem. 2010, 114, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Nevin, S.T.; Inserra, M.; Lewis, R.J.; Adams, D.J.; Alewood, P. On-resin strategy to label α-conotoxins: Cy5-RgIA, a potent α9α10 nicotinic acetylcholine receptor imaging probe. Aust. J. Chem. 2020, 73, 327–333. [Google Scholar] [CrossRef]
- Bernstein, G.M.; Mendonça, A.; Wadia, J.; Burnham, W.M.; Jones, O.T. Kindling induces a long-term enhancement in the density of N-type calcium channels in the rat hippocampus. Neuroscience 1999, 94, 1083–1095. [Google Scholar] [CrossRef]
- Jones, O.T.; Bernstein, G.M.; Jones, E.J.; Jugloff, D.G.; Law, M.; Wong, W.; Mills, L.R. N-Type calcium channels in the developing rat hippocampus: Subunit, complex, and regional expression. J. Neurosci. 1997, 17, 6152–6164. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, R.; Adler, E.M.; Charlton, M.P. Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron 1990, 5, 773–779. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Kuzmenkov, A.I.; Kudryavtsev, D.S.; Chudetskiy, I.S.; Shelukhina, I.V.; Barykin, E.P.; Ivanov, I.A.; Siniavin, A.E.; Ziganshin, R.H.; Baranov, M.S.; et al. Snake toxins labeled by green fluorescent protein or its synthetic chromophore are new probes for nicotinic acetylcholine receptors. Front. Mol. Biosci. 2021, 8, 753283. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, D.S.; Shelukhina, I.V.; Son, L.V.; Ojomoko, L.O.; Kryukova, E.V.; Lyukmanova, E.N.; Zhmak, M.N.; Dolgikh, D.A.; Ivanov, I.A.; Kasheverov, I.E.; et al. Neurotoxins from snake venoms and α-conotoxin ImI inhibit functionally active ionotropic γ-aminobutyric acid (GABA) receptors. J. Biol. Chem. 2015, 290, 22747–22758. [Google Scholar] [CrossRef] [PubMed]
- Brun, O.; Zoukimian, C.; Oliveira-Mendes, B.; Montnach, J.; Lauzier, B.; Ronjat, M.; Béroud, R.; Lesage, F.; Boturyn, D.; De Waard, M. Chemical synthesis of a functional fluorescent-tagged α-bungarotoxin. Toxins 2022, 14, 79. [Google Scholar] [CrossRef]
- Bychkov, M.L.; Kirichenko, A.V.; Shulepko, M.A.; Mikhaylova, I.N.; Kirpichnikov, M.P.; Lyukmanova, E.N. Mambalgin-2 inhibits growth, migration, and invasion of metastatic melanoma cells by targeting the channels containing an ASIC1a subunit whose up-regulation correlates with poor survival prognosis. Biomedicines 2021, 9, 1324. [Google Scholar] [CrossRef]
- Beeton, C.; Wulff, H.; Singh, S.; Botsko, S.; Crossley, G.; Gutman, G.A.; Cahalan, M.D.; Pennington, M.; Chandy, K.G. A novel fluorescent toxin to detect and investigate KV1.3 channel up-regulation in chronically activated T lymphocytes. J. Biol. Chem. 2003, 278, 9928–9937. [Google Scholar] [CrossRef]
- Bingham, J.-P.; Bian, S.; Tan, Z.-Y.; Takacs, Z.; Moczydlowski, E. Synthesis of a biotin derivative of iberiotoxin: Binding interactions with streptavidin and the BK Ca2+-activated K+ channel expressed in a human cell line. Bioconjug. Chem. 2006, 17, 689–699. [Google Scholar] [CrossRef]
- Kuzmenkov, A.I.; Nekrasova, O.V.; Kudryashova, K.S.; Peigneur, S.; Tytgat, J.; Stepanov, A.V.; Kirpichnikov, M.P.; Grishin, E.V.; Feofanov, A.V.; Vassilevski, A.A. Fluorescent protein-scorpion toxin chimera is a convenient molecular tool for studies of potassium channels. Sci. Rep. 2016, 6, 33314. [Google Scholar]
- Kittle, D.S.; Mamelak, A.; Parrish-Novak, J.E.; Hansen, S.; Patil, R.; Gangalum, P.R.; Ljubimova, J.; Black, K.L.; Butte, P. Fluorescence-guided tumor visualization using the tumor paint BLZ-100. Cureus 2014, 6, e210. [Google Scholar] [CrossRef]
- Kovar, J.L.; Curtis, E.; Othman, S.F.; Simpson, M.A.; Olive, D.M. Characterization of IRDye 800CW chlorotoxin as a targeting agent for brain tumors. Anal. Biochem. 2013, 440, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, M.; Gabikian, P.; Bahrami, S.-B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar] [CrossRef]
- Massensini, A.R.; Suckling, J.; Brammer, M.J.; Moraes-Santos, T.; Gomez, M.V.; Romano-Silva, M.A. Tracking sodium channels in live cells: Confocal imaging using fluorescently labeled toxins. J. Neurosci. Methods 2002, 116, 189–196. [Google Scholar] [CrossRef]
