2.1. HGT-Finder: A New Tool to Find Horizontal Gene Transfer
The algorithm behind HGT-Finder is provided in the Methods section. The inputs to this software include: (i) the BLAST search result (tabular format-outfmt 6) of a query set (e.g., proteins of a genome) against the NCBI nonredundant protein (NCBI-nr) database and (ii) the NCBI Taxonomy database. The output of this program is a tabular format file containing the following key information: protein ID, X value (transfer index value), P value and Q value. X is calculated using a mathematical formula detailed in Methods. In brief, for each pair of query and BLAST subject species, a novel taxonomic distance D is calculated such that D ∈ [0, 1], and a BLAST similarity measure R (BLAST bit score ratio relative to the self-hit, see Methods) is calculated such that R ∈ [0, 1]. The X for each query considers D and R values of all of its BLAST subjects. The P value is calculated according to the statistical distribution of the X for all query proteins.
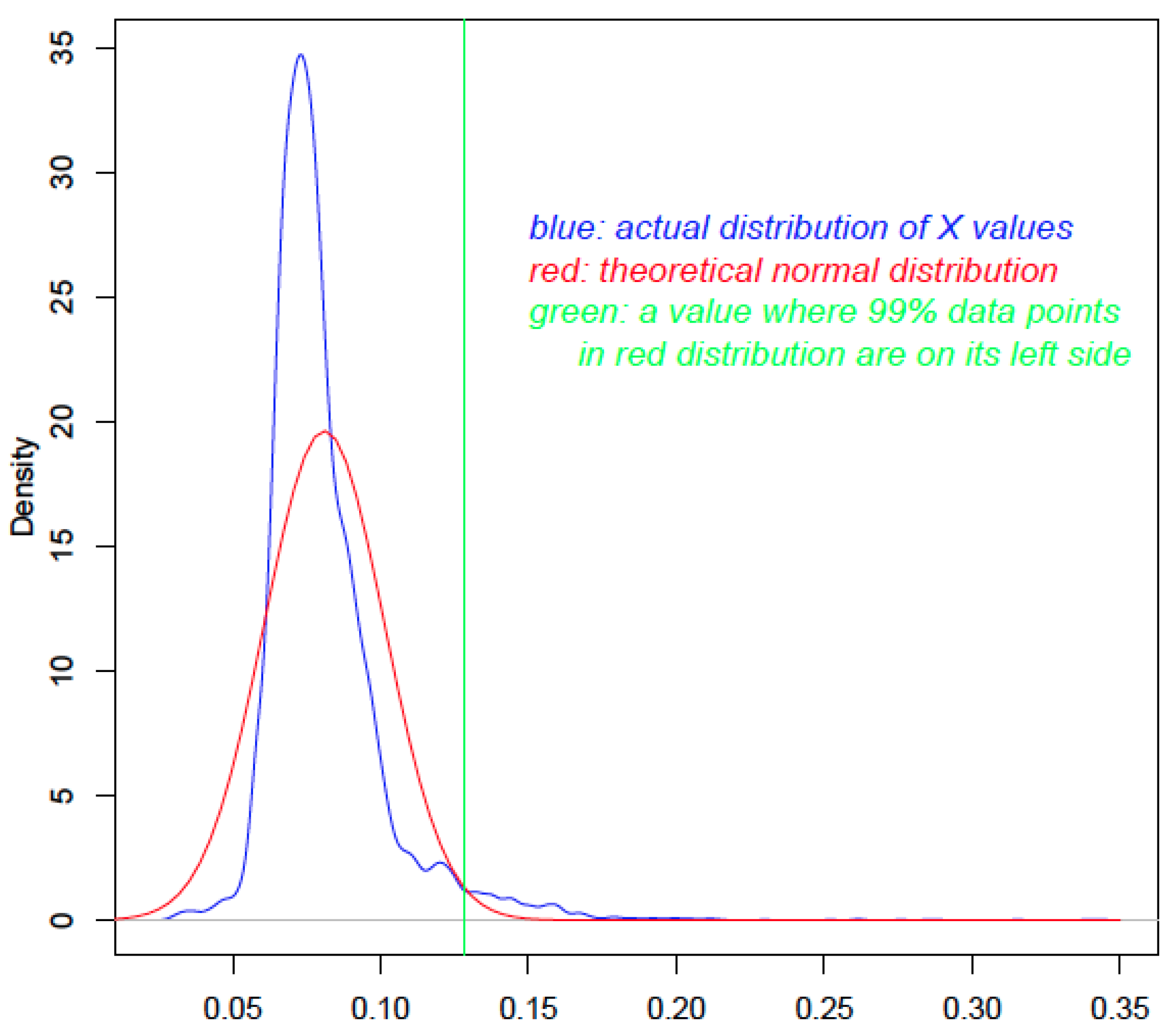
More specifically, the
X values for all query proteins are plotted (blue curve in
Figure 1). The mean and standard deviation values are calculated, which are used to generate a theoretical normal distribution (red curve in
Figure 1). The actual distribution and the theoretical normal distribution are then compared to calculate a probability value for each query protein using the
pnorm function of the R software (R Development Core Team) (
www.r-project.org). The
P value is used to reject the null hypothesis that the to-be-tested value from the actual distribution is smaller than a particular value in the normal distribution (green vertical line in
Figure 1). Thus, proteins with a higher
X will typically have smaller
P values and are more likely to be true HTGs. Since the number of statistical tests to be done is equal to the number of genes in the query set, there will be multiple testing errors that are to be corrected [
30]. The qvalue package of the Bioconductor software (
https://www.bioconductor.org) is used to convert the
P value to a corrected
Q value, which is a more accurate metric to determine statistical significance.
Figure 1.
The use of statistical distribution to calculate P values. The x-axis shows the X value. The blue curve is the distribution of the X values of 9577 Aspfu proteins. The red curve is the theoretical distribution that has the same mean and standard deviation as the blue curve. The green line is drawn to indicate the cutoff value; any X value larger than that in the blue curve will have a P value <0.01.
Figure 1.
The use of statistical distribution to calculate P values. The x-axis shows the X value. The blue curve is the distribution of the X values of 9577 Aspfu proteins. The red curve is the theoretical distribution that has the same mean and standard deviation as the blue curve. The green line is drawn to indicate the cutoff value; any X value larger than that in the blue curve will have a P value <0.01.
