
Uptake of Clostridium botulinum C3 Exoenzyme into Intact HT22 and J774A.1 Cells

Abstract
:1. Introduction
2. Results
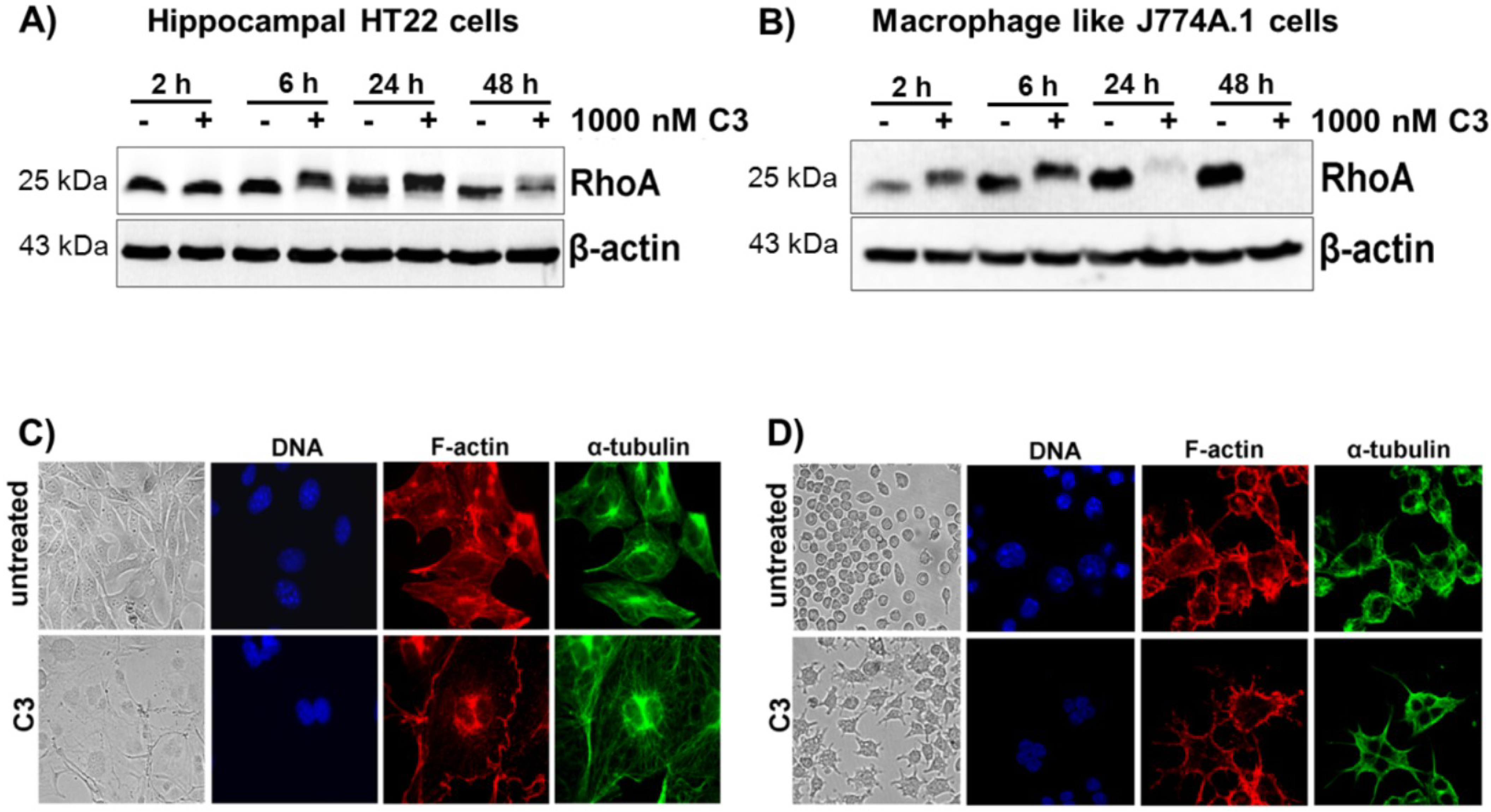
2.1. Cellular Susceptibility of HT22 and J774A.1 Cells towards the C3 Exoenzyme

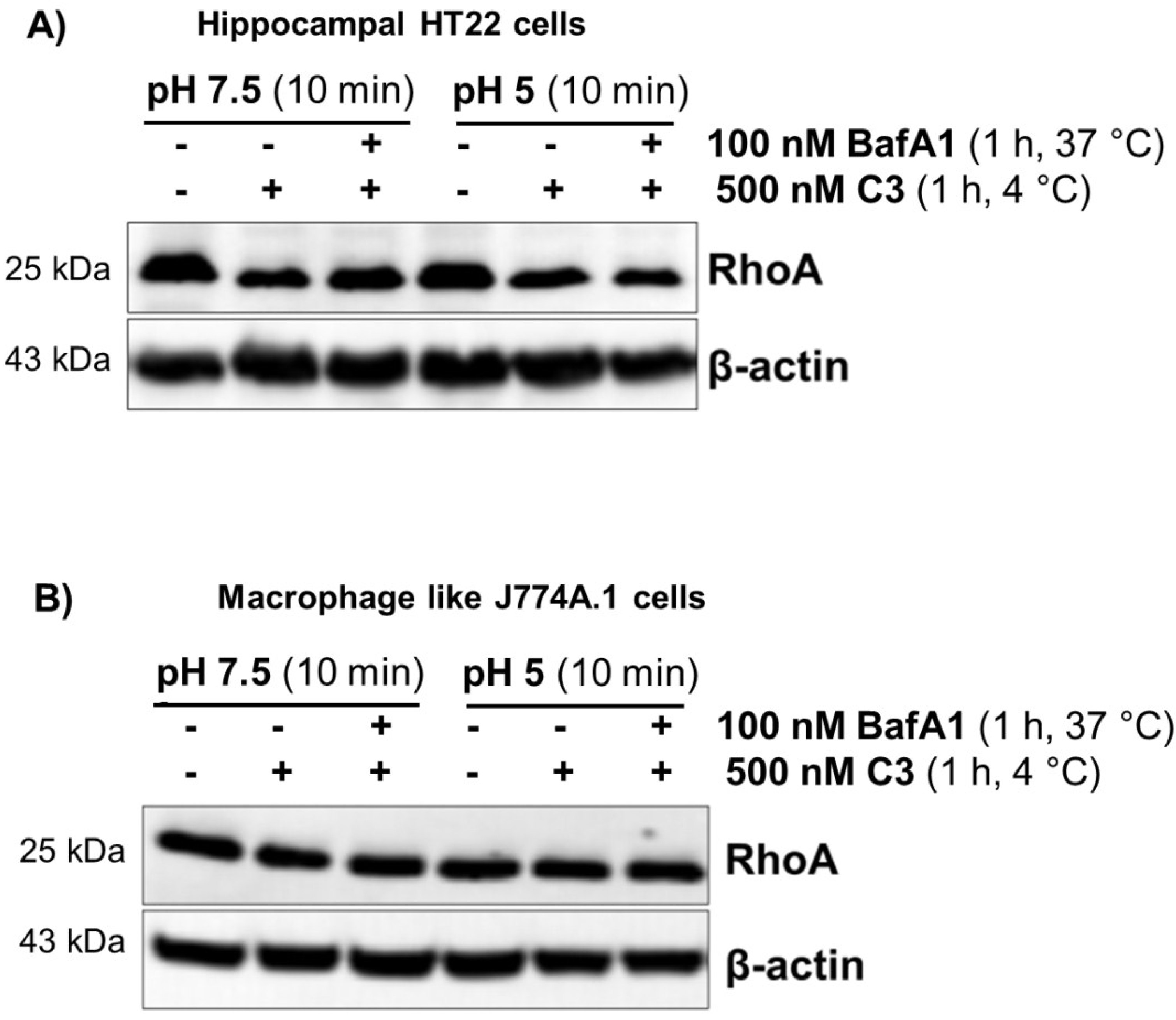
2.2. Acidification of Endosomes is not Essential for the Uptake of C3

2.3. Cholesterol is not Involved in the Uptake of C3


2.4. Intermediate Filaments are Involved in the Uptake of C3

2.5. Dynasore Inhibits the Uptake of C3

3. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism | HT22 | J774A.1 |
|---|---|---|---|
| Bafilomycin A1 | blocks endosomal acidification | − | − |
| Brefeldin A | blocks protein trafficking | − | − |
| Methyl-β-cyclodextrin (MBCD) | blocks caveolin-dependent endocytosis | − | − |
| Filipin | blocks caveolin-dependent endocytosis | − | − |
| Nystatin | blocks caveolin-dependent endocytosis | − | − |
| Chlorpromazine | blocks clathrin-dependent endocytosis | − | − |
| Latrunculin B | blocks actin polymerization | − | − |
| Nocodazole | blocks polymerization of microtubules | − | − |
| Acrylamide | interrupts vimentin filament network | + | + |
| Dynasore | blocks dynamin-dependent endocytosis | + | + |

4. Experimental Section
4.1. Cell Culture
4.2. Expression and Purification of Recombinant C3 Protein
4.3. Western Blot Analysis
4.4. Immunocytochemistry
4.5. Confocal Laser Scanning Microscopy
4.6. ADP-Ribosylation of Rho in Murine Cells
4.7. Reproducibility of the Experiments and Statistics
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Aktories, K.; Frevert, J. ADP-ribosylation of a 21–24 kDa eukaryotic protein(s) by C3, a novel botulinum ADP-ribosyltransferase, is regulated by guanine nucleotide. Biochem. J. 1987, 247, 363–368. [Google Scholar]
- Just, I.; Selzer, J.; Jung, M.; van Damme, J.; Vandekerckhove, J.; Aktories, K. Rho-ADP-ribosylating exoenzyme from Bacillus cereus. Purification, characterization, and identification of the NAD-binding site. Biochemistry 1995, 34, 334–340. [Google Scholar] [CrossRef]
- Wilde, C.; Chhatwal, G.S.; Schmalzing, G.; Aktories, K.; Just, I. A Novel C3-like ADP-ribosyltransferase from Staphylococcus aureus Modifying RhoE and Rnd3. J. Biol. Chem. 2001, 276, 9537–9542. [Google Scholar] [CrossRef] [PubMed]
- Wilde, C.; Vogelsgesang, M.; Aktories, K. Rho-specific Bacillus cereus ADP-Ribosyltransferase C3cer cloning and characterization. Biochemistry 2003, 42, 9694–9702. [Google Scholar] [CrossRef] [PubMed]