- Angelides, K.J.; Nutter, T.J. Preparation and characterization of fluorescent scorpion toxins from Leiurus quinquestriatus quinquestriatus as probes of the sodium channel of excitable cells. J. Biol. Chem. 1983, 258, 11948–11957. [Google Scholar] [CrossRef]
- Tilley, D.C.; Eum, K.S.; Fletcher-Taylor, S.; Austin, D.C.; Dupré, C.; Patrón, L.A.; Garcia, R.L.; Lam, K.; Yarov-Yarovoy, V.; Cohen, B.E.; et al. Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc. Natl. Acad. Sci. USA 2014, 111, E4789–E4796. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Mishra, S.; Nisal, R.; Majhi, S.; Shrivas, R.; Singh, Y.; Anusree, V.S.; Kalia, J. Site-specific fluorescent labeling of the cysteine-rich toxin, DkTx, for TRPV1 ion channel imaging and membrane binding ctudies. Bioconjug. Chem. 2022, 33, 1761–1770. [Google Scholar] [CrossRef]
- Montnach, J.; De Waard, S.; Nicolas, S.; Burel, S.; Osorio, N.; Zoukimian, C.; Mantegazza, M.; Boukaiba, R.; Béroud, R.; Partiseti, M.; et al. Fluorescent- and tagged-protoxin II peptides: Potent markers of the NaV1.7 channel pain target. Br. J. Pharmacol. 2021, 178, 2632–2650. [Google Scholar] [CrossRef]
- Gonzales, J.; Pirovano, G.; Chow, C.Y.; Demétrio De Souza França, P.; Carter, L.M.; Klint, J.K.; Guru, N.; Lewis, J.S.; King, G.F.; Reiner, T. Fluorescence labeling of a NaV1.7-targeted peptide for near-infrared nerve visualization. EJNMMI Res. 2020, 10, 49. [Google Scholar] [CrossRef]
- Hernández-Gil, J.; Chow, C.Y.; Chatras, H.; Demétrio De Souza França, P.; Samuels, Z.V.; Cornejo, M.; King, G.F.; Lewis, J.S.; Reiner, T.; Gonzales, J. Development and validation of nerve-targeted bacteriochlorin sensors. J. Am. Chem. Soc. 2023, 145, 14276–14287. [Google Scholar] [CrossRef]
- Adilbay, D.; Gonzales, J.; Zazhytska, M.; Demétrio De Souza França, P.; Roberts, S.; Viray, T.; Artschwager, R.; Patel, S.; Kodra, A.; Overdevest, J.B.; et al. Non-invasive diagnostic method to objectively measure olfaction and diagnose smell disorders by molecularly targeted fluorescent imaging agent. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gonzales, J.; Demétrio De Souza França, P.; Jiang, Y.; Pirovano, G.; Kossatz, S.; Guru, N.; Yarilin, D.; Agwa, A.J.; Schroeder, C.I.; Patel, S.G.; et al. Fluorescence imaging of peripheral nerves by a NaV1.7-targeted inhibitor cystine knot peptide. Bioconjug. Chem. 2019, 30, 2879–2888. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, J.; Hernández-Gil, J.; Wilson, T.C.; Adilbay, D.; Cornejo, M.; Demétrio De Souza França, P.; Guru, N.; Schroeder, C.I.; King, G.F.; Lewis, J.S.; et al. Bimodal imaging of mouse peripheral nerves with chlorin tracers. Mol. Pharm. 2021, 18, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Naumova, A.V.; Modo, M.; Moore, A.; Murry, C.E.; Frank, J.A. Clinical imaging in regenerative medicine. Nat. Biotechnol. 2014, 32, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Zelmer, A.; Ward, T.H. Noninvasive fluorescence imaging of small animals. J. Microsc. 2013, 252, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Bremer, C.; Ntziachristos, V.; Weissleder, R. Optical-based molecular imaging: Contrast agents and potential medical applications. Eur. Radiol. 2003, 13, 231–243. [Google Scholar] [CrossRef]
- Hong, G.; Antaris, A.L.; Dai, H. Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 2017, 1, 0010. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Tsien, R.Y. Fluorescence-guided surgery with live molecular navigation—A new cutting edge. Nat. Rev. Cancer 2013, 13, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Mieog, J.S.D.; Achterberg, F.B.; Zlitni, A.; Hutteman, M.; Burggraaf, J.; Swijnenburg, R.-J.; Gioux, S.; Vahrmeijer, A.L. Fundamentals and developments in fluorescence-guided cancer surgery. Nat. Rev. Clin. Oncol. 2022, 19, 9–22. [Google Scholar] [CrossRef]