2.2. Use Different R Thresholds to Detect Horizontally Transferred Genes (HTGs)
One very important parameter in running HGT-Finder is the R threshold (see Methods), which is used, prior to the calculation of
X, to remove BLAST hits that are less similar to the query. For example, one can use
R > 0.2, meaning that only hits with
R > 0.2 will be used for the
X calculation. In order to study the impact of this R threshold on HGT predictions, we have run HGT-Finder using
Q value <0.01 and a range of R thresholds from 0.2 to 0.9 on the
Aspergillus fumigatus Af293 (
Aspfu),
Aspergillus flavus NRRL3357 (
Aspfl), and
Aspergillus nidulans FGSC A4 (
Aspni) protein sets to predict HTGs. Hence we obtained eight HTGs sets for each species (
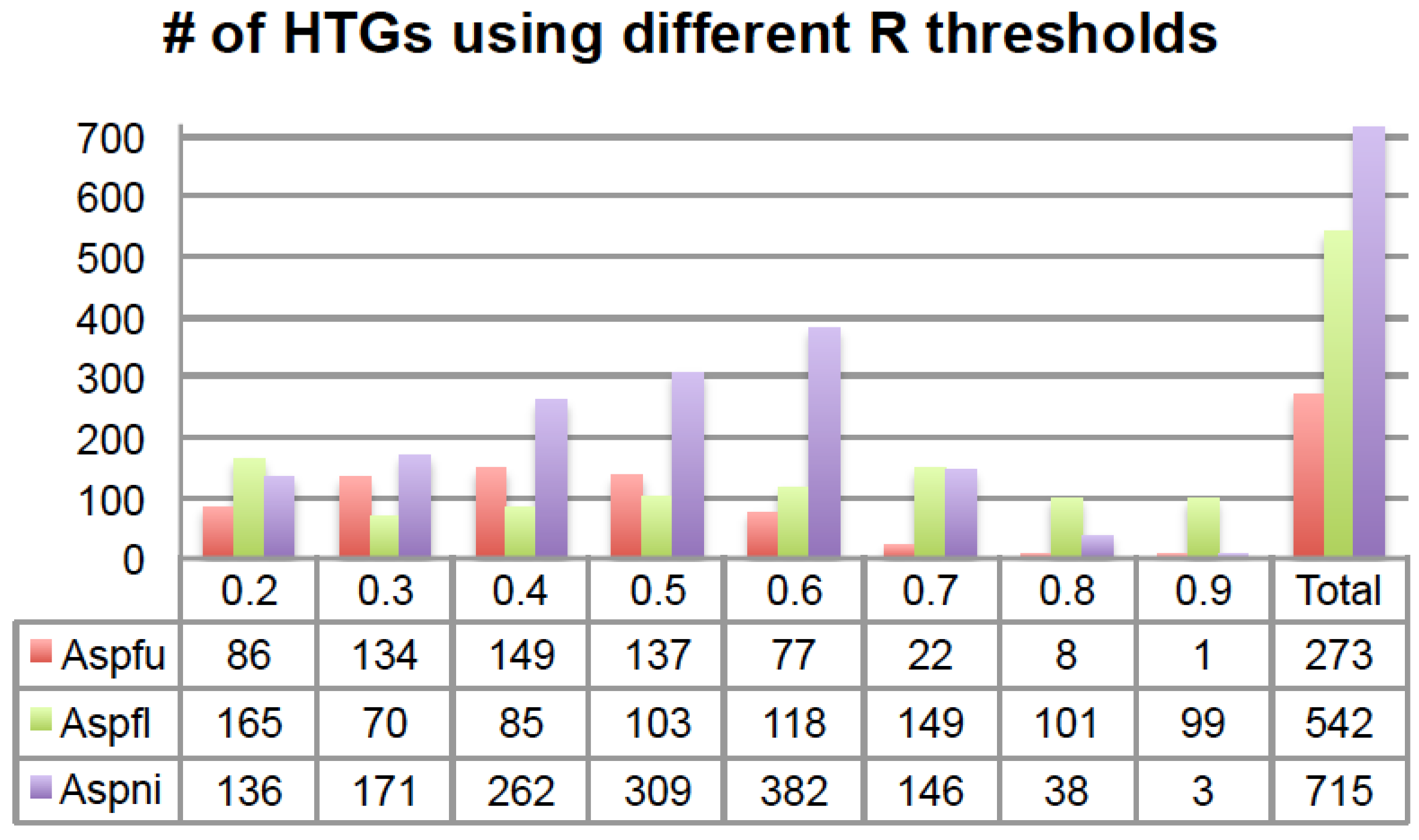
Figure 2).
Figure 2.
The number of HTGs predicted using different R thresholds. The x-axis is the R threshold and the y-axis is the number of HTGs. The last column shows the total number of HTGs after removing overlaps. # means “number”.
Figure 2.
The number of HTGs predicted using different R thresholds. The x-axis is the R threshold and the y-axis is the number of HTGs. The last column shows the total number of HTGs after removing overlaps. # means “number”.
Some HTGs were predicted with multiple R thresholds. For example, in total 273 Aspfu proteins were found in at least one of the eight sets and 47 of the 273 were found in at least four of the eight sets (
Table S1); among those 47 genes, 45 of them were found in the
R > 0.5 set. Similarly, for Aspfl in total 542 proteins were found in at least one of the eight sets and 49 were found in at least four of the eight sets (
Table S1); among those 49 genes, 43 of them were found in the
R > 0.5 set. For Aspni, a total of 715 proteins were found in at least one of the eight sets and 101 were found in at least four of the eight sets (
Table S1); among those 101 genes, 100 of them were found in the
R > 0.5 set. Therefore, for all the three genomes, it is always the
R > 0.5 set that contains the most genes that are shared by the other R threshold sets.
However, using a single R threshold will certainly result in a loss of many HTGs. The higher the R threshold that is used, the fewer BLAST hits that will be considered in the
X calculation. For example, if
R > 0.5 is used, BLAST hits with an
R less than 0.5 will be removed prior to calculating
X. Moreover, a higher R threshold will result in more query proteins that will fail to have an
X calculation. For example, if
R > 0.9 is used, those query proteins that do not have very similar hits in the database will not have an
X value calculated. This explains why, in
Figure 2, there are fewer HTGs predicted for
R > 0.8 and
R > 0.9 sets for all three genomes.
Lastly, a lower R threshold tends to predict more ancient HGTs while a higher R threshold tends to predict more recent HGTs. To verify this, using different R thresholds, we calculated the percentage of HTGs having over 50% of BLAST hits from non-Eukaryotes (
i.e., Bacteria, Archaea and Viruses).
Figure 3 shows that, for all the three genomes, there is a clear trend that when using lower R thresholds, a higher percentage of HTGs are found to have more than 50% of their BLAST hits from different domains of life. This indicates that they might be derived from more ancient HGTs when assuming very recent inter-domain transfers (
i.e., with high
R) are rare. If an HTG has more inter-domain BLAST hits, it is more likely to be an ancient HTG. On the other hand, if an HTG has all of its BLAST hits within the same taxonomic group, e.g., phylum, it is a more recent HTG. Therefore, by default, our HGT-Finder program runs all eight R thresholds and the users are advised to combine the HTGs from all these eight runs to obtain a complete list of HTGs.
Figure 3.
The percentage of HTGs that have more than 50% BLAST hits from non-eukaryotic species using different R thresholds. The x-axis is the R thresholds and the y-axis is the percentage of HTGs.
Figure 3.
The percentage of HTGs that have more than 50% BLAST hits from non-eukaryotic species using different R thresholds. The x-axis is the R thresholds and the y-axis is the percentage of HTGs.
Figure 2 and
Figure 3 also show that although Aspni has more HTGs than the other genomes, Aspfl has a higher percentage of inter-domain HTGs, which agrees with another recent report [
14].
2.3. Verify HTGs Using an Approximate Method and Phylogenetic Analysis
In order to quickly confirm the HGT predictions, we have examined the non-self best hit of the HTG candidates, where “non-self” means that the BLAST subject protein is from a species with a different taxonomy ID. The complete data for
R > 0.5 for the three genomes are available in
Table S2. The NCBI-nr database contains protein sequences from 15 sequenced nuclear genomes of the
Aspergillus genus (
Table S3). If an HTG candidate were transferred from outside of the genus, then its top BLAST hits (here we use the best hit for simplicity) would be from a different genus, family, order, class, phylum or kingdom with an increasing evolutionary distance to the recipient.