- Wilde, C.; Genth, H.; Aktories, K.; Just, I. Recognition of RhoA by Clostridium botulinum C3 exoenzyme. J. Biol. Chem. 2000, 275, 16478–16483. [Google Scholar] [CrossRef] [PubMed]
- Sekine, A.; Fujiwara, M.; Narumiya, S. Asparagine residue in the rho gene product is the modification site for botulinum ADP-ribosyltransferase. J. Biol. Chem. 1989, 264, 8602–8605. [Google Scholar] [PubMed]
- Wiegers, W.; Just, I.; Müller, H.; Hellwig, A.; Traub, P.; Aktories, K. Alteration of the cytoskeleton of mammalian cells cultured in vitro by Clostridium botulinum C2 toxin and C3 ADP-ribosyltransferase. Eur. J. Cell Biol. 1991, 54, 237–245. [Google Scholar] [PubMed]
- Rohrbeck, A.; Kolbe, T.; Hagemann, S.; Genth, H.; Just, I. Distinct biological activities of C3 and ADP-ribosyltransferase-deficient C3-E174Q. FEBS J. 2012, 279, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Höltje, M.; Just, I.; Ahnert-Hilger, G. Clostridial C3 proteins: Recent approaches to improve neuronal growth and regeneration. Ann. Anat. 2011, 193, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Hauser, D.; Boquet, P.; Eklund, M.W.; Gill, D.M. Characterization of the C3 gene of Clostridium botulinum types C and D and its expression in Escherichia coli. Infect. Immun. 1991, 59, 3673–3679. [Google Scholar] [PubMed]
- Han, S.; Arvai, A.S.; Clancy, S.B.; Tainer, J.A. Crystal structure and novel recognition motif of rho ADP-ribosylating C3 exoenzyme from Clostridium botulinum: Structural insights for recognition specificity and catalysis. J. Mol. Biol. 2001, 305, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Rotsch, J.; Rohrbeck, A.; May, M.; Kolbe, T.; Hagemann, S.; Schelle, I.; Just, I.; Genth, H.; Huelsenbeck, S. Inhibition of macrophage migration by C. botulinum exoenzyme C3. Naunyn. Schmiedebergs. Arch. Pharmacol. 2012, 385, 883–890. [Google Scholar]
- Ahnert-Hilger, G.; Höltje, M.; Große, G.; Pickert, G.; Mucke, C.; Nixdorf-Bergweiler, B.; Boquet, P.; Hofmann, F.; Just, I. Differential effects of Rho GTPases on axonal and dendritic development in hippocampal neurones. J. Neurochem. 2004, 90, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; van der Goot, F.G. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010, 6, e1000792. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Zamboglou, C.; Genisyuerek, S.; Guttenberg, G.; Aktories, K. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 2010, 5, e10673. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.; Kannan, T.R.; Baseman, J.B. Mycoplasma pneumoniae CARDS Toxin Is Internalized via Clathrin-Mediated Endocytosis. PLoS One 2013, 8, e62706. [Google Scholar] [CrossRef] [PubMed]
