Insights into the Protein–Lipid Interaction of Perivitellin-2, an Unusual Snail Pore-Forming Toxin

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
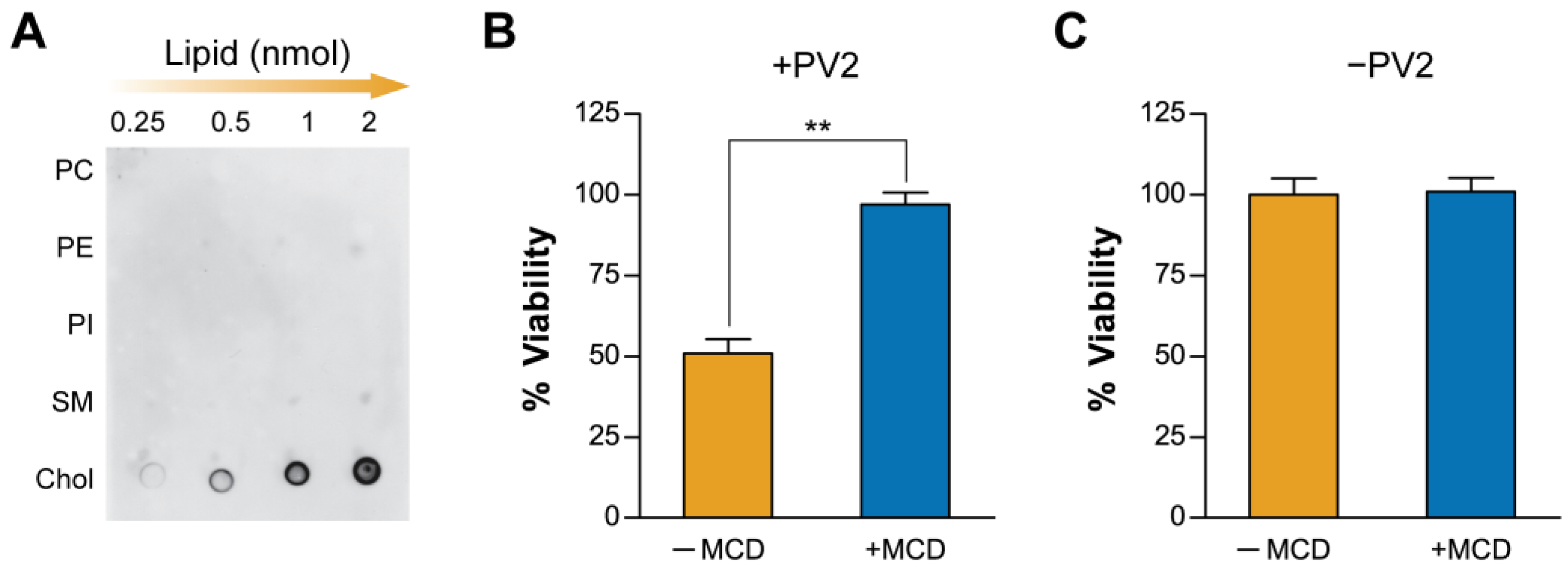
2.1. Plasma Membrane Cholesterol Levels Modulate PmPV2 Toxicity in Gut Cells
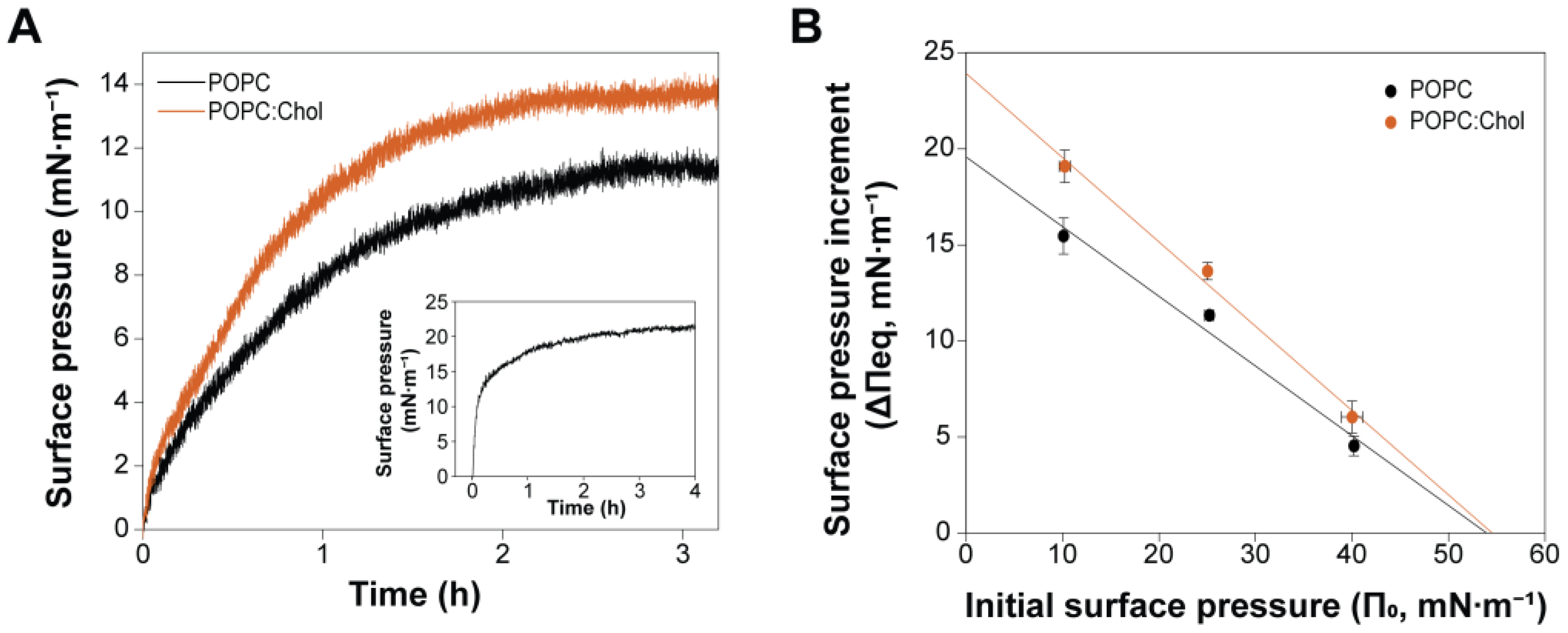
2.2. Cholesterol Enhances PmPV2–Membrane Interaction but Is Not Essential for Toxin Association
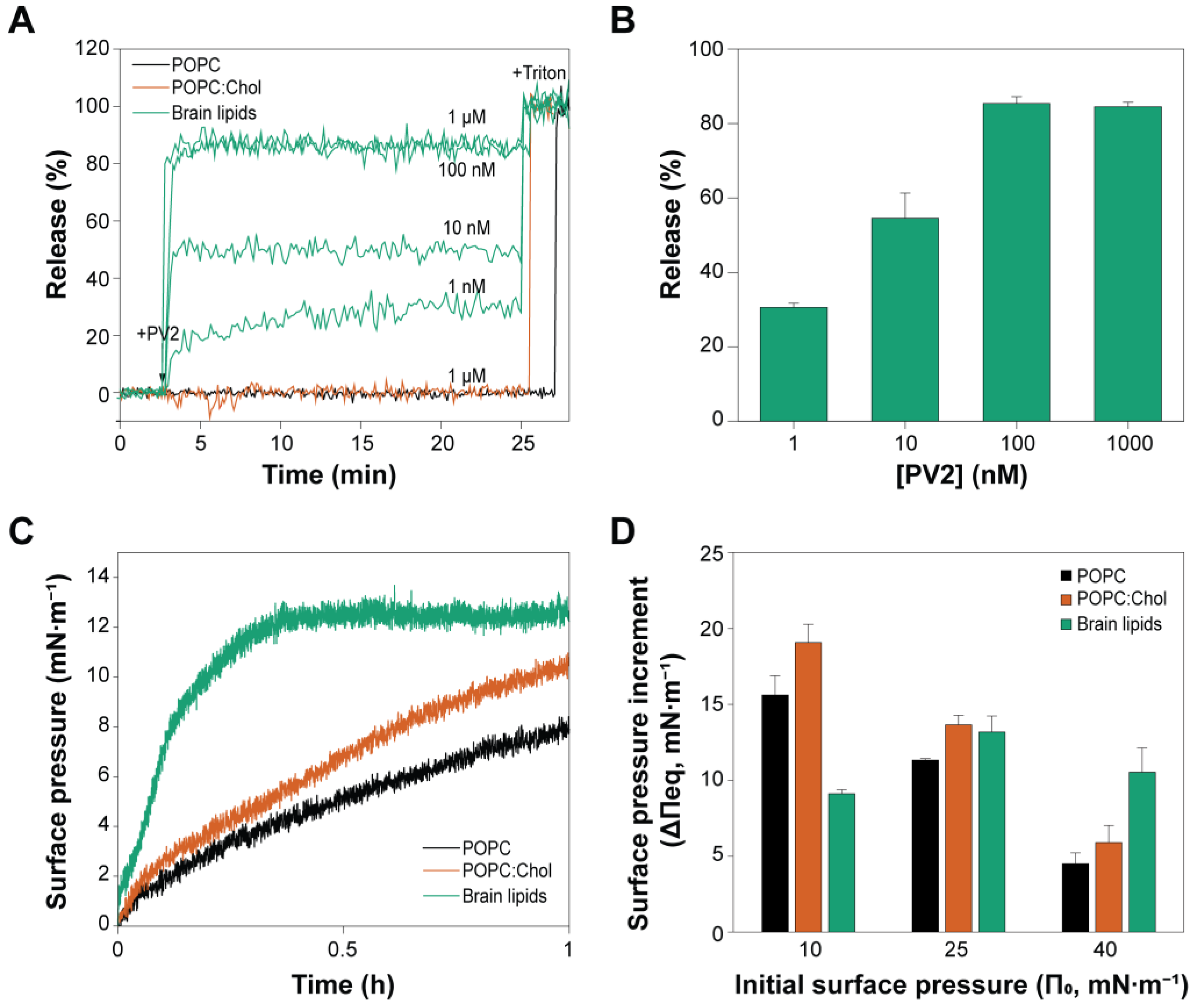
2.3. PmPV2 Does Not Permeabilize POPC/Chol Vesicles but Induces Content Release with Strong Affinity in Brain Lipid Models
2.4. Brain Lipids Promote an Irreversible Association of PmPV2 with Lipid Bilayers
2.5. Brain Lipids Drive PmPV2 Pore Formation
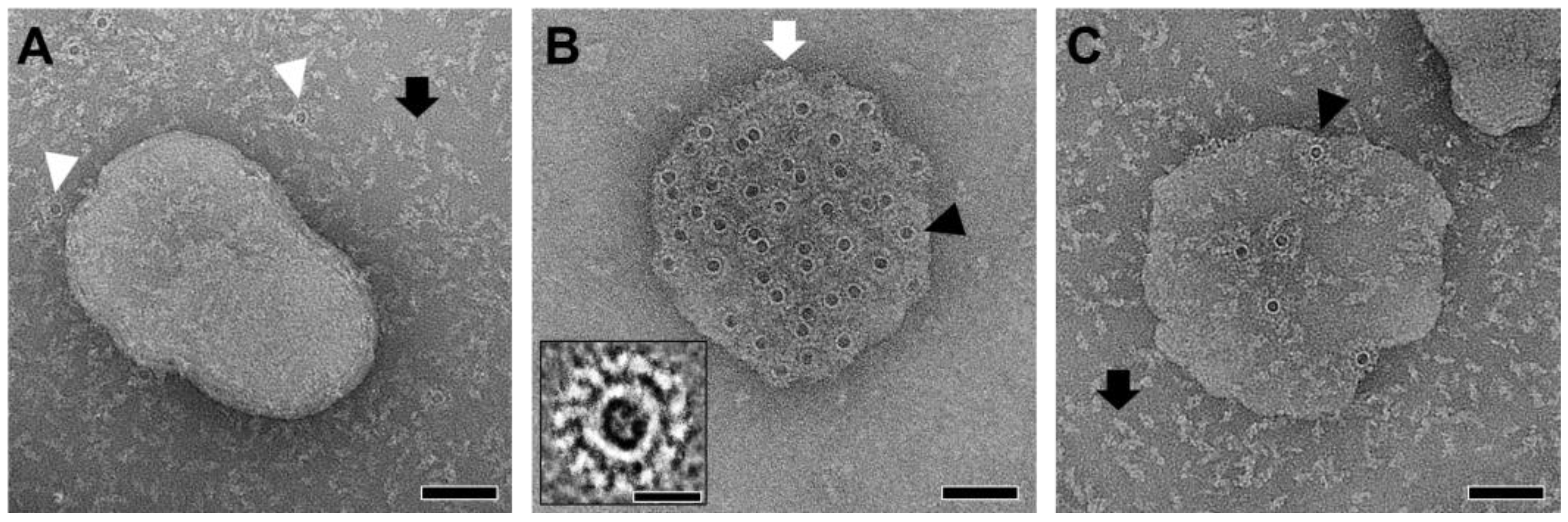
2.6. AFM Imaging of PmPV2 Pores Suggests Lectin–Glycolipid Interactions at the Bilayer Surface
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Protein Purification
4.3. Brain Lipids Extraction
4.4. Lipid Dot Blot
4.5. Influence of Membrane Cholesterol on Cytotoxicity
4.6. Liposome Preparation
4.7. Interaction with Lipid Monolayers
4.8. Permeabilization Assays
4.9. Surface Plasmon Resonance (SPR) Measurements
4.10. Negative-Stain Transmission Electron Microscopy (NS-TEM)
4.11. Atomic Force Microscopy (AFM)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AFM | Atomic Force Microscopy |
| ANTS | 8-aminonaphthalene-1,3,6-trisulfonic-acid |
| CDC | cholesterol-dependent cytolysins |
| Chol | cholesterol |
| DPX | N,N′-p-xylene-bis-pyridinium-bromide |
| HBS | HEPES buffered saline |
| LUVs | large unilamellar vesicles |
| MACPF | membrane attack complex and perforin family |
| MCD | methyl-β-cyclodextrin |
| MIP | maximum insertion pressure |
| MLVs | multilamellar vesicles |
| NS-TEM | Negative-stain Transmission Electron Microscopy |
| PFT | pore-forming toxins |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PI | phosphatidylinositol |
| POPC | 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine |
| PmPV2 | Perivitellin-2 |
| SLBs | supported lipid bilayers |
| SM | sphingomyelin |
| SPR | Surface Plasmon Resonance |
| SUVs | small unilamellar vesicles |
