DNA Repair and Mutagenesis of ADP-Ribosylated DNA by Pierisin

Abstract
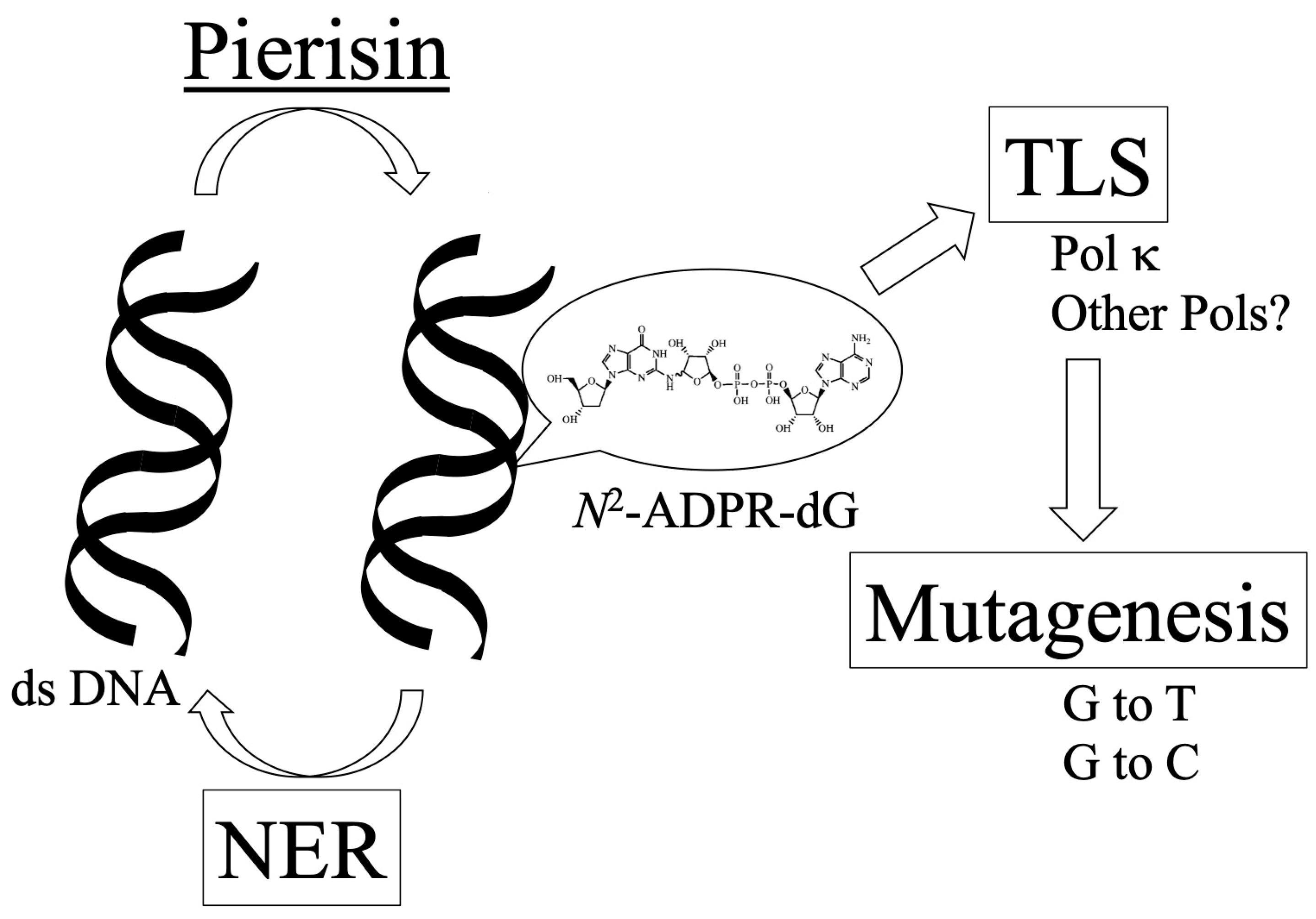
1. Introduction
2. Nucleotide Excision Repair of ADP-Ribosylated DNA by Pierisin-1
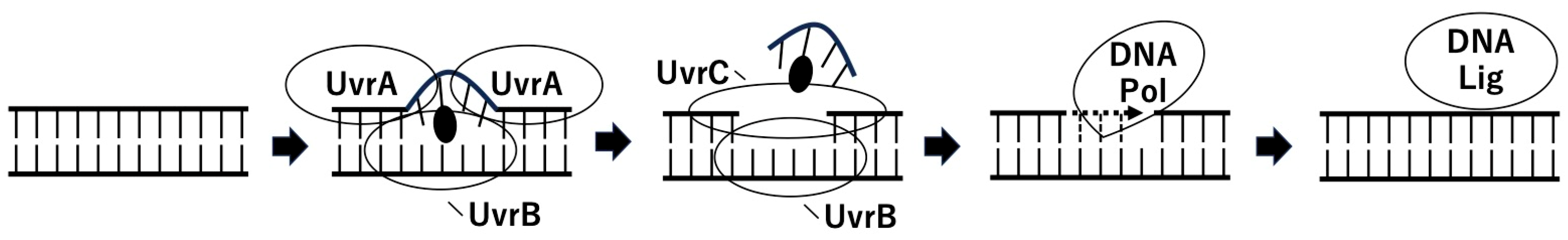

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHO Cell Line | AA8 (Wild) | UV5 (XPD−) | UV20 (ERCC1−) | UV41 (XPF−) | UV135 (XPG−) |
|---|---|---|---|---|---|
| IC50 [ng/mL] | 650 | 230 | 190 | 260 | 240 |
3. Translesion DNA Synthesis across Mono-ADP-Ribosylated Deoxyguanosine by Y-Family DNA Polymerases
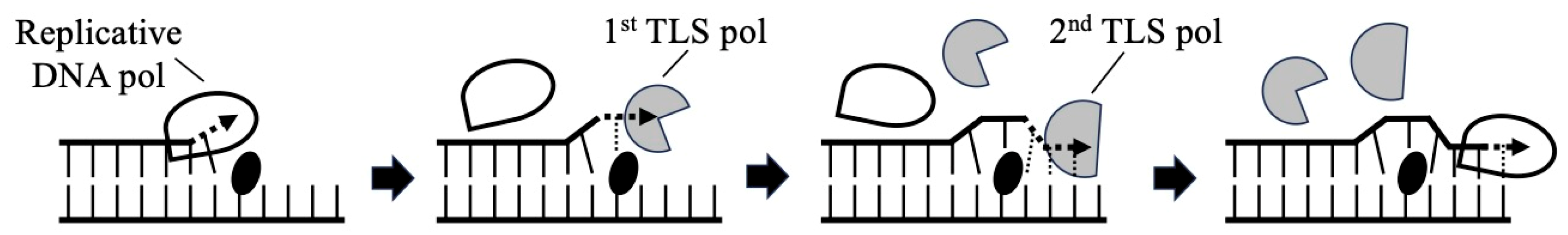
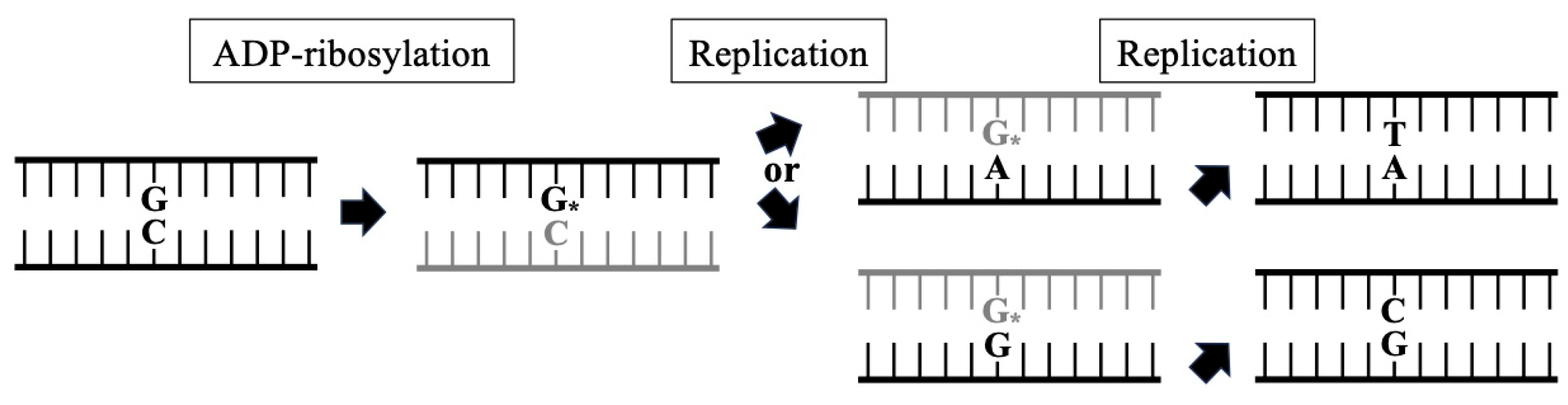
4. Mutagenesis by Mono-ADP-Ribosylated Deoxyguanosine in Mammalian Cell Lines
5. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoshida, T.; Tsuge, H. N2 atom recognition mechanism in pierisin family DNA-targeting, guanine-specific ADP-ribosyltransferase ScARP. J. Biol. Chem. 2018, 293, 13768–13774. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Wakabayashi, K.; Masutani, M.; Koiwai, K.; Watanabe, M.; Yamazaki, S.; Kono, T.; Miki, K.; Sugimura, T. Presence in Pieris rapae of cytotoxic activity against human carcinoma cells. Jpn. J. Cancer Res. 1996, 87, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kono, T.; Koyama, K.; Sugimura, T.; Wakabayashi, K. Purification of pierisin, an inducer of apoptosis in human gastric carcinoma cells, from cabbage butterfly, Pieris rapae. Jpn. J. Cancer Res. 1998, 89, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Takamura-Enya, T.; Watanabe, M.; Totsuka, Y.; Kanazawa, T.; Matsu-shima-Hibiya, Y.; Koyama, K.; Sugimura, T.; Wakabayashi, K. Mono(ADP-ribosyl)ation of 2′-deoxyguanosine residue in DNA by an apoptosis-inducing protein, pierisin-1, from cabbage butterfly. Proc. Natl. Acad. Sci. USA 2001, 98, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Takamura-Enya, T.; Watanabe, M.; Koyama, K.; Sugimura, T.; Wakabayashi, K. Mono(ADP-ribosyl)ation of the N2 amino groups of guanine residues in DNA by pierisin-2, from the cabbage butterfly, Pieris brassicae. Biochem. Biophys. Res. Commun. 2004, 323, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Nakaguchi, A.; Horiuchi, Y.; Yamamoto, M.; Totsuka, Y.; Wakabayashi, K. Pierisin, cytotoxic and apoptosis-inducing DNA ADP-ribosylating protein in cabbage butterfly. Toxins 2024, 16, 270. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Thapa, M.J.; Fabros, R.M.; Alasmar, S.; Chan, K. Analyses of mutational patterns induced by formaldehyde and acetaldehyde reveal similarity to a common mutational signature. G3 2022, 12, jkac238. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef]
- Dip, R.; Camenisch, U.; Naegeli, H. Mechanisms of DNA damage recognition and strand discrimination in human nucleotide excision repair. DNA Repair 2004, 3, 1409–1423. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, T.M.; Geacintov, N.E.; Van Houten, B. Interaction of the UvrABC nuclease system with a DNA duplex containing a single stereoisomer of dG-(+)- or dG-(-)-anti-BPDE. Biochemistry 1995, 34, 13582–13593. [Google Scholar] [CrossRef]
- Zou, Y.; Van Houten, B. Strand opening by the UvrA2B complex allows dynamic recognition of DNA damage. EMBO J. 1999, 18, 4889–4901. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Matsukawa, K.; Kuraoka, I.; Takamura-Enya, T.; Totsuka, Y.; Matsumoto, Y.; Watanabe, M.; Zou, Y.; Tanaka, K.; Sugimura, T.; et al. Molecular evidence of involvement of nucleotide excision repair (NER) system in repair of the mono ADP-ribosylated DNA adduct produced by pierisin-1, an apoptosis-inducing protein from cabbage butterfly. Chem. Res. Toxicol. 2007, 20, 694–700. [Google Scholar] [CrossRef]
- Shiotani, B.; Watanabe, M.; Totsuka, Y.; Sugimura, T.; Wakabayashi, K. Involvement of nucleotide excision repair (NER) system in repair of mono ADP-ribosylated dG adducts produced by pierisin-1, a cytotoxic protein from cabbage butterfly. Mutat. Res. 2005, 572, 150–155. [Google Scholar] [CrossRef]
- De Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef]
- Plosky, B.S.; Woodgate, R. Switching from high-fidelity replicases to low-fidelity lesion-bypass polymerases. Curr. Opin. Genet. Dev. 2004, 14, 113–119. [Google Scholar] [CrossRef]
- Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002, 71, 17–50. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Morrison, A.; Christensen, R.B.; Alley, J.; Beck, A.K.; Bernstine, E.G.; Lemontt, J.F.; Lawrence, C.W. REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J. Bacteriol. 1989, 171, 5659–5667. [Google Scholar] [CrossRef]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef]