- Mettlen, M.; Pucadyil, T.; Ramachandran, R.; Schmid, S.L. Dissecting dynamin’s role in clathrin-mediated endocytosis. Biochem. Soc. Trans. 2009, 37, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Rappuoli, R.; Murgia, M.; Montecucco, C. Cell penetration of diphtheria toxin: Reduction of the interchain disulfide bridge is the rate-limiting step of translocation in the cytosol. J. Biol. Chem. 1993, 268, 1567–1574. [Google Scholar] [PubMed]
- Lemichez, E.; Bomsel, M.; Devilliers, G.; van der Spek, J.; Murphy, J.; Lukianov, E.; Olsnes, S.; Boquet, P. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol. Microbiol. 1997, 23, 445–457. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, P.A.; Fishman, P.H. Filipin-dependent inhibition of cholera toxin: Evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 1998, 141, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Le, P.U.; Nabi, I.R. Ganglioside GM1 levels are a determinant of the extent of caveolae/raft-dependent endocytosis of cholera toxin to the Golgi apparatus. J. Cell Sci. 2004, 117, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Gibert, M.; Monier, M.N.; Ruez, R.; Lamaze, C.; Popoff, M. Endocytosis and toxicity of clostridial binary toxins depend on a clathrin-independent pathway regulated by Rho-GDI. Cell. Microbiol. 2011, 13, 154–170. [Google Scholar] [CrossRef]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Giesemann, T.; Jank, T.; Gerhard, R.; Aktories, K. Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 2006, 281, 10808–10815. [Google Scholar] [CrossRef]
- Sager, P.R. Cytoskeletal effects of acrylamide and 2,5-hexanedione: selective aggregation of vimentin filaments. Toxicol. Appl. Pharmacol. 1989, 97, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Eckert, B.S. Alteration of intermediate filament distribution in PtK1 cells by acrylamide. Eur. J. Cell Biol. 1985, 37, 169–174. [Google Scholar] [PubMed]
- Eckert, B.S. Alteration of the distribution of intermediate filaments in PtK1 cells by acrylamide. II: Effect on the organization of cytoplasmic organelles. Cell Motil. Cytoskelet. 1986, 6, 15–24. [Google Scholar] [CrossRef]
- Chardin, P.; McCormick, F. Brefeldin A: The advantage of being uncompetitive. Cell 1999, 97, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.J.; Kirchhausen, T.; Murthy, V.N. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 17955–17960. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Schmidt, G.; Just, I. Rho GTPases as Targets of Bacterial Protein Toxins. Biol. Chemistry 2000, 381, 421–426. [Google Scholar] [CrossRef]