| TBS | Tris-buffered saline |
References
- Bischofberger, M.; Gonzalez, M.R.; van der Goot, F.G. Membrane injury by pore-forming proteins. Curr. Opin. Cell Biol. 2009, 21, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Peraro, M.D.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Kafsack, B.F.; Carruthers, V.B. Apicomplexan perforin-like proteins. Commun. Integr. Biol. 2010, 3, 18–23. [Google Scholar] [CrossRef]
- Nayak, A.P.; Green, B.J.; Beezhold, D.H. Fungal hemolysins. Med. Mycol. 2013, 51, 1–16. [Google Scholar] [CrossRef]
- Rosado, C.J.; Kondos, S.; Bull, T.E.; Kuiper, M.J.; Law, R.H.; Buckle, A.M.; Voskoboinik, I.; Bird, P.I.; Trapani, J.A.; Whisstock, J.C.; et al. The MACPF/CDC family of pore-forming toxins. Cell. Microbiol. 2008, 10, 1765–1774. [Google Scholar] [CrossRef]
- Ellisdon, A.M.; Reboul, C.F.; Panjikar, S.; Huynh, K.; Oellig, C.A.; Winter, K.L.; Dunstone, M.A.; Hodgson, W.C.; Seymour, J.; Dearden, P.K.; et al. Stonefish toxin defines an ancient branch of the perforin-like superfamily. Proc. Natl. Acad. Sci. USA 2015, 112, 15360–15365. [Google Scholar] [CrossRef]
- Rojko, N.; Dalla Serra, M.; Maček, P.; Anderluh, G. Pore formation by actinoporins, cytolysins from sea anemones. Biochim. Biophys. Acta 2016, 1858, 446–456. [Google Scholar] [CrossRef]
- Giglio, M.L.; Ituarte, S.; Ibañez, A.E.; Dreon, M.S.; Prieto, E.; Fernández, P.E.; Heras, H. Novel Role for Animal Innate Immune Molecules: Enterotoxic Activity of a Snail Egg MACPF-Toxin. Front. Immunol. 2020, 11, 428. [Google Scholar] [CrossRef]
- Anderluh, G.; Lakey, J. Proteins: Membrane Binding and Pore Formation; Springer Science: New York, NY, USA, 2010; pp. 1–172. [Google Scholar]
- Heuck, A.P.; Johnson, A.E. Membrane Recognition and Pore Formation by Bacterial Pore-forming Toxins. In Protein–Lipid Interactions; Tamm, L.K., Ed.; Wiley: Hoboken, NJ, USA, 2005; pp. 163–186. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Mengist, H.M.; Shi, C.; Zhang, C.; Wang, B.; Li, T.; Huang, Y.; Xu, Y.; Jin, T. Structural Basis of the Pore-Forming Toxin/Membrane Interaction. Toxins 2021, 13, 128. [Google Scholar] [CrossRef]