- Johnson, R.E.; Washington, M.T.; Haracska, L.; Prakash, S.; Prakash, L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 2000, 406, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Woodgate, R. Evolution of the two-step model for UV-mutagenesis. Mutat. Res. 2001, 485, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (Xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Araki, M.; Yamada, A.; Kusumoto, R.; Nogimori, T.; Maekawa, T.; Iwai, S.; Hanaoka, F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999, 18, 3491–3501. [Google Scholar] [CrossRef]
- Johnson, R.E.; Kondratick, C.M.; Prakash, S.; Prakash, L. hRAD30 mutations in the variant form of Xeroderma pigmentosum. Science 1999, 285, 263–265. [Google Scholar] [CrossRef]
- Trincao, J.; Johnson, R.E.; Escalante, C.R.; Prakash, S.; Prakash, L.; Aggarwal, A.K. Structure of the catalytic core of S. cerevisiae DNA polymerase η: Implications for translesion DNA synthesis. Mol. Cell 2001, 8, 417–426. [Google Scholar] [CrossRef]
- Zhou, B.L.; Pata, J.D.; Steitz, T.A. Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol. Cell 2001, 8, 427–437. [Google Scholar] [CrossRef]
- Ling, H.; Boudsocq, F.; Woodgate, R.; Yang, W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell 2001, 107, 91–102. [Google Scholar] [CrossRef]
- Silvian, L.F.; Toth, E.A.; Pham, P.; Goodman, M.F.; Ellenberger, T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat. Struct. Biol. 2001, 8, 984–989. [Google Scholar] [CrossRef]
- Ling, H.; Boudsocq, F.; Plosky, B.S.; Woodgate, R.; Yang, W. Replication of a cis-syn thymine dimer at atomic resolution. Nature 2003, 424, 1083–1087. [Google Scholar] [CrossRef]
- Jansen, J.G.; de Wind, N. Biological functions of translesion synthesis proteins in vertebrates. DNA Repair 2003, 2, 1075–1085. [Google Scholar] [CrossRef]
- Kawanishi, M.; Matsukawa, K.; Ohashi, E.; Takamura, T.; Totsuka, Y.; Watanabe, M.; Sugimura, T.; Wakabayashi, K.; Hanaoka, F.; Ohmori, H.; et al. Translesion DNA synthesis across mono ADP-ribosylated deoxyguanosine by Y-family DNA polymerases. In New Developments in Mutation Research; Valon, C., Ed.; Nova Science Publishers: New York, NY, USA, 2006; pp. 133–148. [Google Scholar]