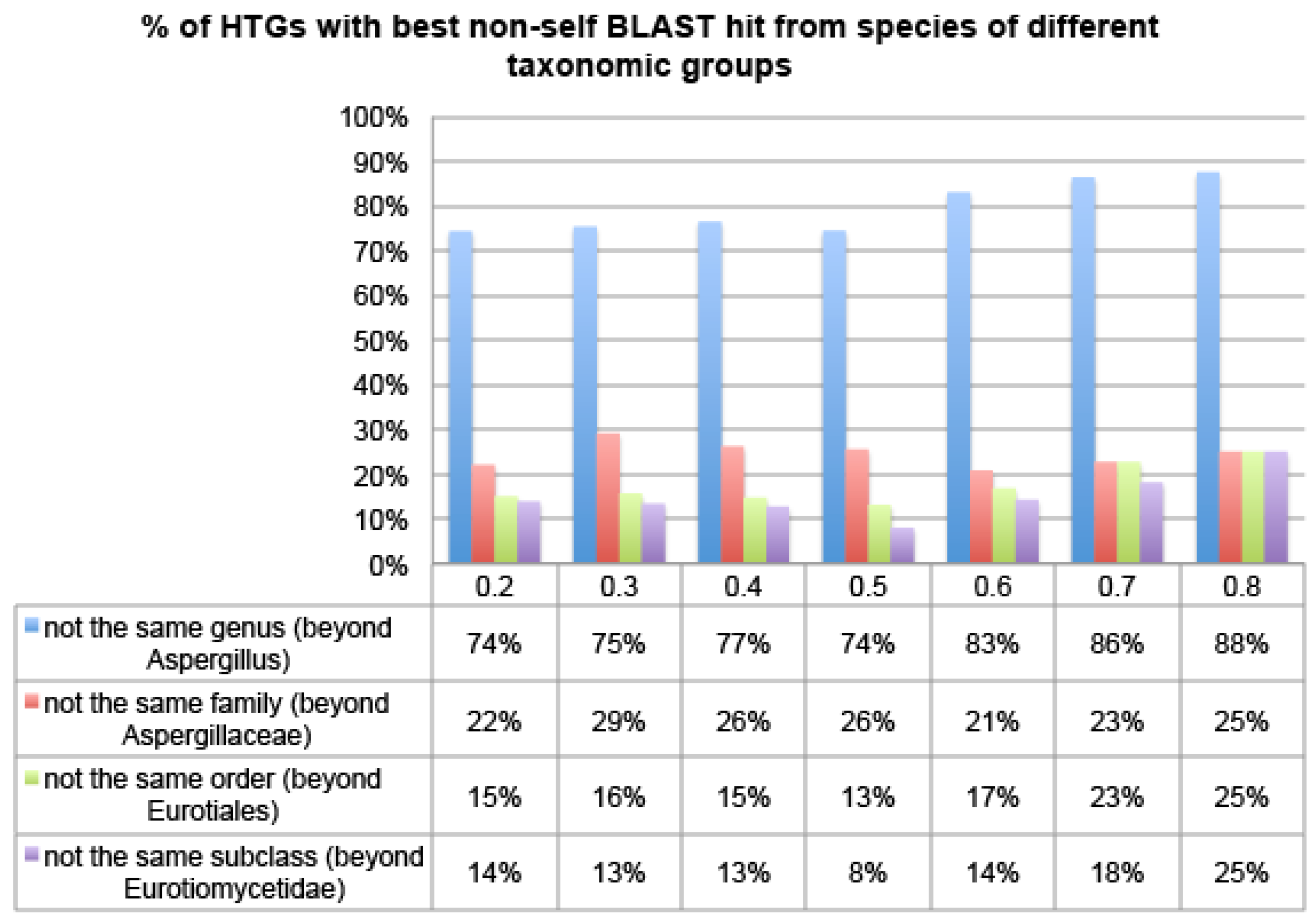
Figure 4 shows that over 74% of HTG candidates of Aspfu have their best non-self hit from species of different genera, over 20% from even different families, and over 15% from even different orders, irrespective of which R thresholds were used. This pattern is even more pronounced in the other two genomes, Aspfl and Aspni (
Figures S1 and S2). This suggests that HGT-Finder does succeed in making meaningful predictions.
Figure 4.
The percentage of Aspfu HTGs that have more than 50% BLAST hits from non-Eukaryotic species using different R thresholds. The x-axis is the R threshold and the y-axis is the percentage of HTGs.
Figure 4.
The percentage of Aspfu HTGs that have more than 50% BLAST hits from non-Eukaryotic species using different R thresholds. The x-axis is the R threshold and the y-axis is the percentage of HTGs.
This simple and approximate method, although very informative, fast, and easy to execute, cannot conclusively verify HTGs because a best hit from a distant organism could also be due to other reasons such as: (i) the subject gene may be recently transferred from (not to) the query genome; (ii) the query gene evolved very rapidly so it becomes very different from its orthologous genes in closely related species; (iii) the orthologous genes in closely related species were independently lost during evolution.
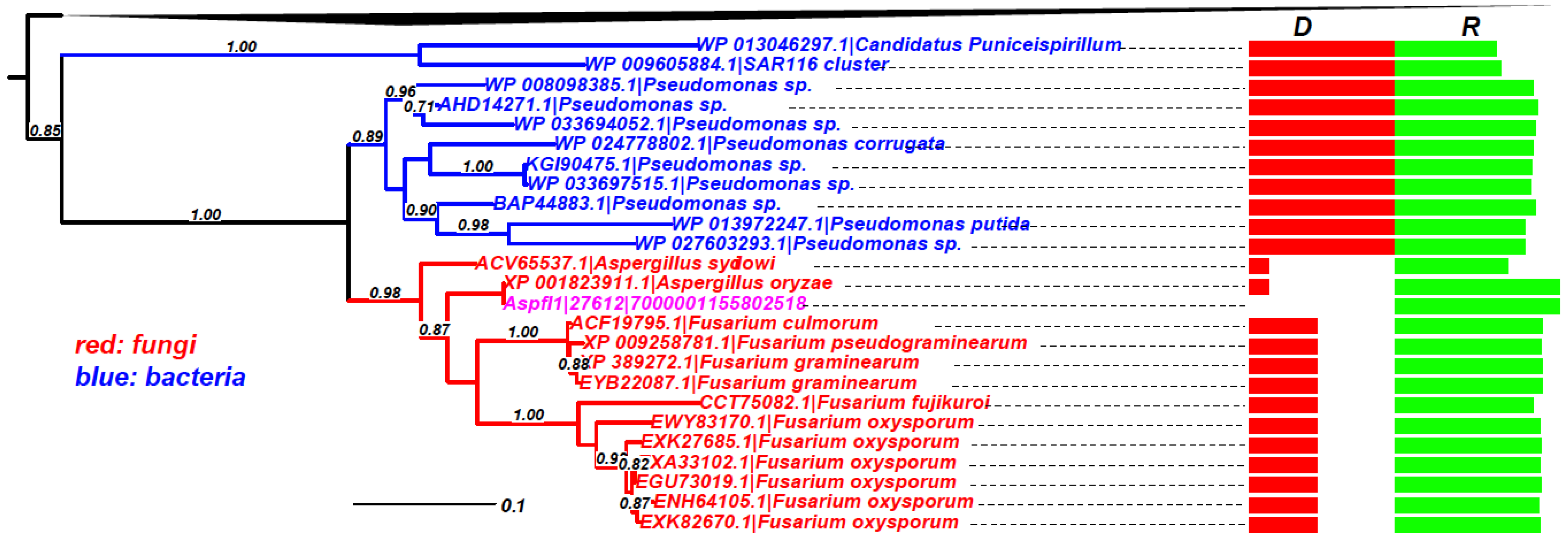
As mentioned above, the gene-by-gene phylogenetic analysis, although not computationally suitable for large-scale analysis, is the golden standard method to claim a gene is a HTG. We have performed phylogenetic analyses on the
R > 0.5 set for Aspfl. In total, there are 103 HTGs predicted in Aspfl by HGT-Finder (
Figure 2), 73 of which have at least four BLAST hits in other taxa and thus applicable for building phylogenies.