- Wang, K.; Du, Y.; Zhang, Z.; He, K.; Cheng, Z.; Yin, L.; Dong, D.; Li, C.; Li, W.; Hu, Z.; et al. Fluorescence image-guided tumour surgery. Nat Rev Bioeng 2023, 1, 161–179. [Google Scholar] [CrossRef]
- Lauwerends, L.J.; van Driel, P.; Baatenburg de Jong, R.J.; Hardillo, J.A.U.; Koljenovic, S.; Puppels, G.; Mezzanotte, L.; Löwik, C.; Rosenthal, E.L.; Vahrmeijer, A.L.; et al. Real-time fluorescence imaging in intraoperative decision making for cancer surgery. Lancet Oncol. 2021, 22, e186–e195. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ahluwalia, M.; Baehring, J.; Brem, H.; Brem, S.; Butowski, N.; Campian, J.L.; Clark, S.W.; Fabiano, A.J.; et al. Central nervous system cancers, version 3.2020, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 1537–1570. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Lippens, G.; Najib, J.; Wodak, S.J.; Tartar, A. NMR sequential assignments and solution structure of chlorotoxin, a small scorpion toxin that blocks chloride channels. Biochemistry 1995, 34, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Dash, T.S.; Shafee, T.; Harvey, P.J.; Zhang, C.; Peigneur, S.; Deuis, J.R.; Vetter, I.; Tytgat, J.; Anderson, M.A.; Craik, D.J.; et al. A centipede toxin family defines an ancient class of Csαβ defensins. Structure 2019, 27, 315–326. [Google Scholar] [CrossRef] [PubMed]
- DeBin, J.A.; Maggio, J.E.; Strichartz, G.R. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am. J. Physiol. 1993, 264, C361–C369. [Google Scholar] [CrossRef]
- DeBin, J.A.; Strichartz, G.R. Chloride channel inhibition by the venom of the scorpion Leiurus quinquestriatus. Toxicon 1991, 29, 1403–1408. [Google Scholar] [CrossRef]
- Ullrich, N.; Sontheimer, H. Biophysical and pharmacological characterization of chloride currents in human astrocytoma cells. Am. J. Physiol. 1996, 270, C1511–C1521. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.; Bordey, A.; Gillespie, G.Y.; Sontheimer, H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience 1998, 83, 1161–1173. [Google Scholar] [PubMed]
- Olsen, M.L.; Schade, S.; Lyons, S.A.; Amaral, M.D.; Sontheimer, H. Expression of voltage-gated chloride channels in human glioma cells. J. Neurosci. 2003, 23, 5572–5582. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar] [PubMed]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, K.; Ratliff, J.; Johnson, E.W.; Dahlberg, W.; Asara, J.M.; Misra, P.; Frangioni, J.V.; Jacoby, D.B. Annexin A2 is a molecular target for TM601, a peptide with tumor-targeting and anti-angiogenic effects. J. Biol. Chem. 2010, 285, 4366–4374. [Google Scholar] [CrossRef] [PubMed]
- Dardevet, L.; Rani, D.; Aziz, T.A.; Bazin, I.; Sabatier, J.M.; Fadl, M.; Brambilla, E.; De Waard, M. Chlorotoxin: A helpful natural scorpion peptide to diagnose glioma and fight tumor invasion. Toxins 2015, 7, 1079–1101. [Google Scholar] [CrossRef] [PubMed]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Stroud, M.R.; Hansen, S.J.; Olson, J.M. In vivo bio-imaging using chlorotoxin-based conjugates. Curr. Pharm. Des. 2011, 17, 4362–4371. [Google Scholar] [CrossRef]
- Hockaday, D.C.; Shen, S.; Fiveash, J.; Raubitschek, A.; Colcher, D.; Liu, A.; Alvarez, V.; Mamelak, A.N. Imaging glioma extent with 131I-TM-601. J. Nucl. Med. 2005, 46, 580–586. [Google Scholar] [PubMed]
- Mamelak, A.N.; Rosenfeld, S.; Bucholz, R.; Raubitschek, A.; Nabors, L.B.; Fiveash, J.B.; Shen, S.; Khazaeli, M.B.; Colcher, D.; Liu, A.; et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J. Clin. Oncol. 2006, 24, 3644–3650. [Google Scholar] [CrossRef] [PubMed]