- Meehan, T.; Straub, K. Double stranded DNA stereo-selectively binds benzo[a]pyrene diol epoxides. Nature 1979, 277, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Hilton, B.D.; Roman, J.M.; Dipple, A. DNA adducts from carcinogenic and noncarcinogenic enantiomers of benzo[a]pyrene dihydrodiol epoxide. Chem. Res. Toxicol. 1989, 2, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Dasaradhi, L.; Shibutani, S. Identification of tamoxifen-DNA adducts formed by R-sulfate tamoxifen and R-acetoxytamoxifen. Chem. Res. Toxicol. 1997, 10, 189–196. [Google Scholar] [CrossRef]
- Osborne, M.R.; Hewer, A.; Hardcastle, I.R.; Carmichael, P.L.; Phillips, D.H. Identification of the major tamoxifen-deoxyguanosine adduct formed in the liver DNA of rats treated with tamoxifen. Cancer Res. 1996, 56, 66–71. [Google Scholar] [PubMed]
- Yang, J.L.; Chen, R.H.; Maher, V.M.; McCormick, J.J. Kinds and location of mutations induced by (+/-)-7 beta, 8 alpha-dihydroxy-9 alpha, 10 alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]-pyrene in the coding region of the hypoxanthine (guanine) phosphoribosyltransferase gene in diploid human fibroblasts. Carcinogenesis 1991, 12, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Maher, V.M.; McCormick, J.J. Kinds of mutations formed when a shuttle vector containing adducts of (+/-)-7 beta, 8 alpha-dihydroxy-9 alpha, 10 alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]-pyrene replicates in human cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3787–3791. [Google Scholar] [CrossRef]
- Mazur, M.; Glickman, B.W. Sequence specificity of mutations induced by benzo[a]pyrene-7,8-diol-9,10-epoxide at endogenous aprt gene in CHO cells. Somat. Cell. Mol. Genet. 1988, 14, 393–400. [Google Scholar] [CrossRef]
- Terashima, I.; Suzuki, N.; Shibutani, S. Mutagenic potential of R-(N2-deoxyguanosinyl)tamoxifen lesions, the major DNA adducts detected in endometrial tissues of patients treated with tamoxifen. Cancer Res. 1999, 59, 2091–2095. [Google Scholar]