Figure 5 shows an example (Aspfl1|27612, Peptidase M24) phylogeny, which clearly indicates that the common ancestor of the fungal proteins, including the query protein, must have been transferred from some
Pseudomonas bacteria (
Gammaproteobacteria). Phylogenies of other genes are combined and made available in Supplemental data file 1. We have manually inspected all these phylogenies containing protein hits with
R > 0.2 in order to determine if they are true HTGs. Among these 73 genes, phylogenies seem to support 68 (93%) of them to be HTGs (
Table S3), which include: (i) 30 that have hits in smaller numbers of fungi but many bacteria, suggesting transfers from bacteria; (ii) 38 have hits in very few
Aspergillus genomes (mostly restricted to Aspfl and the very closely related
A. oryzae) and are phylogenetically clustered with hits of different fungal genera, or even more distant taxonomic groups, suggesting transfers from distant fungi. The remaining five genes do not have a strong phylogenetic signal to suggest that they are HTGs. One of the five is a very conserved ribosomal protein (jgi|Aspfl1|30709), which is restricted to Aspfl and
A. oryzae of the
Aspergillus genus, and further clustered with a termite (
Coptotermes formosanus) protein, suggesting a recent gene transfer into termite (Supplemental data file 1).
Figure 5.
Phylogeny of Aspfl1|27612|7000001155802518 (GenBank ID: AFLA_091530) as an example of HTG. The query protein (in pink)’s homologs are restricted to very few fungal species, two other Aspergillus species and a few Fusarium species. The rest of the homologs are all from Proteobacteria. The boxes on the right show the taxonomic distance (D) in green and sequence similarity (R) in red. The black triangle on the top represents the collapsed Proteobacteria homologs.
Figure 5.
Phylogeny of Aspfl1|27612|7000001155802518 (GenBank ID: AFLA_091530) as an example of HTG. The query protein (in pink)’s homologs are restricted to very few fungal species, two other Aspergillus species and a few Fusarium species. The rest of the homologs are all from Proteobacteria. The boxes on the right show the taxonomic distance (D) in green and sequence similarity (R) in red. The black triangle on the top represents the collapsed Proteobacteria homologs.
It should be noted that some of the 38 fungi-fungi HTG candidates might have very complex evolutionary trajectories. For example, jgi|Aspfl1|31710 (Supplemental data file 1) might have been recently transferred from other fungi (very few
Aspergillus hits); furthermore, all of the fungi hits might have been transferred from bacteria in an earlier event (not many fungi hits but numerous bacteria hits). Compared to bacteria-fungi transfers, fungi-fungi transfers are more difficult to detect, because patchy phyletic distribution of BLAST hits could also be a result of independent gene loss occurred in closely related species [
31]. Even when the phylogeny is available, to reliably distinguish the two possibilities (gene transfer and gene loss) is still not easy, which is complicated by the incompleteness and biased sequence sampling of the BLAST database. It would therefore be safer to conclude that the 38 fungi-fungi HTG candidates have been confirmed to have patchy taxonomic distribution based on phylogenetic analyses. Nevertheless, they have a higher likelihood to be horizontally transferred because independently losing these genes in most of the closely related
Aspergillus genomes (
Table S3) is a less parsimonious explanation than the HGT hypothesis.
We have also manually examined the 30 HTGs that have less than three hits (with
R > 0.5) by inspecting the BLAST output with
R > 0.2 and searching them using NCBI’s Blink service. When relaxing the R threshold to 0.2, most of the 30 HTGs have more hits. We found that: (i) five of the 30 genes must have been transferred from bacteria, (ii) 17 might have been transferred from distant fungi more recently, (iii) three have complex evolution involving possible recent transfers from distant fungi and more ancient transfers from bacteria, (iv) two might have involved
Metazoa in the transfer, and (v) the remaining three seem to be orphan genes. We have made comments on all the 103 Aspfl HTGs in
Table S3 based on our manual curation. Such detailed phylogenetic analyses suggest that our HGT-Finder program indeed performs well in identifying true HTGs. It should be noted that we have used a very stringent
Q value < 0.01 as the cutoff to keep statistically significant candidates. The number of HTGs may thus have been underestimated.
2.5. Function of HGT Genes
Previous studies have suggested that metabolic enzymes are prone to be horizontally transferred [
10,
32,
33], which has never been tested using strict statistical approaches in fungi. In brief,
h is the number of genes with a certain function in the HTG set and
H is the total number of HTGs; this
h/
H ratio has to be compared to the genome background ratio
t/
T, where
t is the number of genes with that function in the genome and
T is the total number of genes in the genome. We have performed hypergeometric enrichment tests on the Gene Ontology (GO) annotations of 273 Aspfu, 542 Aspfl, and 715 Aspni HTGs (numbers from
Figure 2) by comparing them with the genome background.
Table 2,
Table 3 and
Table 4 (complete datasets are in
Table S4) list the top GO functions in the three genomes that have at least 10 assigned HTGs.
Table 2.
GO functional categories having at least 10 assigned HTGs in Aspfu. # means “number”.
Table 2.
GO functional categories having at least 10 assigned HTGs in Aspfu. # means “number”.
| GO Name | GO ID | # of Assigned HTGs | # of Assigned Genes in the Genome | P Value (Red Font if <0.05) |
|---|
| un-annotated by GO | - | 141 | 4008 | 0.0001964405 |
| metabolic process | GO:0008152 | 31 | 893 | 0.1191704 |
| catalytic activity | GO:0003824 | 28 | 883 | 0.2647851 |
| binding | GO:0005488 | 17 | 538 | 0.3332352 |
| ribonuclease H activity | GO:0004523 | 16 | 19 | 7.89 × 10−23 |
| nucleic acid binding | GO:0003676 | 16 | 401 | 0.09561368 |
| oxidoreductase activity | GO:0016491 | 16 | 522 | 0.3859507 |
| RNA-dependent DNA replication | GO:0006278 | 14 | 23 | 8.17 × 10−17 |
| RNA-directed DNA polymerase activity | GO:0003964 | 14 | 23 | 8.17 × 10−17 |
| RNA binding | GO:0003723 | 14 | 102 | 8.23 × 10−7 |
| integral to membrane | GO:0016021 | 13 | 516 | 0.6891403 |
| transport | GO:0006810 | 12 | 467 | 0.6582828 |
| carbohydrate metabolic process | GO:0005975 | 11 | 225 | 0.05062836 |
| transporter activity | GO:0005215 | 11 | 292 | 0.1936068 |
| membrane | GO:0016020 | 11 | 431 | 0.6636644 |
| hydrolase activity, hydrolyzing O-glycosyl compounds | GO:0004553 | 10 | 141 | 0.006067584 |
Table 3.