- Morton, C.J.; Sani, M.A.; Parker, M.W.; Separovic, F. Cholesterol-Dependent Cytolysins: Membrane and Protein Structural Requirements for Pore Formation. Chem. Rev. 2019, 119, 7721–7736. [Google Scholar] [CrossRef] [PubMed]
- Lukoyanova, N.; Kondos, S.C.; Farabella, I.; Law, R.H.; Reboul, C.F.; Caradoc-Davies, T.T.; Spicer, B.A.; Kleifeld, O.; Traore, D.A.; Ekkel, S.M.; et al. Conformational changes during pore formation by the perforin-related protein pleurotolysin. PLoS Biol. 2015, 13, e1002049. [Google Scholar] [CrossRef]
- Lukoyanova, N.; Hoogenboom, B.W.; Saibil, H.R. The membrane attack complex, perforin and cholesterol-dependent cytolysin superfamily of pore-forming proteins. J. Cell Sci. 2016, 129, 2125–2133. [Google Scholar] [CrossRef] [PubMed]
- Praper, T.; Sonnen, A.; Viero, G.; Kladnik, A.; Froelich, C.J.; Anderluh, G.; Dalla Serra, M.; Gilbert, R.J. Human perforin employs different avenues to damage membranes. J. Biol. Chem. 2011, 286, 2946–2955. [Google Scholar] [CrossRef]
- Rosado, C.J.; Buckle, A.M.; Law, R.H.P.; Butcher, R.E.; Kan, W.T.; Bird, C.H.; Ung, K.; Browne, K.A.; Baran, K.; Bashtannyk-Puhalovich, T.A.; et al. A Common Fold Mediates Vertebrate Defense and Bacterial Attack. Science 2007, 317, 1548–1551. [Google Scholar] [CrossRef]
- Antia, R.; Schlegel, R.A.; Williamson, P. Binding of perforin to membranes is sensitive to lipid spacing and not headgroup. Immunol. Lett. 1992, 32, 153–157. [Google Scholar] [CrossRef]
- Hodel, A.W.; Rudd-Schmidt, J.A.; Trapani, J.A.; Voskoboinik, I.; Hoogenboom, B.W. Lipid specificity of the immune effector perforin. Faraday Discuss. 2021, 232, 236–255. [Google Scholar] [CrossRef]
- Heras, H.; Garin, C.F.; Pollero, R.J. Biochemical composition and energy sources during embryo development and in early juveniles of the snail Pomacea canaliculata (Mollusca: Gastropoda). J. Exp. Zool. 1998, 280, 375–383. [Google Scholar] [CrossRef]
- Dreon, M.S.; Frassa, M.V.; Ceolín, M.; Ituarte, S.; Qiu, J.W.; Sun, J.; Fernández, P.E.; Heras, H. Novel Animal Defenses against Predation: A Snail Egg Neurotoxin Combining Lectin and Pore-Forming Chains That Resembles Plant Defense and Bacteria Attack Toxins. PLoS ONE 2013, 8, e63782. [Google Scholar] [CrossRef]