- Molinari, G.; Rohde, M.; Wilde, C.; Just, I.; Aktories, K.; Chhatwal, G.S. Localization of the C3-like ADP-ribosyltransferase from Staphylococcus aureus during baterial invasion of mammalian cells. Inf. Immun. 2006, 74, 3673–3677. [Google Scholar] [CrossRef]
- Fahrer, J.; Kuban, J.; Heine, K.; Rupps, G.; Kaiser, E.; Felder, E.; Benz, R.; Barth, H. Selective and specific internalization of clostridial C3 ADP-ribosyltransferases into macrophages and monocytes. Cell Microbiol. 2010, 12, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Traub, L.M. Tickets to ride: Selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 2009, 10, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases—Nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, T.; Izumi, H.; Ise, T.; Murakami, T.; Uramoto, H.; Ishiguchi, H.; Yoshida, Y.; Tanabe, M.; Nomoto, M.; Kohno, K. Vacuolar H(+)-ATPase: Functional mechanisms and potential as a target for cancer chemotherapy. Anticancer Drugs 2002, 13, 237–243. [Google Scholar] [CrossRef]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Bowman, E.J.; Graham, L.A.; Stevens, T.H.; Bowman, B.J. The bafilomycin/concanamycin binding site in subunit c of the V-ATPases from Neurospora crassa and Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 33131–33138. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Nakano, Y.; Yokomakura, A.; Ishihara, K.; Kim, S.; Kang, Y.S.; Ohuchi, K. Nitric oxide production by the vacuolar-type (H+)-ATPase inhibitors bafilomycin A1 and concanamycin A and its possible role in apoptosis in RAW 264.7 cells. J. Pharmacol. Exp. Ther. 2006, 319, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Characterization of a nonclathrin endocytic pathway: Membrane cargo and lipid requirements. Mol. Biol. Cell 2004, 15, 3542–3552. [Google Scholar] [CrossRef] [PubMed]
- Eyster, C.A.; Higginson, J.D.; Porat-shliom, N.; Weigert, R.; Wu, W.W.; Shen, R.; Donaldson, J.G. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic 2009, 10, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Helenius, A. Endocytosis via caveolae. Traffic 2002, 3, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G.W. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Schröder, A.; Hagemann, S.; Pich, A.; Höltje, M.; Ahnert-Hilger, G.; Just, I. Vimentin mediates uptake of C3bot exoenzyme. PLoS One 2014, 9, e101071. [Google Scholar] [CrossRef] [PubMed]