GO functional categories having at least 10 assigned HTGs in Aspfl.
Table 3.
GO functional categories having at least 10 assigned HTGs in Aspfl.
| GO Name | GO ID | # of Assigned HTGs | # of Assigned Genes in the Genome | P Value (Red Font if <0.05) |
|---|
| Un-annotated by GO | - | 279 | 5513 | 3.32823 × 10−6 |
| catalytic activity | GO:0003824 | 74 | 1309 | 0.00360352 |
| metabolic process | GO:0008152 | 73 | 1304 | 0.004876894 |
| oxidoreductase activity | GO:0016491 | 40 | 820 | 0.1634237 |
| binding | GO:0005488 | 39 | 788 | 0.1454589 |
| electron transport | GO:0006118 | 23 | 494 | 0.3167226 |
| ATP binding | GO:0005524 | 19 | 633 | 0.9504606 |
| transport | GO:0006810 | 18 | 684 | 0.9885602 |
| integral to membrane | GO:0016021 | 16 | 700 | 0.997863 |
| membrane | GO:0016020 | 15 | 553 | 0.9738793 |
| hydrolase activity | GO:0016787 | 13 | 208 | 0.09374698 |
| iron ion binding | GO:0005506 | 13 | 232 | 0.1696784 |
| transporter activity | GO:0005215 | 13 | 476 | 0.9629008 |
| proteolysis | GO:0006508 | 12 | 227 | 0.2362409 |
| nucleus | GO:0005634 | 12 | 631 | 0.9995651 |
| pyridoxal phosphate binding | GO:0030170 | 11 | 100 | 0.002880844 |
| biosynthetic process | GO:0009058 | 10 | 94 | 0.005585243 |
| DNA binding | GO:0003677 | 10 | 435 | 0.9879336 |
Table 4.
GO functional categories having at least 10 assigned HTGs in Aspni.
Table 4.
GO functional categories having at least 10 assigned HTGs in Aspni.
| GO Name | GO ID | # of Assigned HTGs | # of Assigned Genes in the Genome | P Value (Red Font if <0.05) |
|---|
| Un-annotated by GO | - | 242 | 4367 | 0.9999737 |
| metabolic process | GO:0008152 | 147 | 1058 | 6.12 × 10−19 |
| catalytic activity | GO:0003824 | 136 | 1046 | 4.77 × 10−15 |
| oxidoreductase activity | GO:0016491 | 106 | 668 | 1.06 × 10−17 |
| binding | GO:0005488 | 84 | 633 | 6.18 × 10−10 |
| electron transport | GO:0006118 | 73 | 411 | 5.94 × 10−15 |
| transport | GO:0006810 | 72 | 560 | 4.38 × 10−8 |
| integral to membrane | GO:0016021 | 67 | 573 | 4.20 × 10−6 |
| transporter activity | GO:0005215 | 60 | 382 | 3.42 × 10−10 |
| heme binding | GO:0020037 | 43 | 183 | 1.85 × 10−13 |
| membrane | GO:0016020 | 43 | 470 | 0.02210711 |
| monooxygenase activity | GO:0004497 | 39 | 165 | 2.07 × 10−12 |
| iron ion binding | GO:0005506 | 37 | 175 | 2.47 × 10−10 |
| nucleus | GO:0005634 | 32 | 671 | 0.987011 |
| carbohydrate metabolic process | GO:0005975 | 25 | 215 | 0.004820523 |
| DNA binding | GO:0003677 | 23 | 449 | 0.9321919 |
| L-arabinose isomerase activity | GO:0008733 | 22 | 97 | 3.05 × 10−7 |
| hydrolase activity | GO:0016787 | 22 | 172 | 0.002504542 |
| zinc ion binding | GO:0008270 | 22 | 696 | 0.9999923 |
| carbohydrate transport | GO:0008643 | 18 | 92 | 3.16 × 10−5 |
| sugar:hydrogen symporter activity | GO:0005351 | 18 | 94 | 4.27 × 10−5 |
| hydrolase activity, hydrolyzing O-glycosyl compounds | GO:0004553 | 16 | 132 | 0.01493863 |
| cofactor binding | GO:0048037 | 15 | 71 | 5.65 × 10−5 |
| FAD binding | GO:0050660 | 15 | 138 | 0.04270059 |
| transcription factor activity | GO:0003700 | 15 | 409 | 0.9974484 |
| regulation of transcription, DNA-dependent | GO:0006355 | 15 | 456 | 0.999605 |
| proteolysis | GO:0006508 | 14 | 184 | 0.3491031 |
| phosphopantetheine binding | GO:0031177 | 13 | 50 | 1.71 × 10−5 |
| unspecific monooxygenase activity | GO:0050381 | 12 | 44 | 2.13 × 10−5 |
| nucleic acid binding | GO:0003676 | 12 | 394 | 0.9996286 |
| F420H2 dehydrogenase activity | GO:0043738 | 11 | 47 | 0.000211107 |
| malolactic enzyme activity | GO:0043883 | 11 | 47 | 0.000211107 |
| regulation of oxidoreductase activity | GO:0051341 | 11 | 47 | 0.000211107 |
| sulfur oxygenase reductase activity | GO:0043826 | 11 | 47 | 0.000211107 |
| DNA integration | GO:0015074 | 10 | 26 | 3.37 × 10−6 |
| peroxidase activity | GO:0004601 | 10 | 29 | 1.06 × 10−5 |
| aromatic compound metabolic process | GO:0006725 | 10 | 66 | 0.01182745 |
| ATP binding | GO:0005524 | 10 | 554 | 1 |
Not all genes could be annotated by GO (only 56% Aspfl, 59% Aspfu and 59% Aspni have GO annotations), which are listed by the “un-annotated by GO” category in the first line of each table. The tables show that un-annotated genes are enriched in the HTG sets for Aspfu and Aspfl, but not in the HTG set for Aspni. “Metabolic process” and “catalytic activity,” the two high-level GO categories that involve most enzymes in the genome, are enriched in the HTG sets for Aspfl and Aspni, but not in the HTG set for Aspfu. Compared to the other two genomes, Aspfu has four unique GO categories: “ribonuclease H activity,” “RNA-dependent DNA replication,” “RNA-directed DNA polymerase activity,” and “RNA binding” that have the lowest P values (most enriched). These four categories are very much redundant with each other sharing 14 HTGs. A keyword search of these 14 genes at NCBI found that these genes were annotated as “reverse transcriptase, RNaseH.” They are now labeled as “discontinued.” This is probably because they were contaminants or mistakenly predicted genes originally submitted by the data producer, but were later removed by NCBI. Since the genome data used in this paper was downloaded from JGI, these genes were included in our analyses. In Aspni, most of the top GO categories are enriched in HTGs, which is not surprising because Aspni has a higher percentage of HTGs (6.7%) than Aspfl (4.2%) and Aspfu (2.8%).
We have performed the similar hypergeometric enrichment tests on the KEGG (Kyoto Encyclopedia of Genes and Genomes pathway) and KOG (Eukaryotic Orthologous Groups of proteins) annotations for the HTGs by comparing them with the genome background. The results also showed that more functional categories of KEGG and KOG are enriched in HTGs of Aspni than Aspfu and Aspfl. Interestingly, the “Biosynthesis of Secondary Metabolites” category is enriched in HTGs of Aspni (P value = 0.002) and Aspfu (P value = 0.04). In Aspni, “Carbohydrate Metabolism” (P value = 0.0007), “Lipid Metabolism” (P value = 0.002), and “Metabolism of Other Amino Acids” (P value = 0.04) are all enriched in HTGs.