- Giglio, M.L.; Ituarte, S.; Pasquevich, M.Y.; Heras, H. The eggs of the apple snail Pomacea maculata are defended by indigestible polysaccharides and toxic proteins. Can. J. Zool. 2016, 94, 777–785. [Google Scholar] [CrossRef]
- Giglio, M.L.; Ituarte, S.; Milesi, V.; Dreon, M.S.; Brola, T.R.; Caramelo, J.; Ip, J.C.H.; Maté, S.; Qiu, J.W.; Otero, L.H.; et al. Exaptation of two ancient immune proteins into a new dimeric pore-forming toxin in snails. J. Struct. Biol. 2020, 211, 107531. [Google Scholar] [CrossRef] [PubMed]
- Brola, T.R.; Dreon, M.S.; Fernández, P.E.; Portiansky, E.L.; Heras, H. Ingestion of Poisonous Eggs of the Invasive Apple Snail Pomacea canaliculata Adversely Affects Bullfrog Lithobathes catesbeianus Intestine Morphophysiology. Malacologia 2021, 63, 171–182. [Google Scholar] [CrossRef]
- Fernández, P.; Frassa, V.; Gimeno, E.; Dreon, M.; Heras, H. Changes in carbohydrate expression in the cervical spinal cord of mice intoxicated with perivitellin PV2 from Pomacea canaliculata. In Poisoning by Plants, Mycotoxins and Related Toxins; CABI Digital Library: Wallingford, UK, 2011; pp. 482–488. [Google Scholar] [CrossRef]
- Giglio, M.; Garro, C.; Caviedes-Vidal, E.; Heras, H. Egg perivitelline fluid of the invasive snail Pomacea canaliculata affects mice gastrointestinal function and morphology. PeerJ 2018, 6, e5314. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C.; Graham, D.E.; Hauser, H. Lateral compressibility and penetration into phospholipid monolayers and bilayer membranes. Nature 1975, 254, 154–156. [Google Scholar] [CrossRef]
- Marsh, D. Lateral pressure in membranes. Biochim. Biophys. Acta 1996, 1286, 183–223. [Google Scholar] [CrossRef]
- Calvez, P.; Bussières, S.; Eric, D.; Salesse, C. Parameters modulating the maximum insertion pressure of proteins and peptides in lipid monolayers. Biochimie 2009, 91, 718–733. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar] [CrossRef]
