
ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus ?-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes

Abstract
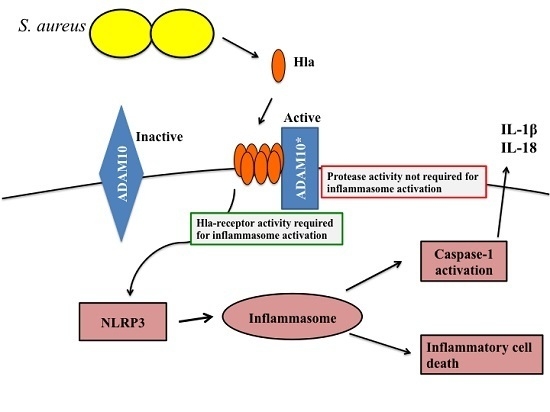
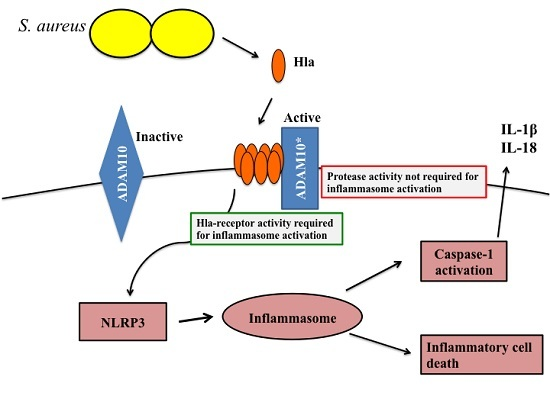
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
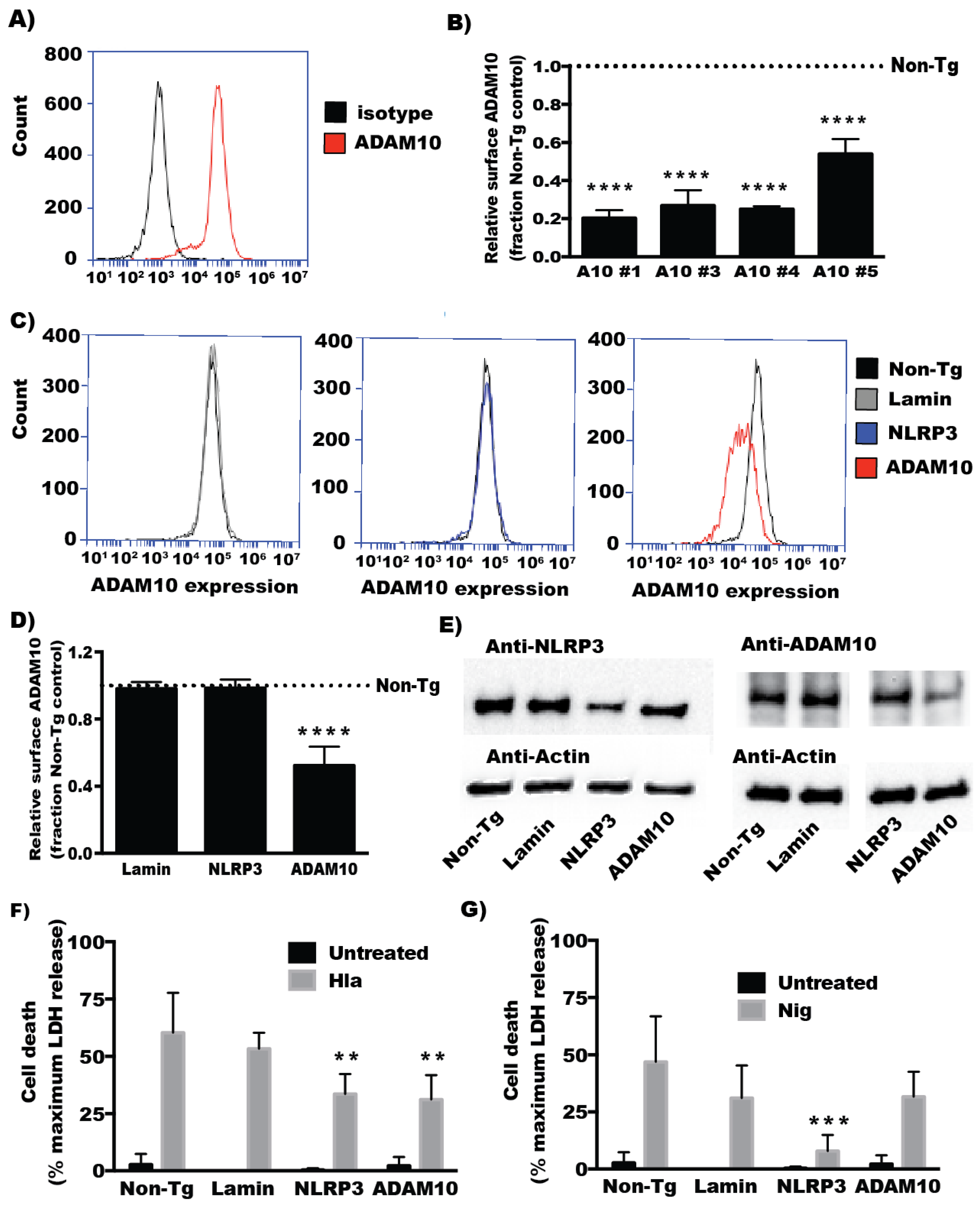
2.1. ADAM10 Expression Is Required for α-Hemolysin Induced Cell Death in Human Monocyte-Derived Cells
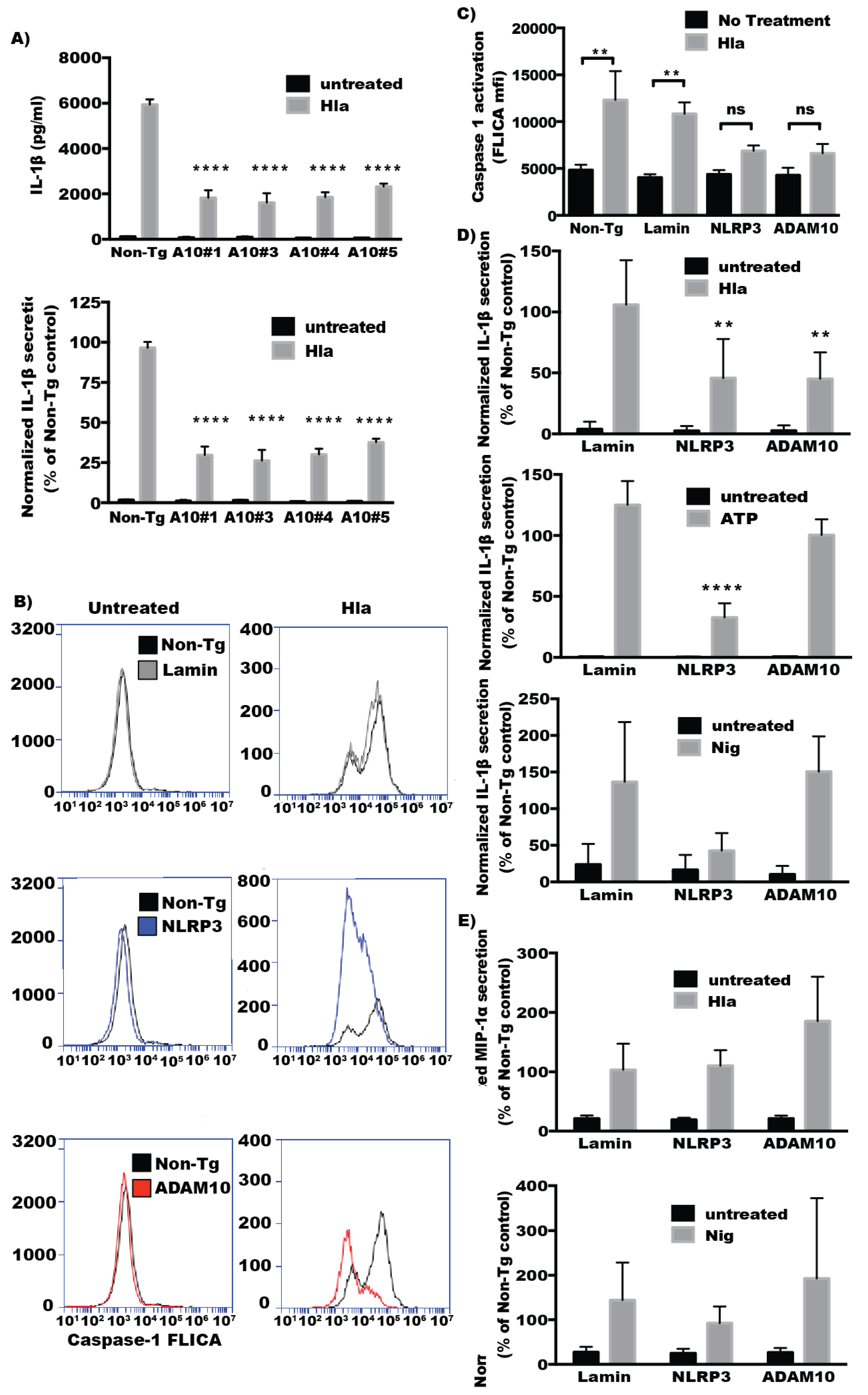
2.2. ADAM10 Expression Is Required for Hla-Mediated NLRP3 Inflammasome Activation in Monocytes
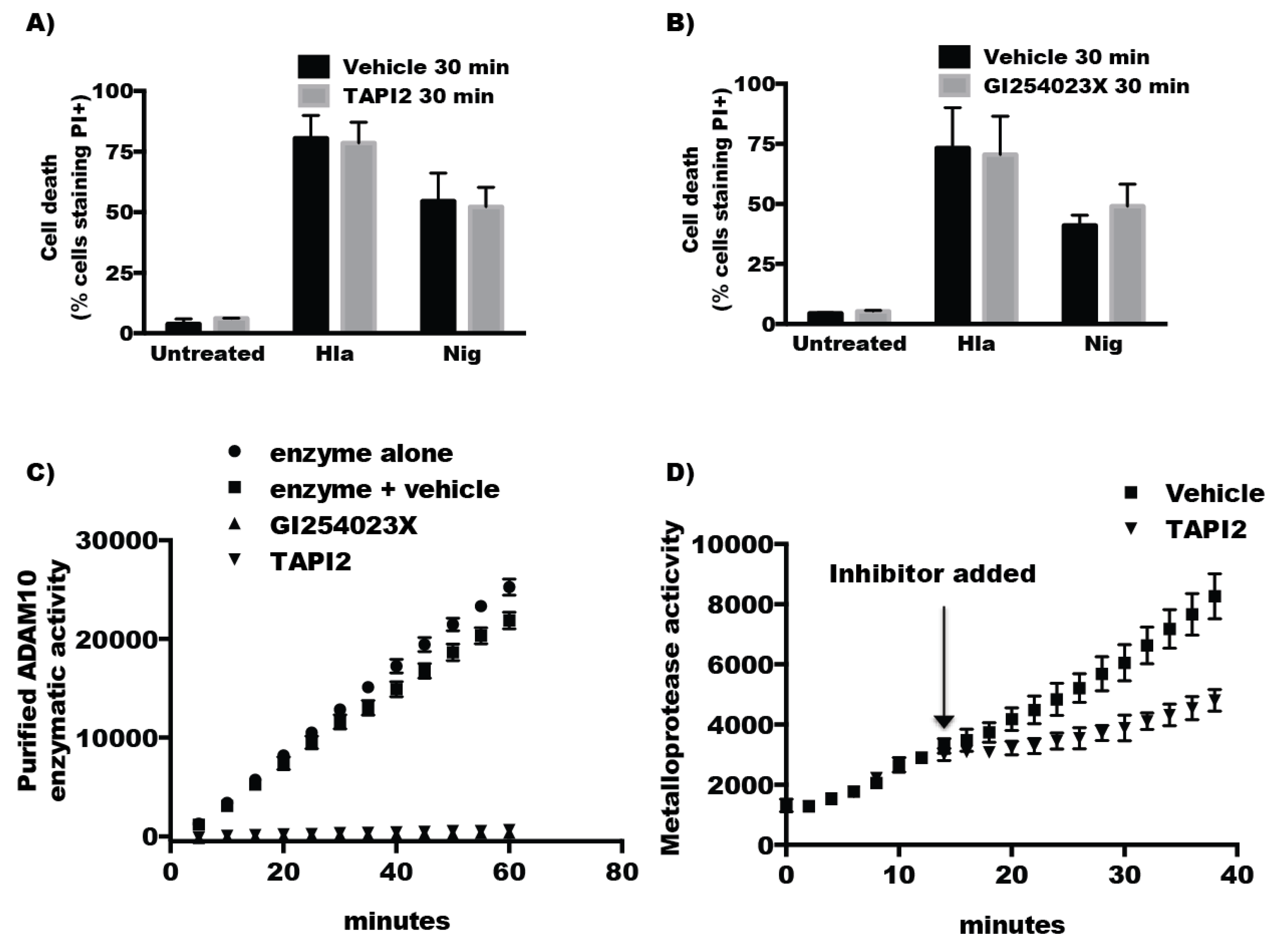
2.3. The Protease Activity of ADAM10 Is Not Required for Hla-Mediated Activation of NLRP3-Induced Cell Death
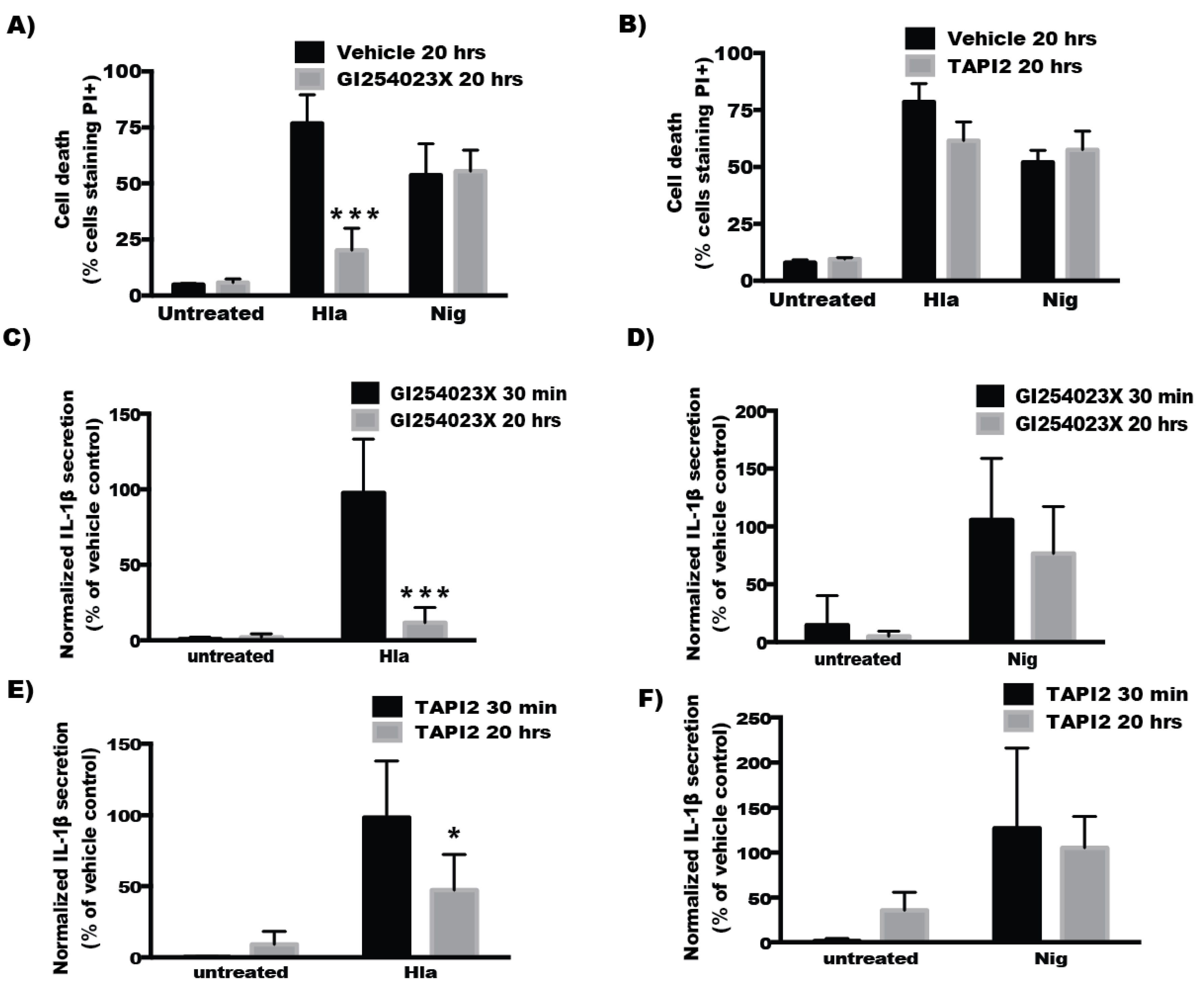
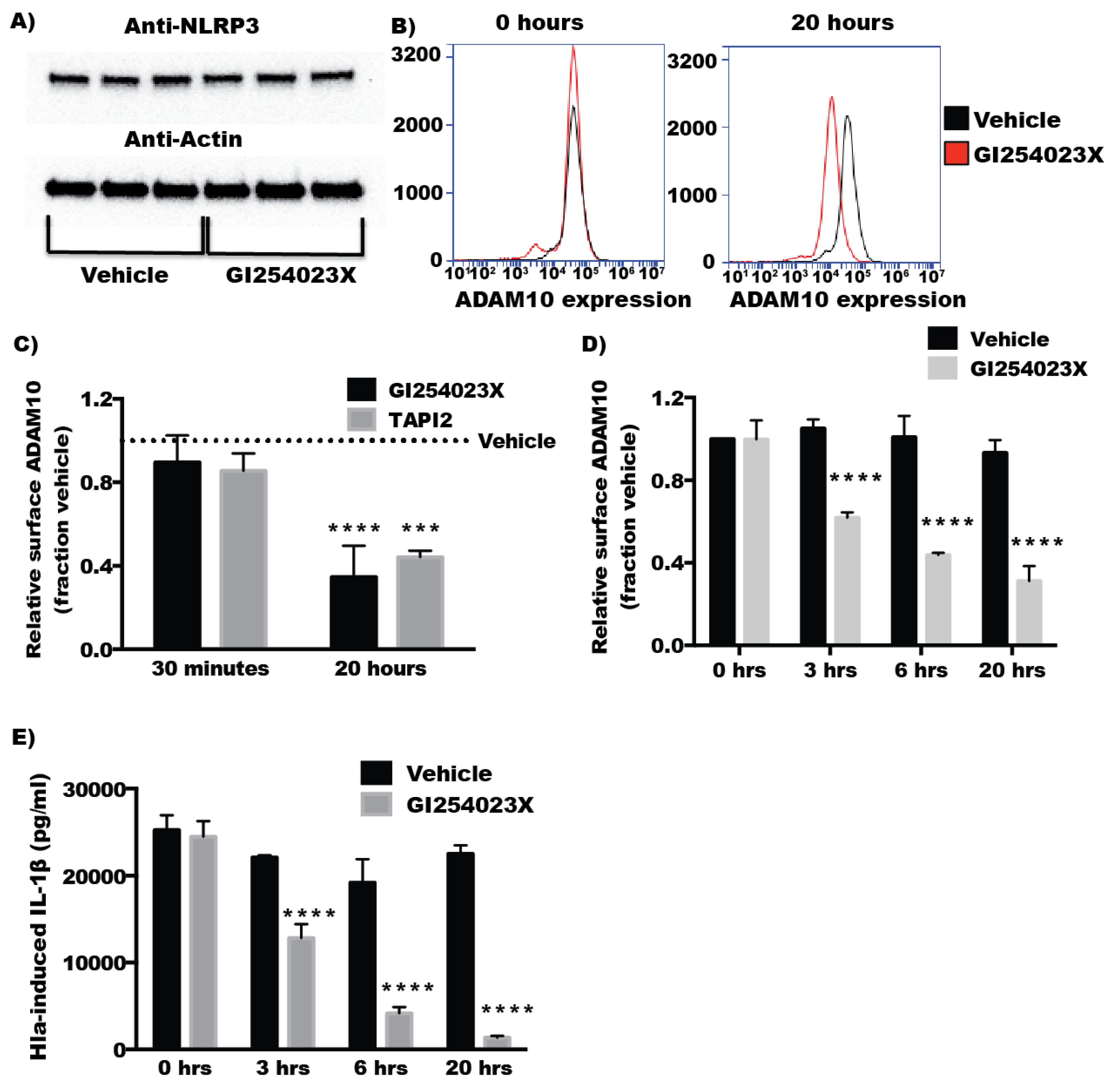
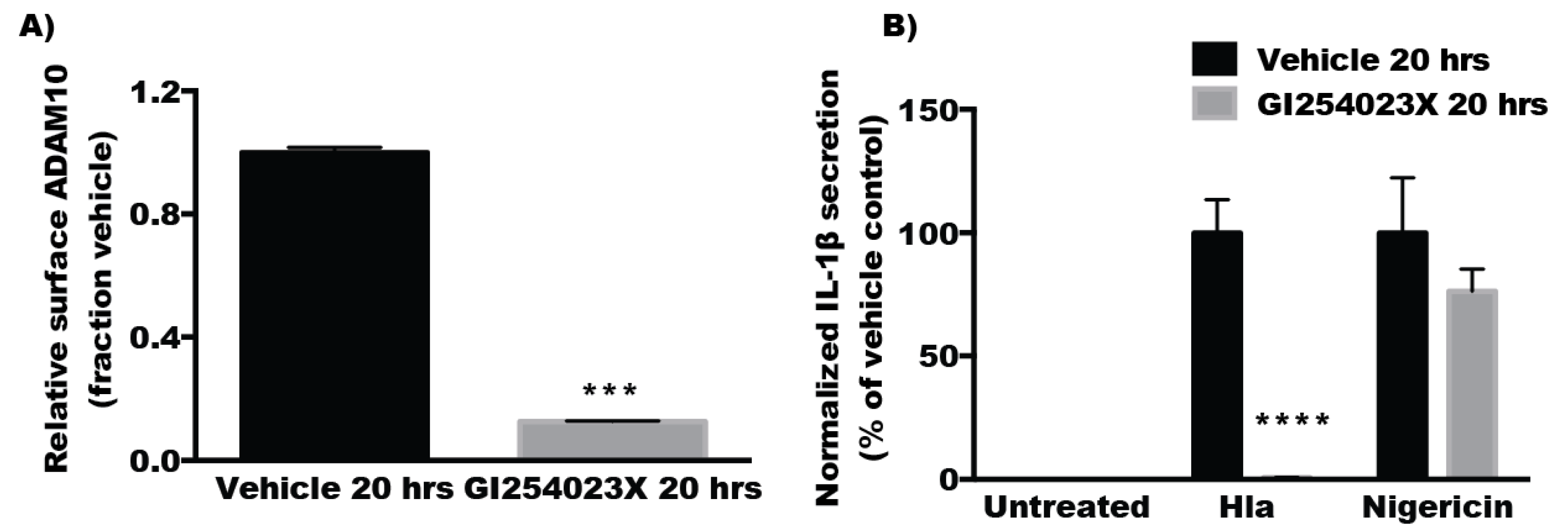
2.4. Inhibitors of ADAM10 Protease Activity Reduce Hla-Mediated Activation of the NLRP3 Inflammasome through down Regulation of Surface ADAM10 Levels
3. Experimental Section
3.1. siRNA Transfection of THP1 Cells
Immunotblot Analysis
3.2. Treatment of Cells with Inhibitors