- Totsuka, Y.; Kawanishi, M.; Nishigaki, R.; Matsukawa, K.; Yagi, T.; Takamura-Enya, T.; Watanabe, M.; Sugimura, T.; Wakabayashi, K. Analysis of HPRT and supF mutations caused by pierisin-1, a guanine specific ADP-ribosylating toxin derived from the cabbage butterfly. Chem. Res. Toxicol. 2003, 16, 945–952. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Seidman, M.M. Use of supF, an Escherichia coli tyrosine suppressor transfer-RNA gene as a mutagenic target in shuttle-vector plasmids. Mutat. Res. 1989, 220, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Vrieling, H.; Zeeland, A.A.V.; Jenssen, D. Spectrum of spontaneously occurring mutations in the hprt gene of V79 Chinese hamster cells. J. Mol. Biol. 1992, 223, 627–635. [Google Scholar] [CrossRef]
- Lewis, P.D.; Harvey, J.S.; Waters, E.M.; Skibinski, D.O.F.; Parry, J.M. Spontaneous mutation spectra in supF: Comparative analysis of mammalian cell line base substitution spectra. Mutagenesis 2001, 16, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Wang, X.; Miyata, Y.; Saeki, K.; Kohara, A.; Kawazoe, Y.; Hayashi, M.; Sofuni, T. Hepatocarcinogen quinoline induces G:C to C:G transversions in cII gene in the liver of lambda/lacZ transgenic mice (MutaMouse). Mutat. Res. 2000, 456, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.; Hunter, W.N.; Kneale, G.; Kennard, O. Molecular structure of the G‚A base pair in DNA and its implication for the mechanism of transversion mutations. Proc. Natl. Acad. Sci. USA 1986, 83, 2402–2406. [Google Scholar] [CrossRef]
- Norman, D.; Abuaf, P.; Hingerty, B.E.; Live, D.; Grunberger, D.; Broyde, S.; Patel, D.J. NMR and computational characterization of the N-(deoxyguanosine-8-yl)aminofluorene adduct [AF(G)] opposite adenosine in DNA: (AF)G[syn]‚A[anti] pair formation and its pH dependence. Biochemistry 1989, 28, 7462–7467. [Google Scholar] [CrossRef]
- Kadlubar, F.F. A transversion mutation hypothesis for chemical carcinogenesis by N2-substitution of guanine in DNA. Chem.-Biol. Interact. 1980, 31, 255–263. [Google Scholar] [CrossRef]
- Goodman, M.F.; Woodgate, R. Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Guo, D.; Rechkoblit, O.; Geacintov, N.E.; Wang, Z. Two-step error-prone bypass of the (+)- and (-)-trans-anti-BPDE-N2-dG adducts by human DNA polymerases η and κ. Mutat. Res. 2002, 510, 23–35. [Google Scholar] [CrossRef]