- Sych, T.; Mély, Y.; Römer, W. Lipid self-assembly and lectin-induced reorganization of the plasma membrane. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170117. [Google Scholar] [CrossRef]
- Jiao, F.; Dehez, F.; Ni, T.; Yu, X.; Dittman, J.S.; Gilbert, R.; Chipot, C.; Scheuring, S. Perforin-2 clockwise hand-over-hand pre-pore to pore transition mechanism. Nat. Commun. 2022, 13, 5039. [Google Scholar] [CrossRef]
- Zhang, Y.; Appelkvist, E.L.; Kristensson, K.; Dallner, G. The lipid compositions of different regions of rat brain during development and aging. Neurobiol. Aging 1996, 17, 869–875. [Google Scholar] [CrossRef]
- Surolia, A.; Bachhawat, B.K. The effect of lipid composition on liposome-lectin interaction. Biochem. Biophys. Res. Commun. 1978, 83, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.W.M.; Peters, M.W. Lectin-membrane interactions information from model systems. Biochim. Biophys. Acta 1984, 779, 403–422. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.X.; Hotze, E.M.; Rouiller, I.; Tweten, R.K.; Wilson-Kubalek, E.M. Prepore to pore transition of a cholesterol-dependent cytolysin visualized by electron microscopy. J. Struct. Biol. 2005, 150, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.J.; Orlova, E.V.; Gilbert, R.J.; Andrew, P.W.; Saibil, H.R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 2005, 121, 247–256. [Google Scholar] [CrossRef]
- Leung, C.; Hodel, A.W.; Brennan, A.J.; Lukoyanova, N.; Tran, S.; House, C.M.; Kondos, S.C.; Whisstock, J.C.; Dunstone, M.A.; Trapani, J.A.; et al. Real-time visualization of perforin nanopore assembly. Nat. Nanotechnol. 2017, 12, 467–473. [Google Scholar] [CrossRef]
- Ota, K.; Leonardi, A.; Mikelj, M.; Skočaj, M.; Wohlschlager, T.; Künzler, M.; Aebi, M.; Narat, M.; Križaj, I.; Anderluh, G.; et al. Membrane cholesterol and sphingomyelin, and ostreolysin A are obligatory for pore-formation by a MACPF/CDC-like pore-forming protein, pleurotolysin B. Biochimie 2013, 95, 1855–1864. [Google Scholar] [CrossRef]
- Panevska, A.; Hodnik, V.; Skočaj, M.; Novak, M.; Modic, Š.; Pavlic, I.; Podržaj, S.; Zarić, M.; Resnik, N.; Maček, P.; et al. Pore-forming protein complexes from Pleurotus mushrooms kill western corn rootworm and Colorado potato beetle through targeting membrane ceramide phosphoethanolamine. Sci. Rep. 2019, 9, 5073. [Google Scholar] [CrossRef]