2.6. Sequence Properties of HTGs: Guanine Cytosine, Length, Ka, Ks
In bacteria, HTGs were shown to have a lower GC content and more relaxed selection [
34,
35,
36]. In
Table 5, we have compared the sequence properties of HTGs and non-HTGs in the three fungi. We found that, in all three fungi, HTGs have significantly shorter length, higher GC content at the third position of codons (GC3), higher
Ka (the number of nonsynonymous substitutions per non-synonymous site), higher
Ks (the number of synonymous substitutions per synonymous site), and higher
Ka/
Ks ratio. We used GC at the third position of codons because the third position is more freely changeable and less affected by translational selection than the other two positions. In bacteria, the lower GC content of HTGs might be related to the suppression of gene expression of HTGs [
37]. Hence, it is surprising that, in opposition to what is found in bacteria, fungi HTGs have higher GC content than non-HTGs. The shorter length of HTGs might be due to the simpler protein domain architectures [
38] in HTGs.
Table 5.
Sequence properties of HTGs vs. non-HTGs in the three genomes.
Table 5.
Sequence properties of HTGs vs. non-HTGs in the three genomes.
| In Parentheses are Hypotheses Supported by the Wilcoxon Rank Tests | Aspfu | Aspfl | Aspni |
|---|
| Length median/mean | HTG | 861/1143 | 620/824 | 1170/1312 |
| non-HTG | 1257/1487 | 1185/1408 | 1239/1466 |
| P value (shorter in HTG) | <2.2 × 10−16 | <2.2 × 10−16 | 4.32 × 10−6 |
| GC3 median/mean | HTG | 0.64/0.63 | 0.57/0.57 | 0.62/0.61 |
| non-HTG | 0.59/0.60 | 0.56/0.57 | 0.58/0.59 |
| P value (higher in HTG) | 2.29 × 10−7 | 0.003305 | <2.2 × 10−16 |
| Ka median/mean | HTG | 0.04/0.11 | 0.41/0.42 | 2.21/2.27 |
| non-HTG | 0.02/0.06 | 0.20/0.28 | 1.53/1.66 |
| P value (higher in HTG) | 5.10 × 10−15 | 6.684 × 10−13 | <2.2 × 10−16 |
| Ks median/mean | HTG | 0.17/0.48 | 1.86/2.09 | 0.62/0.58 |
| non-HTG | 0.12/0.22 | 1.72/1.83 | 0.19/0.27 |
| P value (higher in HTG) | <2.2 × 10−16 | 0.01375 | <2.2 × 10−16 |
| Ka/Ks median/mean | HTG | 0.23/0.30 | 0.25/0.35 | 0.26/0.27 |
| non-HTG | 0.20/0.24 | 0.12/0.16 | 0.13/0.15 |
| P value (higher in HTG) | 0.0004822 | 3.264 × 10−13 | <2.2 × 10−16 |
Ka measures the nucleotide substitutions that cause amino acid changes, which are under very strong selection pressure, while Ks measures the nucleotide substitutions that do not lead to amino acid changes, which are more neutral to selection. The Ka/Ks ratio is widely used as a proxy to evaluate the intensity of selection. For most genes, this ratio should be close to 0 due to purifying selection (most nucleotide substitutions in the coding regions do not change the protein products). For genes that are newly incorporated into the host genome, it is not surprising that, in order to explore the new environment and network, they are allowed to have more freedom to change, in sequence, under a more relaxed selection pressure.
2.7. Horizontally Transferred Gene Clusters (HTGCs)
The evolution of metabolic gene clusters (MGCs), especially those involved in secondary metabolism, are affected by HGT [
15]. A dozen HGT cases in MGCs have been summarized in a recent review [
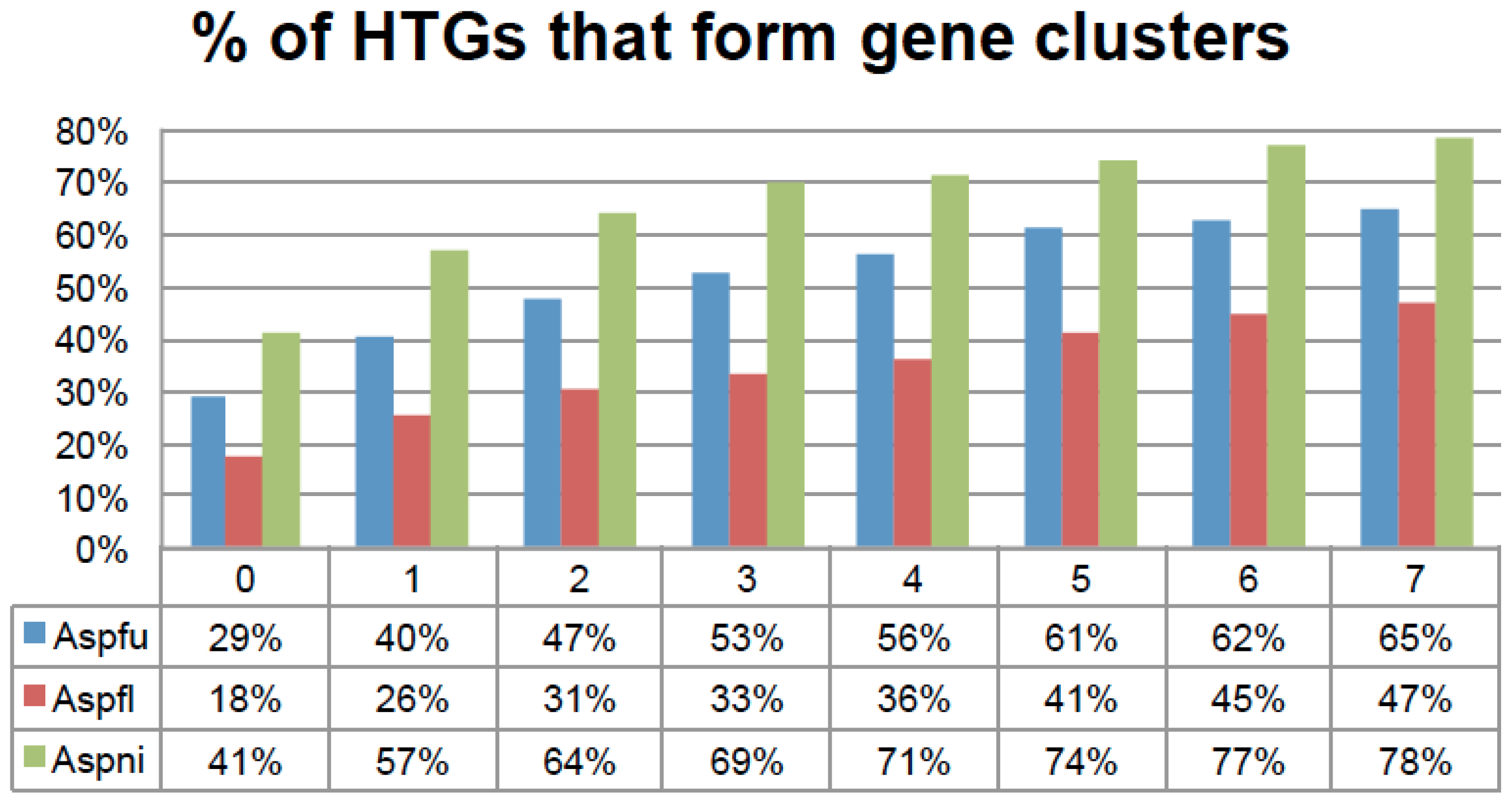
39]. We have implemented a program in the HGT-Finder software to examine the genomic locations of HTGs and further derive horizontally transferred gene clusters (HTGCs). We defined an HTGC as a group of physically linked genes containing at least two HTGs separated by less than
N non-HTGs, where
N was explored from 0 to 7 (
Figure 6 and
Table S5). We have also tried to add another restriction: the base pair distance between two adjacent genes in the HTGC should be less than 10 kb, which appeared to have little effect on the results (
Table S5).