- Akcan, M.; Stroud, M.R.; Hansen, S.J.; Clark, R.J.; Daly, N.L.; Craik, D.J.; Olson, J.M. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J. Med. Chem. 2011, 54, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Troyan, S.L.; Kianzad, V.; Gibbs-Strauss, S.L.; Gioux, S.; Matsui, A.; Oketokoun, R.; Ngo, L.; Khamene, A.; Azar, F.; Frangioni, J.V. The FLARE intraoperative near-infrared fluorescence imaging system: A first-in-human clinical trial in breast cancer sentinel lymph node mapping. Ann. Surg. Oncol. 2009, 16, 2943–2952. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, E.; Chen, F.Y.; Flaumenhaft, R.; Graham, G.J.; Laurence, R.G.; Frangioni, J.V. Real-time assessment of cardiac perfusion, coronary angiography, and acute intravascular thrombi using dual-channel near-infrared fluorescence imaging. J. Thorac. Cardiovasc. Surg. 2009, 138, 133–140. [Google Scholar] [CrossRef] [PubMed]
- van der Vorst, J.R.; Schaafsma, B.E.; Hutteman, M.; Verbeek, F.P.; Liefers, G.J.; Hartgrink, H.H.; Smit, V.T.; Löwik, C.W.; van de Velde, C.J.; Frangioni, J.V.; et al. Near-infrared fluorescence-guided resection of colorectal liver metastases. Cancer 2013, 119, 3411–3418. [Google Scholar] [CrossRef] [PubMed]
- Butte, P.V.; Mamelak, A.; Parrish-Novak, J.; Drazin, D.; Shweikeh, F.; Gangalum, P.R.; Chesnokova, A.; Ljubimova, J.Y.; Black, K. Near-infrared imaging of brain tumors using the tumor paint BLZ-100 to achieve near-complete resection of brain tumors. Neurosurg. Focus 2014, 36, E1. [Google Scholar] [CrossRef] [PubMed]
- Parrish-Novak, J.; Byrnes-Blake, K.; Lalayeva, N.; Burleson, S.; Fidel, J.; Gilmore, R.; Gayheart-Walsten, P.; Bricker, G.A.; Crumb, W.J.; Tarlo, K.S.; et al. Nonclinical profile of BLZ-100, a tumor-targeting fluorescent imaging agent. Int. J. Toxicol. 2017, 36, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Patil, C.G.; Walker, D.G.; Miller, D.M.; Butte, P.; Morrison, B.; Kittle, D.S.; Hansen, S.J.; Nufer, K.L.; Byrnes-Blake, K.A.; Yamada, M.; et al. Phase 1 safety, pharmacokinetics, and fluorescence imaging study of Tozuleristide (BLZ-100) in adults with newly diagnosed or recurrent gliomas. Neurosurgery 2019, 85, E641–E649. [Google Scholar] [CrossRef]
- Dintzis, S.M.; Hansen, S.; Harrington, K.M.; Tan, L.C.; Miller, D.M.; Ishak, L.; Parrish-Novak, J.; Kittle, D.; Perry, J.; Gombotz, C.; et al. Real-time visualization of breast carcinoma in pathology specimens from patients receiving fluorescent tumor-marking agent tozuleristide. Arch. Pathol. Lab. Med. 2018, 143, 1076–1083. [Google Scholar] [CrossRef]
- Yamada, M.; Miller, D.M.; Lowe, M.; Rowe, C.; Wood, D.; Soyer, H.P.; Byrnes-Blake, K.; Parrish-Novak, J.; Ishak, L.; Olson, J.M.; et al. A first-in-human study of BLZ-100 (tozuleristide) demonstrates tolerability and safety in skin cancer patients. Contemp. Clin. Trials. Commun. 2021, 23, 100830. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Perlas, C.; Varese, M.; Guardiola, S.; García, J.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. From venoms to BBB-shuttles. MiniCTX3: A molecular vector derived from scorpion venom. Chem. Commun. 2018, 54, 12738–12741. [Google Scholar] [CrossRef] [PubMed]
- Maravilla, K.R.; Bowen, B.C. Imaging of the peripheral nervous system: Evaluation of peripheral neuropathy and plexopathy. AJNR Am. J. Neuroradiol. 1998, 19, 1011–1023. [Google Scholar]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Qasim, M.; Zafar, S.; Aziz, N.; Razzaq, A.; Hussain, R.; de Aguilar, J.G.; et al. Current status of therapeutic approaches against peripheral nerve injuries: A detailed story from injury to recovery. Int. J. Biol. Sci. 2020, 16, 116–134. [Google Scholar] [CrossRef]
- Lopes, B.; Sousa, P.; Alvites, R.; Branquinho, M.; Sousa, A.C.; Mendonça, C.; Atayde, L.M.; Luís, A.L.; Varejão, A.S.P.; Maurício, A.C. Peripheral nerve injury treatments and advances: One health perspective. Int. J. Mol. Sci. 2022, 23, 918. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Rivlin, M.; Graham, J.G.; Beredjiklian, P.K. Peripheral nerve injury, scarring, and recovery. Connect. Tissue Res. 2019, 60, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Crombie, I.K.; Davies, H.T.; Macrae, W.A. Cut and thrust: Antecedent surgery and trauma among patients attending a chronic pain clinic. Pain 1998, 76, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.F.; Malahias, M.; Hindocha, S.; Khan, W.S. Peripheral nerve injury: Principles for repair and regeneration. Open Orthop. J. 2014, 8, 199–203. [Google Scholar]