- Chiapperino, D.; Kroth, H.; Kramarczuk, I.H.; Sayer, J.M.; Masutani, C.; Hanaoka, F.; Jerina, D.M.; Cheh, A.M. Preferential misincorporation of purine nucleotides by human DNA polymerase η opposite benzo[a]pyrene 7,8-diol 9,10-epoxide deoxyguanosine adducts. J. Biol. Chem. 2002, 277, 11765–11771. [Google Scholar] [CrossRef]
- Rechkoblit, O.; Zhang, Y.; Guo, D.; Wang, Z.; Amin, S.; Krzeminsky, J.; Louneva, N.; Geacintov, N.E. Trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 2002, 277, 30488–30494. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.T.; Johnson, R.E.; Prakash, L.; Prakash, S. Human DINB1-encoded DNA polymerase κ is a promiscuous extender of mispaired primer termini. Proc. Natl. Acad. Sci. USA 2002, 99, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Weixler, L.; Scharinger, K.; Momoh, J.; Luscher, B.; Feijs, K.L.H.; Zaja, R. ADP-ribosylation of RNA and DNA: From in vitro characterization to in vivo function. Nucleic Acids Res. 2021, 49, 3634–3650. [Google Scholar] [CrossRef]
- de Moraes, M.H.; Hsu, F.; Huang, D.; Bosch, D.E.; Zeng, J.; Radey, M.C.; Simon, N.; Ledvina, H.E.; Frick, J.P.; Wiggins, P.A.; et al. An interbacterial DNA deaminase toxin directly mutagenizes surviving target populations. eLife 2021, 10, e62967. [Google Scholar] [CrossRef]
- De Jong, M.; Alto, N.M. Toxins, mutations and adaptations. eLife 2021, 10, e66676. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawanishi, M.; Yagi, T.; Totsuka, Y.; Wakabayashi, K. DNA Repair and Mutagenesis of ADP-Ribosylated DNA by Pierisin. Toxins 2024, 16, 331. https://doi.org/10.3390/toxins16080331
Kawanishi M, Yagi T, Totsuka Y, Wakabayashi K. DNA Repair and Mutagenesis of ADP-Ribosylated DNA by Pierisin. Toxins. 2024; 16(8):331. https://doi.org/10.3390/toxins16080331
Chicago/Turabian StyleKawanishi, Masanobu, Takashi Yagi, Yukari Totsuka, and Keiji Wakabayashi. 2024. "DNA Repair and Mutagenesis of ADP-Ribosylated DNA by Pierisin" Toxins 16, no. 8: 331. https://doi.org/10.3390/toxins16080331
APA StyleKawanishi, M., Yagi, T., Totsuka, Y., & Wakabayashi, K. (2024). DNA Repair and Mutagenesis of ADP-Ribosylated DNA by Pierisin. Toxins, 16(8), 331. https://doi.org/10.3390/toxins16080331