Figure 6.
The percentage of HTGs that form physically linked gene clusters on chromosomes. The x-axis is the N thresholds and the y-axis is the percentage of HTGs. N is used to define gene clusters. For example if two HTGs are separated by less than N non-HTGs, these N + 2 genes will belong to one gene cluster. More HTGs will be included until the N threshold is not met. N is explored from 0 to 7 in this figure.
Figure 6.
The percentage of HTGs that form physically linked gene clusters on chromosomes. The x-axis is the N thresholds and the y-axis is the percentage of HTGs. N is used to define gene clusters. For example if two HTGs are separated by less than N non-HTGs, these N + 2 genes will belong to one gene cluster. More HTGs will be included until the N threshold is not met. N is explored from 0 to 7 in this figure.
For N = 5, the 273 Aspfu, 542 Aspfl, and 715 Aspni HTGs yielded 57, 84, and 129 HTGCs, respectively, which encompass 326, 421, and 1034 genes in total including 167 (61%), 215 (41%), and 530 (74%) HTGs. A permutation experiment that randomly selected (100 times) the same amount of genes from the genome and then ran our gene clustering program suggested that such gene clustering of HTGs is not random but statistically significant (P value = 4.1 × 10−246 for Aspfu, P value = 0.01 for Aspfl, and P value = 3.7 × 10−23 for Aspni when N = 5). This is an indication that these HTGs have a very strong tendency to form physically linked gene clusters.
2.8. Overlap between Horizontally Transferred Gene Clusters (HTGCs) and Secondary Metabolism Gene Clusters
We went further to investigate how our predicted HTGCs overlap with the secondary metabolism gene clusters (SMGCs). We obtained a list of manually curated SMGCs for Aspfu (251 genes of 33 clusters) and Aspni (458 genes of 65 clusters) from [
40]. Comparing these genes with the HTGs in the HTGCs of the two genomes, we found that: (i) in Aspni, 98 of the 458 SMGs are HTGs (
Table S6), a hypergeometric test returned a
P value = 8.6 × 10
−27, suggesting SMGs are very much enriched in the HTG set; and similarly (ii) in Aspfu, 22 of the 251 SMGs are HTGs (
Table S6) with a hypergeometric test
P value = 1.9 × 10
−6, also supporting that SMGs are enriched in the HTG set.
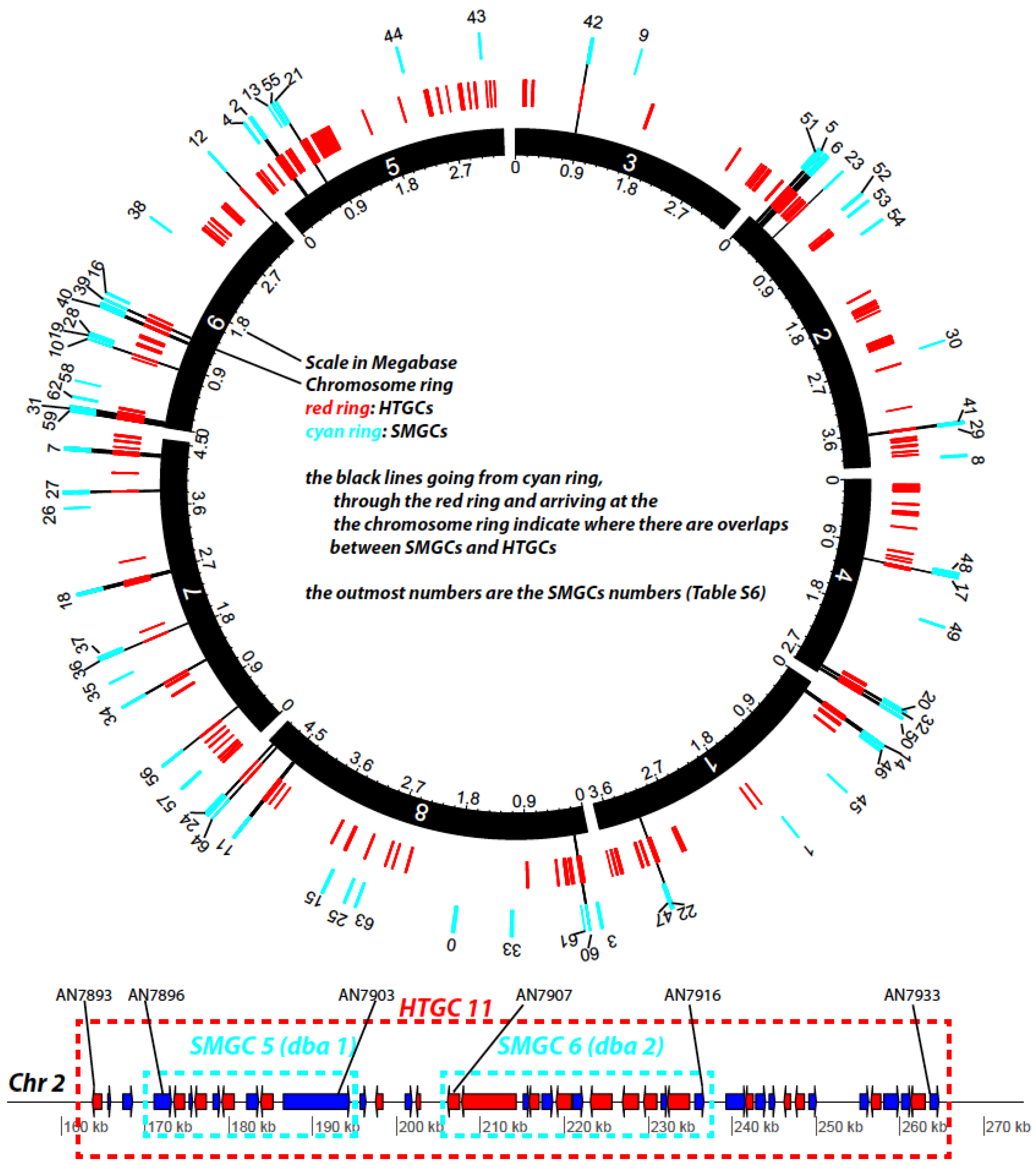
Figure 7 provides an overall representation of the 65 SMGCs (cyan ring) and 129 HTGCs (red ring) on the Aspni chromosomes. The bottom of the figure shows an example of two adjacent SMGCs, Derivative of Benzaldehyde1 (dba) and F9775 hybrid cluster 1 (named dba 1 in the figure, nine genes) and Derivative of Benzaldehyde1 (dba) and F9775 hybrid cluster 2 (named dba 2, 10 genes), being enclosed by the large HTGC 11 (42 genes). All nine genes in dba 1 are HTGs and eight of the 10 genes in dba 2 are HTGs. The detailed information about the component genes is provided in
Table S6. A hypothesis was proposed 15 years ago that HGTs play a significant role in the evolution of SMCs in fungi [
41]. Our genome-wide analysis presented here provides very strong evidence to support this hypothesis.