- Styers, M.L.; Salazar, G.; Love, R.; Peden, A.A.; Kowalczyk, A.P.; Faundez, V. The endo-lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol. Biol. Cell 2004, 15, 5369–5382. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.K.; Connelly, R.J.; Pennathur, S.; Dubrovsky, L.; Haffar, O.K.; Bukrinsky, M.I. Anti-idiotypic antibody to the V3 domain of gp120 binds to vimentin: A possible role of intermediate filaments in the early steps of HIV-1 infection cycle. Viral Immunol. 1996, 9, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Risco, C.; Rodriguez, J.R.; Lopez-Iglesias, C.; Carrascosa, J.L.; Esteban, M.; Rodriguez, D. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 2002, 76, 1839–1855. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, J.J.; Inda, C.; Senin, S. B-Raf and CRHR1 internalization mediate biphasic ERK1/2 activation by CRH in hippocampal HT22 Cells. Mol. Endocrinol. 2013, 27, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Czerkies, M.; Borzecka, K.; Zdioruk, M.I.; Płóciennikowska, A.; Sobota, A.; Kwiatkowska, K. An interplay between scavenger receptor A and CD14 during activation of J774 cells by high concentrations of LPS. Immunobiology 2013, 218, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Lauvrak, S.U.; Torgersen, M.L.; Sandvig, K. Efficient endosome-to-Golgi transport of Shiga toxin is dependent on dynamin and clathrin. J. Cell Sci. 2004, 117, 2321–2331. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandvig, K.; Olsnes, S.; Brown, J.E.; Petersen, O.W.; van Deurs, B. Endocytosis from coated pits of Shiga toxin: A glycolipid-binding protein from Shigella dysenteriae 1. J. Cell Biol. 1989, 108, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [PubMed]
- Sandvig, K.; Grimmer, S.; Lauvrak, S. Pathways followed by ricin and Shiga toxin into cells. Histochem. Cell Biol. 2002, 117, 131–141. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohrbeck, A.; Von Elsner, L.; Hagemann, S.; Just, I. Uptake of Clostridium botulinum C3 Exoenzyme into Intact HT22 and J774A.1 Cells. Toxins 2015, 7, 380-395. https://doi.org/10.3390/toxins7020380
Rohrbeck A, Von Elsner L, Hagemann S, Just I. Uptake of Clostridium botulinum C3 Exoenzyme into Intact HT22 and J774A.1 Cells. Toxins. 2015; 7(2):380-395. https://doi.org/10.3390/toxins7020380
Chicago/Turabian StyleRohrbeck, Astrid, Leonie Von Elsner, Sandra Hagemann, and Ingo Just. 2015. "Uptake of Clostridium botulinum C3 Exoenzyme into Intact HT22 and J774A.1 Cells" Toxins 7, no. 2: 380-395. https://doi.org/10.3390/toxins7020380
APA StyleRohrbeck, A., Von Elsner, L., Hagemann, S., & Just, I. (2015). Uptake of Clostridium botulinum C3 Exoenzyme into Intact HT22 and J774A.1 Cells. Toxins, 7(2), 380-395. https://doi.org/10.3390/toxins7020380