- Lopez, J.A.; Susanto, O.; Jenkins, M.R.; Lukoyanova, N.; Sutton, V.R.; Law, R.H.P.; Johnston, A.; Bird, C.H.; Bird, P.I.; Whisstock, J.C.; et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013, 121, 2659–2668. [Google Scholar] [CrossRef]
- Rudd-Schmidt, J.A.; Hodel, A.W.; Noori, T.; Lopez, J.A.; Cho, H.J.; Verschoor, S.; Ciccone, A.; Trapani, J.A.; Hoogenboom, B.W.; Voskoboinik, I. Lipid order and charge protect killer T cells from accidental death. Nat. Commun. 2019, 10, 5396. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hotze, E.M.; Shao, Z.; Tweten, R.K. Vertical collapse of a cytolysin prepore moves its transmembrane beta-hairpins to the membrane. EMBO J. 2004, 23, 3206–3215. [Google Scholar] [CrossRef]
- Unno, H.; Goda, S.; Hatakeyama, T. Hemolytic Lectin CEL-III Heptamerizes via a Large Structural Transition from α-Helices to a β-Barrel during the Transmembrane Pore Formation Process. J. Biol. Chem. 2014, 289, 12805–12812. [Google Scholar] [CrossRef] [PubMed]
- Kouzuma, Y.; Suzuki, Y.; Nakano, M.; Matsuyama, K.; Tojo, S.; Kimura, M.; Yamasaki, T.; Aoyagi, H.; Hatakeyama, T. Characterization of functional domains of the hemolytic lectin CEL-III from the marine invertebrate Cucumaria echinata. J. Biochem. 2003, 134, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, D.; Zhang, F.H.; Gan, Y. AFM tip-sample convolution effects for cylinder protrusions. Appl. Surf. Sci. 2017, 422, 482–491. [Google Scholar] [CrossRef]
- Giddings, K.S.; Johnson, A.E.; Tweten, R.K. Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc. Natl. Acad. Sci. USA 2003, 100, 11315–11320. [Google Scholar] [CrossRef]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef]
- Farrand, S.; Hotze, E.; Friese, P.; Hollingshead, S.K.; Smith, D.F.; Cummings, R.D.; Dale, G.L.; Tweten, R.K. Characterization of a streptococcal cholesterol-dependent cytolysin with a lewis y and b specific lectin domain. Biochemistry 2008, 47, 7097–7107. [Google Scholar] [CrossRef]