- Kretschmer, T.; Antoniadis, G.; Braun, V.; Rath, S.A.; Richter, H.P. Evaluation of iatrogenic lesions in 722 surgically treated cases of peripheral nerve trauma. J. Neurosurg. 2001, 94, 905–912. [Google Scholar] [CrossRef]
- Joliat, G.R.; Guarnero, V.; Demartines, N.; Schweizer, V.; Matter, M. Recurrent laryngeal nerve injury after thyroid and parathyroid surgery: Incidence and postoperative evolution assessment. Medicine 2017, 96, e6674. [Google Scholar] [CrossRef]
- Walsh, P.C.; Donker, P.J. Impotence following radical prostatectomy: Insight into etiology and prevention. J. Urol. 2017, 197, S165–S170. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.A.; Ariyan, S.; Restifo, R.; Sasaki, C.T. Use of the operating microscope and loupes for head and neck free microvascular tissue transfer: A retrospective comparison. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.R. The basics of electromyography. J. Neurol. Neurosurg. Psychiatry 2005, 76, ii32–ii35. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Wilder-Smith, E.; Bendszus, M. Imaging of the peripheral nervous system. Handb. Clin. Neurol. 2013, 115, 137–153. [Google Scholar] [PubMed]
- Strakowski, J.A. Ultrasound-Guided Peripheral Nerve Procedures. Phys. Med. Rehabil. Clin. N. Am. 2016, 27, 687–715. [Google Scholar] [CrossRef]
- Stone, J.J.; Graffeo, C.S.; de Ruiter, G.C.W.; Rock, M.G.; Spinner, R.J. Intraoperative intravenous fluorescein as an adjunct during surgery for peroneal intraneural ganglion cysts. Acta Neurochir. (Wien.) 2018, 160, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, I.G.; Acerbi, F.; Falco, J.; Devigili, G.; Rinaldo, S.; Messina, G.; Prada, F.; D’Ammando, A.; Nazzi, V. Fluorescein-guided removal of peripheral nerve sheath tumors: A preliminary analysis of 20 cases. J. Neurosurg. 2019, 134, 260–269. [Google Scholar] [CrossRef]
- Chen, S.-C.; Wang, M.-C.; Wang, W.-H.; Lee, C.-C.; Yang, T.-F.; Lin, C.-F.; Wang, J.-T.; Liao, C.-H.; Chang, C.-C.; Chen, M.-H.; et al. Fluorescence-assisted visualization of facial nerve during mastoidectomy: A novel technique for preventing iatrogenic facial paralysis. Auris Nasus Larynx 2015, 42, 113–118. [Google Scholar] [CrossRef]
- Mangano, M.S.; De Gobbi, A.; Beniamin, F.; Lamon, C.; Ciaccia, M.; Maccatrozzo, L. Robot-assisted nerve-sparing radical prostatectomy using near-infrared fluorescence technology and indocyanine green: Initial experience. Urologia 2018, 85, 29–31. [Google Scholar] [CrossRef]
- He, K.; Li, P.; Zhang, Z.; Liu, J.; Liu, P.; Gong, S.; Chi, C.; Liu, P.; Chen, C.; Tian, J. Intraoperative near-infrared fluorescence imaging can identify pelvic nerves in patients with cervical cancer in real time during radical hysterectomy. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 2929–2937. [Google Scholar] [CrossRef]
- Wagner, O.J.; Louie, B.E.; Vallières, E.; Aye, R.W.; Farivar, A.S. Near-infrared fluorescence imaging can help identify the contralateral phrenic nerve during robotic thymectomy. Ann. Thorac. Surg. 2012, 94, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.M.; Cole, D.; Tipirneni, K.E.; Bland, K.I.; Udayakumar, N.; Kasten, B.B.; Bevans, S.L.; McGrew, B.M.; Kain, J.J.; Nguyen, Q.T.; et al. Fluorescence imaging of nerves during surgery. Ann. Surg. 2019, 270, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Enquist, L.W.; Card, J.P. Recent advances in the use of neurotropic viruses for circuit analysis. Curr. Opin. Neurobiol. 2003, 13, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, S.L.; Xie, Y.; Goodwill, H.L.; Nasr, K.A.; Ashitate, Y.; Madigan, V.J.; Siclovan, T.M.; Zavodszky, M.; Tan Hehir, C.A.; Frangioni, J.V. Structure-activity relationship of nerve-highlighting fluorophores. PLoS ONE 2013, 8, e73493. [Google Scholar] [CrossRef]