3.3. Treatment of Cultured Cells with Inflammasome Activators for Cytokine Studies
3.4. Treatment of Cultured Cells with Inflammasome Activator for Cell Death Studies
3.5. Propidium Iodide Studies
3.6. Measurement of Caspase-1 Activity in Treated Cells
3.7. Measurement of ADAM10 Cell Surface Immunofluorescence Staining Protocol
3.8. Measurement of Metalloprotease Activity in Cells Treated with TAPI2
3.9. Purified ADAM10 Activity Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Franklin, D.L. Staphylococcus aureus infections. N. Eng. J. Med. 1998, 339, 520–532. [Google Scholar]
- Miller, L.; Perdreau-Remington, F.; Rieg, G.; Mehdi, S.; Perlroth, J.; Bayer, A.; Tang, A.; Phung, T.; Spellberg, B. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N. Eng. J. Med. 2005, 352, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Price, C.S. Community-acquired methicillin-resistant Staphylococcus aureus emerging as an important cause of necrotizing fasciitis. Surg. Infect. 2008, 9, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.; Doherty, M.; Lopatin, U.; Johnston, C.; Sinha, G.; Ross, T.; Cai, M.; Hansel, N.; Perl, T.; Ticehurst, J.; et al. Severe community-onset pneumonia in healthy adults caused by methicillin-resistant Staphylococcus aureus carrying the panton-valentine leukocidin genes. Clin. Infect. Dis. 2005, 40, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Bubeck Wardenburg, J.; Bae, T.; Otto, M.; Deleo, F.; Schneewind, O. Poring over pores: Alpha-hemolysin and panton-valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 2007, 13, 1405–1406. [Google Scholar] [CrossRef] [PubMed]
- Bubeck Wardenburg, J.; Patel, R.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.; Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Inoshima, I.; Inoshima, N.; Wilke, G.; Powers, M.; Frank, K.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Inoshima, N.; Wang, Y.; Bubeck Wardenburg, J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J. Investig. Dermatol. 2012, 132, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.; Kim, H.; Wang, Y.; Bubeck Wardenburg, J. ADAM10 mediates vascular injury induced by Staphylococcus aureus α-hemolysin. J. Infect. Dis. 2012, 206, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.E.; Berube, B.J.; Sampedro, G.R.; Dedent, A.C.; Bubeck Wardenburg, J. Tissue-specific patterning of host innate immune responses by Staphylococcus aureus α-toxin. J. Innate Immun. 2014, 6, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.E.; Becker, R.E.N.; Sailer, A.; Turner, J.R.; Bubeck Wardenburg, J. Synergistic action of Staphylococcus aureus α-toxin on platelets and myeloid lineage cells contributes to lethal sepsis. Cell Host Microbe 2015, 17, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.; Gao, X.; Allen, I.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.; Duncan, J. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Franchi, L.; Miller, L.; Núñez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the NLRP3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in muckle-wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Willingham, S.; Bergstralh, D.; O’Connor, W.; Morrison, A.; Taxman, D.; Duncan, J.; Barnoy, S.; Venkatesan, M.; Flavell, R.; Deshmukh, M.; et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2007, 2, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.; Allen, I.; Bergstralh, D.; Brickey, W.; Huang, M.; Taxman, D.; Duncan, J.; Ting, J. NLRP3 (NALP3, cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 2009, 183, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Malireddi, R.; Kanneganti, T.D. Role of the NLRP3 inflammasome in microbial infection. Front. Microbiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Kebaier, C.; Chamberland, R.; Allen, I.; Gao, X.; Broglie, P.; Hall, J.; Jania, C.; Doerschuk, C.; Tilley, S.; Duncan, J. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]
- John, S.C.; Yi, G.; Romela Irene, R.; Frank, H.; Seema, B.P.; Caiyun, X.; Jennifer, L.G.; Hironori, M.; Akira, T.; Yoichiro, I.; et al. Neutrophil-derived IL-1β is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012. [Google Scholar] [CrossRef]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [PubMed]
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.; Curti, A.; Idzko, M.; Panther, E.; di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Melehani, J.H.; James, D.B.; DuMont, A.L.; Torres, V.J.; Duncan, J.A. Staphylococcus aureus leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog. 2015. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. Regulation of the proteolytic disintegrin metalloproteinases, the “sheddases”. Semin. Cell Dev. Biol. 2009, 20, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Perret, M.; Badiou, C.; Lina, G.; Burbaud, S.; Benito, Y.; Bes, M.; Cottin, V.; Couzon, F.; Juruj, C.; Dauwalder, O.; et al. Cross-talk between Staphylococcus aureus leukocidins-intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome-dependent manner. Cell. Microbiol. 2012, 14, 1019–1036. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.P.; Ajao, A.O.; Aman, M.J.; Karauzum, H.; Sarwar, J.; Lydecker, A.D.; Johnson, J.K.; Nguyen, C.; Chen, W.H.; Roghmann, M.C. Lower antibody levels to Staphylococcus aureus exotoxins are associated with sepsis in hospitalized adults with invasive S. aureus infections. J. Infect. Dis. 2012, 206, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.A.; Tiemann, K.M.; Hogan, P.G.; Epplin, E.K.; Rodriguez, M.; Al-Zubeidi, D.N.; Wardenburg, J.B.; Hunstad, D.A. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin. Infect. Dis. 2013, 56, 1554–1561. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezekwe, E.A.D.; Weng, C.; Duncan, J.A. ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus ?-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes. Toxins 2016, 8, 95. https://doi.org/10.3390/toxins8040095
Ezekwe EAD, Weng C, Duncan JA. ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus ?-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes. Toxins. 2016; 8(4):95. https://doi.org/10.3390/toxins8040095
Chicago/Turabian StyleEzekwe, Ejiofor A.D., Chengyu Weng, and Joseph A. Duncan. 2016. "ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus ?-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes" Toxins 8, no. 4: 95. https://doi.org/10.3390/toxins8040095
APA StyleEzekwe, E. A. D., Weng, C., & Duncan, J. A. (2016). ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus ?-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes. Toxins, 8(4), 95. https://doi.org/10.3390/toxins8040095