2.9. Comparisons with Published Results and Tools
In
Section 2.3, we showed that most predictions made by HGT-Finder are supported by phylogenetic analysis. One question remains: how do HGT-Finder predictions compare with published results? We have compared the predictions of HGT-Finder with published HTG sets for the three
Aspergillus genomes.
Aspfu has been surveyed previously for HGTs using a composition-based method [
17] where 214 genes were reported to be horizontally transferred. Aspfu, Aspfl, and Aspni have also been studied for prokaryotes-fungi gene transfers using a phyletic distribution method followed by phylogenetic analysis [
14]; 20 Aspfu, 45 Aspfl, and 14 Aspni genes were found to be HTGs from prokaryotes (named TIG2010 set here). For Aspfu, we have compared our 273 HTG set with the composition-based 214 HTG set, and found that 16 (7.5% of 214) HTGs were shared by both sets. This is not surprising because it is known that composition-based methods tend to identify different HTGs compared to other methods [
16,
24,
25]. We have also compared our HGT-Finder sets (273 Aspfu, 542 Aspfl, and 715 Aspni) against the TIG2010 sets. We found that six (30% of 20) Aspfu, 12 (26.7% of 45) Aspfl, and four (28.6% of 14) Aspni were shared between the HGT-Finder and the TIG2010 sets. These percentages suggest that HGT-Finder might have missed many prokaryotes-fungi HTGs. Another explanation is that, with new genome data added to the database, many HTGs found in TIG2010 now turned out to be non-HTGs. It should be noted that TIG2010 just focused on prokaryotes-fungi gene transfers and our HGT-Finder can find transfers from all kinds of organisms.
We have further compared HGT-Finder with DarkHorse [
28], one of the four published phyletic distribution-based softwares. DarkHorse was selected for comparison because it was a relatively recent development and the easiest to install and run based on our own experience. Other tools are either very difficult to install or require extensive human intervention to run. DarkHorse ranked genes based on a “lineage probability index” (
LPI) that has a range between 0 and 1. Although it does not provide a statistical distribution-based probability value for each gene, according to its tutorial, an empirical LPI score <0.6 is recommended to be a safe cutoff to call HTGs. After running DarkHorse on the three fungal genomes with an LPI threshold of
LPI < 0.6 and default parameters, we found 231 Aspfl and 397 Aspni HTGs, but only three Aspfu HTGs. Overlapping these DarkHorse sets with our HGT-Finder sets revealed that no Aspfu HTGs, 102 (44% of 231) Aspfl HTGs, and 74 (19% of 397) Aspni HTGs are shared by the two programs.
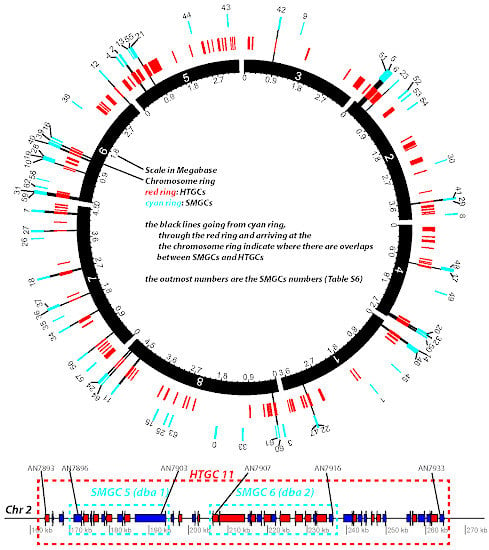
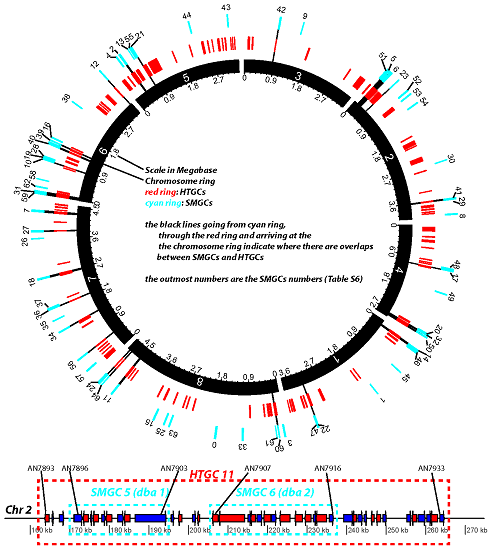
Figure 7.
Diagram representation of HTGCs and SMGCs in Aspni. The top graph is a Circos plot [
42] of the chromosomal distribution of HTGCs and SMGCs in Aspni. The outmost numbers are the IDs of SMGCs, which were extracted from [
40]. The functional descriptions of these SMGCs are available in
Table S6. The bottom linear graph, as an example of overlapping between HTGCs and SMGCs, shows the detailed genomic neighborhood of SMGC 5 and 6 (cyan frames) as well as the overlapping HTGC 11 (red frame).
Figure 7.
Diagram representation of HTGCs and SMGCs in Aspni. The top graph is a Circos plot [
42] of the chromosomal distribution of HTGCs and SMGCs in Aspni. The outmost numbers are the IDs of SMGCs, which were extracted from [
40]. The functional descriptions of these SMGCs are available in
Table S6. The bottom linear graph, as an example of overlapping between HTGCs and SMGCs, shows the detailed genomic neighborhood of SMGC 5 and 6 (cyan frames) as well as the overlapping HTGC 11 (red frame).
This surprising finding suggests that, just like the composition-based method, different phyletic distribution-based methods also produce very different HTG predictions. Therefore, it is not wise to use one surrogate method’s prediction to evaluate the other surrogate method’s performance. Gene-by-gene phylogenetic analysis, although performed at a much lower throughput, is the only gold-standard method to evaluate any HGT detection programs. A common practice in the literature is to take a two-step approach: run surrogate tools to narrow down to a short list of HTG candidates (e.g., from 10,000 to a few hundred genes for a typical fungal genome), and then use phylogenetic analysis to verify the candidates on a gene-by-gene basis [
10,
19]. Since different phyletic distribution-based tools tend to identify different sets of HTGs, our recommendation is to combine outputs from multiple tools and then perform phylogenetic analysis. HGT-Finder will be a very valuable addition to the toolbox of HGT research because many of its predictions can be verified by phylogenetic analysis, it is fully automated, and much easier to install and run.
HGT-Finder requires pre-annotated genomes (
i.e., protein-coding genes should be predicted prior to the HGT-Finder run). Because a statistical distribution of
X values is needed for the
P and
Q value calculation, HGT-Finder will work best for genome-scale HGT detection and may not work for individual genes. The HGT-Finder program with source code, example files (with Aspfl BLAST output), and documents are freely available at
http://cys.bios.niu.edu/HGTFinder/HGTFinder.tar.gz, which can be run on command-line terminals of OS X and Linux computers.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