- Park, M.H.; Hyun, H.; Ashitate, Y.; Wada, H.; Park, G.; Lee, J.H.; Njiojob, C.; Henary, M.; Frangioni, J.V.; Choi, H.S. Prototype nerve-specific near-infrared fluorophores. Theranostics 2014, 4, 823–833. [Google Scholar] [CrossRef]
- Wang, C.; Wu, C.; Zhu, J.; Miller, R.H.; Wang, Y. Design, synthesis, and evaluation of coumarin-based molecular probes for imaging of myelination. J. Med. Chem. 2011, 54, 2331–2340. [Google Scholar] [CrossRef]
- Wang, L.G.; Barth, C.W.; Kitts, C.H.; Mebrat, M.D.; Montaño, A.R.; House, B.J.; McCoy, M.E.; Antaris, A.L.; Galvis, S.N.; McDowall, I.; et al. Near-infrared nerve-binding fluorophores for buried nerve tissue imaging. Sci. Transl. Med. 2020, 12, eaay0712. [Google Scholar] [CrossRef]
- Whitney, M.A.; Crisp, J.L.; Nguyen, L.T.; Friedman, B.; Gross, L.A.; Steinbach, P.; Tsien, R.Y.; Nguyen, Q.T. Fluorescent peptides highlight peripheral nerves during surgery in mice. Nat. Biotechnol. 2011, 29, 352–356. [Google Scholar] [CrossRef]
- Hussain, T.; Mastrodimos, M.B.; Raju, S.C.; Glasgow, H.L.; Whitney, M.; Friedman, B.; Moore, J.D.; Kleinfeld, D.; Steinbach, P.; Messer, K.; et al. Fluorescently labeled peptide increases identification of degenerated facial nerve branches during surgery and improves functional outcome. PLoS ONE 2015, 10, e0119600. [Google Scholar]
- Hingorani, D.V.; Whitney, M.A.; Friedman, B.; Kwon, J.K.; Crisp, J.L.; Xiong, Q.; Gross, L.; Kane, C.J.; Tsien, R.Y.; Nguyen, Q.T. Nerve-targeted probes for fluorescence-guided intraoperative imaging. Theranostics 2018, 8, 4226–4237. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Huxley, A.F. The components of membrane conductance in the giant axon of Loligo. J. Physiol. 1952, 116, 473–496. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Frézel, N.; Dib-Hajj, S.D.; Waxman, S.G. Expression of NaV1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol. Pain 2012, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gu, J.; Li, Y.Q.; Tao, Y.X. Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol. Pain 2011, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Yang, Y.; Black, J.A.; Waxman, S.G. The NaV1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 2013, 14, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Vasylyev, D.V.; Han, C.; Zhao, P.; Dib-Hajj, S.; Waxman, S.G. Dynamic-clamp analysis of wild-type human NaV1.7 and erythromelalgia mutant channel L858H. J. Neurophysiol. 2014, 111, 1429–1443. [Google Scholar] [CrossRef]
- Huang, X.; Jin, X.; Huang, G.; Huang, J.; Wu, T.; Li, Z.; Chen, J.; Kong, F.; Pan, X.; Yan, N. Structural basis for high-voltage activation and subtype-specific inhibition of human NaV1.8. Proc. Natl. Acad. Sci. USA 2022, 119, e2208211119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Shi, H.; Tonggu, L.; Gamal El-Din, T.M.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the cardiac sodium channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef]
- Li, X.; Xu, F.; Xu, H.; Zhang, S.; Gao, Y.; Zhang, H.; Dong, Y.; Zheng, Y.; Yang, B.; Sun, J.; et al. Structural basis for modulation of human NaV1.3 by clinical drug and selective antagonist. Nat. Commun. 2022, 13, 1286. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, T.; Huang, B.; Zhou, F.; Peng, C.; Li, X.; Qiu, Y.; Yang, B.; Zhao, Y.; Huang, Z.; et al. Structure of human NaV1.6 channel reveals Na+ selectivity and pore blockade by 4,9-anhydro-tetrodotoxin. Nat. Commun. 2023, 14, 1030. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Huang, X.; Huang, G.; Gao, S.; Shen, H.; Liu, L.; Lei, J.; Yan, N. Molecular basis for pore blockade of human Na+ channel NaV1.2 by the μ-conotoxin KIIIA. Science 2019, 363, 1309–1313. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Jin, X.; Zhao, Y.; Huang, G.; Huang, X.; Shen, Z.; Cao, Y.; Dong, M.; Lei, J.; et al. Comparative structural analysis of human NaV1.1 and NaV1.5 reveals mutational hotspots for sodium channelopathies. Proc. Natl. Acad. Sci. USA 2021, 118, e2100066118. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel NaV1.4 in complex with β1. Science 2018, 362, eaau2486. [Google Scholar] [CrossRef]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Vetter, I. No Gain, No Pain: NaV1.7 as an analgesic target. ACS Chem. Neurosci. 2014, 5, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Salvage, S.C.; Rahman, T.; Eagles, D.A.; Rees, J.S.; King, G.F.; Huang, C.L.; Jackson, A.P. The β3-subunit modulates the effect of venom peptides ProTx-II and OD1 on NaV1.7 gating. J. Cell. Physiol. 2023, 238, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Eagles, D.A.; Chow, C.Y.; King, G.F. Fifteen years of NaV 1.7 channels as an analgesic target: Why has excellent in vitro pharmacology not translated into in vivo analgesic efficacy? Br. J. Pharmacol. 2022, 179, 3592–3611. [Google Scholar] [CrossRef]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Wisedchaisri, G.; Tonggu, L.; Gamal El-Din, T.M.; McCord, E.; Zheng, N.; Catterall, W.A. Structural basis for high-affinity trapping of the NaV1.7 channel in its resting state by tarantula toxin. Mol. Cell 2021, 81, 38–48.e4. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef]
- Tian, C.; Wang, K.; Ke, W.; Guo, H.; Shu, Y. Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front. Cell Neurosci. 2014, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S.; Kenny, C.J.; Ganesh, V.; Jang, A.; Borges-Monroy, R.; Partlow, J.N.; Hill, R.S.; Shin, T.; Chen, A.Y.; Doan, R.N.; et al. Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron 2018, 99, 905–913.e7. [Google Scholar] [CrossRef]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium channel NaV1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Tian, C.; Li, T.; Yang, M.; Hou, H.; Shu, Y. Distinct contributions of NaV1.6 and NaV1.2 in action potential initiation and backpropagation. Nat. Neurosci. 2009, 12, 996–1002. [Google Scholar] [CrossRef]
- Jiang, Y.; Castro, J.; Blomster, L.V.; Agwa, A.J.; Maddern, J.; Schober, G.; Herzig, V.; Chow, C.Y.; Cardoso, F.C.; Demétrio De Souza França, P.; et al. Pharmacological inhibition of the voltage-gated sodium channel NaV1.7 alleviates chronic visceral pain in a rodent model of irritable bowel syndrome. ACS Pharmacol. Transl. Sci. 2021, 4, 1362–1378. [Google Scholar] [CrossRef]
- Gonzales, J.; Adilbay, D.; Demétrio De Souza França, P.; Artschwager, R.; Chow, C.Y.; Viray, T.; Delissa, S.J.; Jiang, Y.; Patel, S.G.; Ganly, I.; et al. NaV1.7 targeted fluorescence imaging agents for nerve identification during intraoperative procedures. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ahn, H.S.; Black, J.A.; Zhao, P.; Tyrrell, L.; Waxman, S.G.; Dib-Hajj, S.D. NaV1.7 is the predominant sodium channel in rodent olfactory sensory neurons. Mol. Pain 2011, 7, 32. [Google Scholar] [CrossRef]
- Weiss, J.; Pyrski, M.; Jacobi, E.; Bufe, B.; Willnecker, V.; Schick, B.; Zizzari, P.; Gossage, S.J.; Greer, C.A.; Leinders-Zufall, T.; et al. Loss-of-function mutations in sodium channel NaV1.7 cause anosmia. Nature 2011, 472, 186–190. [Google Scholar] [CrossRef]
- Rupasinghe, D.B.; Knapp, O.; Blomster, L.V.; Schmid, A.B.; Adams, D.J.; King, G.F.; Ruitenberg, M.J. Localization of Nav 1.7 in the normal and injured rodent olfactory system indicates a critical role in olfaction, pheromone sensing and immune function. Channels 2012, 6, 103–110. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Lei, Z.; Zhang, F. Recent advances in intraoperative nerve bioimaging: Fluorescence-guided surgery for nerve preservation. Small Struct. 2020, 1, 2000036. [Google Scholar] [CrossRef]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and treatment of multiple sclerosis: A review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Kepe, V.; Huang, S.C.; Small, G.W.; Satyamurthy, N.; Barrio, J.R. Visualizing pathology deposits in the living brain of patients with Alzheimer’s disease. Methods Enzymol. 2006, 412, 144–160. [Google Scholar]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18 kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends. Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Lopresti, B.J.; Wang, G.; Hamilton, R.L.; Mathis, C.A.; Klunk, W.E.; Apte, U.M.; Wiley, C.A. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol. Aging 2009, 30, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Newcombe, J.; Gunn, R.N.; Cagnin, A.; Turkheimer, F.; Heppner, F.; Price, G.; Wegner, F.; Giovannoni, G.; Miller, D.H.; et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: Quantitative in vivo imaging of microglia as a measure of disease activity. Brain 2000, 123, 2321–2337. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Foster, V.S.; Rash, L.D.; King, G.F.; Rank, M.M. Acid-sensing ion channels: Expression and function in resident and infiltrating immune cells in the central nervous dystem. Front. Cell. Neurosci. 2021, 15, 738043. [Google Scholar] [CrossRef]
- Redd, M.A.; Scheuer, S.E.; Saez, N.J.; Yoshikawa, Y.; Chiu, H.S.; Gao, L.; Hicks, M.; Villanueva, J.E.; Joshi, Y.; Chow, C.Y.; et al. Therapeutic inhibition of acid-sensing ion channel 1a recovers heart function after ischemia-reperfusion injury. Circulation 2021, 144, 947–960. [Google Scholar] [CrossRef]
- Chassagnon, I.R.; McCarthy, C.A.; Chin, Y.K.; Pineda, S.S.; Keramidas, A.; Mobli, M.; Pham, V.; De Silva, T.M.; Lynch, J.W.; Widdop, R.E.; et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2017, 114, 3750–3755. [Google Scholar] [CrossRef]
- Redd, M.A.; Yoshikawa, Y.; Khan, N.; Waqar, M.; Saez, N.J.; Russell, J.S.; Chiu, H.S.; Er, S.Y.; Mardon, K.; Fraser, J.F.; et al. Blockade of acid-sensing ion channel 1a is equipotent to sodium-hydrogen exchange inhibition in reducing cardiac injury in rodent models of myocardial infarction. Eur. Heart J. 2023, 45, 1571–1574. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Source | Peptide | Molecular Target | Fluorophore | Reference |
|---|---|---|---|---|
| Cone snail | α-conotoxin TxID | α3β4 nAChR | 5-TAMRA | [27] |
| α-conotoxin ArIB | α7 nAChR | Cy3, Alexa Fluor 546 | [28,29] | |
| α-conotoxin RgIA | α9α10 nAChR | Cy5 | [30] | |
| ω-CgTx | CaV2.2 | TexasRed, fluorescein, Cy3 | [31,32,33] | |
| Snake | α-cobratoxin | α7 and α9/α10 nAChR | Enhanced GFP | [34] |
| α1β3γ2 GABAA receptor | Alexa Fluor 546 | [35] | ||
| α-bungarotoxin | α7 nAChR | Cy5 | [36] | |
| Mambalgin-2 | ASIC1a | CF647 | [37] | |
| Scorpion | ShK | KV1.3 | F6CA | [38] |
| HgTx1 | KV family channels | Cy3, Cy5, Alexa Fluor family | [26] | |
| IbTx | KCa1.1 | Alexa Fluor 488 | [39] | |
| OSK1 | KV1.1–1.3 | Enhanced GFP | [40] | |
| AgTx2 | KV1.1–1.3, 1.6 | RFP | [40] | |
| CTX | Cl– channels | Cy5.5, IR800, ICG | [41,42,43] | |
| TsTx | NaV channels | Alexa Fluor 488 | [44] | |
| TiTx-γ | NaV channels | Alexa Fluor 568 | [44] | |
| LqqV | NaV channels | DACA | [45] | |
| Spider | GsTx | KV2.1 | DyLight 550 | [46] |
| DkTx | TRPV1 | Fluorescein | [47] | |
| ProTx-II | NaV1.2, 1.5, 1.7, 1.8 | ATTO488 | [48] | |
| Hs1a | NaV1.1–1.3, 1.6, 1.7 | Cy7.5, bacteriochlorin | [49,50] | |
| Tsp1a | NaV1.7 | BODIPY, chlorin, IR800 | [49,51,52,53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chow, C.Y.; King, G.F. Shining a Light on Venom-Peptide Receptors: Venom Peptides as Targeted Agents for In Vivo Molecular Imaging. Toxins 2024, 16, 307. https://doi.org/10.3390/toxins16070307
Chow CY, King GF. Shining a Light on Venom-Peptide Receptors: Venom Peptides as Targeted Agents for In Vivo Molecular Imaging. Toxins. 2024; 16(7):307. https://doi.org/10.3390/toxins16070307
Chicago/Turabian StyleChow, Chun Yuen, and Glenn F. King. 2024. "Shining a Light on Venom-Peptide Receptors: Venom Peptides as Targeted Agents for In Vivo Molecular Imaging" Toxins 16, no. 7: 307. https://doi.org/10.3390/toxins16070307
APA StyleChow, C. Y., & King, G. F. (2024). Shining a Light on Venom-Peptide Receptors: Venom Peptides as Targeted Agents for In Vivo Molecular Imaging. Toxins, 16(7), 307. https://doi.org/10.3390/toxins16070307