- Shewell, L.K.; Harvey, R.M.; Higgins, M.A.; Day, C.J.; Hartley-Tassell, L.E.; Chen, A.Y.; Gillen, C.M.; James, D.B.; Alonzo, F., 3rd; Torres, V.J.; et al. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc. Natl. Acad. Sci. USA 2014, 111, E5312–E5320. [Google Scholar] [CrossRef]
- Shewell, L.K.; Day, C.J.; Jen, F.E.; Haselhorst, T.; Atack, J.M.; Reijneveld, J.F.; Everest-Dass, A.; James, D.B.A.; Boguslawski, K.M.; Brouwer, S.; et al. All major cholesterol-dependent cytolysins use glycans as cellular receptors. Sci. Adv. 2020, 6, eaaz4926. [Google Scholar] [CrossRef]
- Stoffel, W.; Bosio, A. Myelin glycolipids and their functions. Curr. Opin. Neurobiol. 1997, 7, 654–661. [Google Scholar] [CrossRef]
- Singh, A.K.; Harrison, S.H.; Schoeniger, J.S. Gangliosides as Receptors for Biological Toxins: Development of Sensitive Fluoroimmunoassays Using Ganglioside-Bearing Liposomes. Anal. Chem. 2000, 72, 6019–6024. [Google Scholar] [CrossRef]
- Kouzel, I.U.; Pohlentz, G.; Storck, W.; Radamm, L.; Hoffmann, P.; Bielaszewska, M.; Bauwens, A.; Cichon, C.; Schmidt, M.A.; Mormann, M.; et al. Association of Shiga toxin glycosphingolipid receptors with membrane microdomains of toxin-sensitive lymphoid and myeloid cells. J. Lipid Res. 2013, 54, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.J.; Farrand, A.J.; Tweten, R.K. The cholesterol-dependent cytolysin signature motif: A critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 2012, 8, e1002787. [Google Scholar] [CrossRef]
- Polekhina, G.; Giddings, K.S.; Tweten, R.K.; Parker, M.W. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. USA 2005, 102, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Dai, X.; Lin, D.S.; Connor, W.E. The lipids of slugs and snails: Evolution, diet and biosynthesis. Lipids 1994, 29, 869–875. [Google Scholar] [CrossRef]
- Heras, H.; Frassa, M.V.; Fernández, P.E.; Galosi, C.M.; Gimeno, E.J.; Dreon, M.S. First egg protein with a neurotoxic effect on mice. Toxicon 2008, 52, 481–488. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloan, E.; Standley, G. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Lambert, D.; O’Neill, C.A.; Padfield, P.J. Depletion of Caco-2 cell cholesterol disrupts barrier function by altering the detergent solubility and distribution of specific tight-junction proteins. Biochem. J. 2005, 387, 553–560. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Vazquez, R.F.; Mate, S.M.; Bakas, L.S.; Fernandez, M.M.; Malchiodi, E.L.; Herlax, V.S. Novel evidence for the specific interaction between cholesterol and alpha-haemolysin of Escherichia coli. Biochem. J. 2014, 458, 481–489. [Google Scholar] [CrossRef]
- Smolarsky, M.; Teitelbaum; Sela, M.D.; Gitler, C. A simple fluorescent method to determine complement-mediated liposome immune lysis. J. Immunol. Methods 1977, 15, 255–265. [Google Scholar] [CrossRef]
- Chen, P.; Toribara, T.; Warner, H. Microdetermination of phosphorus. Anal. Chem. 1956, 28, 1756–1758. [Google Scholar] [CrossRef]
- Daza Millone, M.A.; Vázquez, R.F.; Maté, S.M.; Vela, M.E. Phase-segregated Membrane Model assessed by a combined SPR-AFM Approach. Colloids Surf. B Biointerfaces 2018, 172, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez, R.F.; Daza Millone, M.A.; Giglio, M.L.; Brola, T.R.; Maté, S.M.; Heras, H. Insights into the Protein–Lipid Interaction of Perivitellin-2, an Unusual Snail Pore-Forming Toxin. Toxins 2025, 17, 183. https://doi.org/10.3390/toxins17040183
Vázquez RF, Daza Millone MA, Giglio ML, Brola TR, Maté SM, Heras H. Insights into the Protein–Lipid Interaction of Perivitellin-2, an Unusual Snail Pore-Forming Toxin. Toxins. 2025; 17(4):183. https://doi.org/10.3390/toxins17040183
Chicago/Turabian StyleVázquez, Romina F., M. Antonieta Daza Millone, Matías L. Giglio, Tabata R. Brola, Sabina M. Maté, and Horacio Heras. 2025. "Insights into the Protein–Lipid Interaction of Perivitellin-2, an Unusual Snail Pore-Forming Toxin" Toxins 17, no. 4: 183. https://doi.org/10.3390/toxins17040183
APA StyleVázquez, R. F., Daza Millone, M. A., Giglio, M. L., Brola, T. R., Maté, S. M., & Heras, H. (2025). Insights into the Protein–Lipid Interaction of Perivitellin-2, an Unusual Snail Pore-Forming Toxin. Toxins, 17(4), 183. https://doi.org/10.3390/toxins17040183