
Whole Transcriptomic Analysis Provides Insights into Molecular Mechanisms for Toxin Biosynthesis in a Toxic Dinoflagellate Alexandrium catenella (ACHK-T)


Abstract
:1. Introduction
2. Results
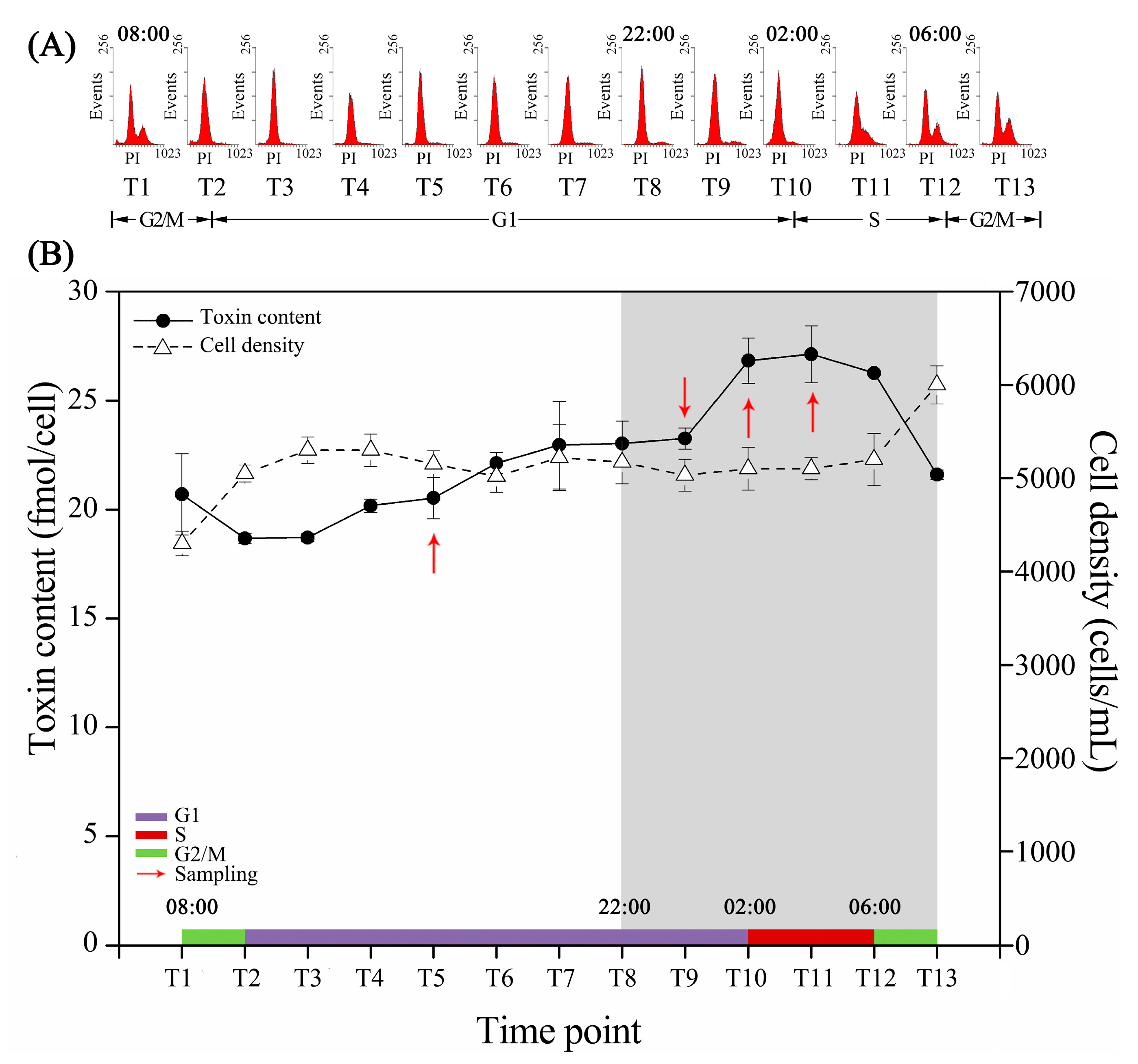
2.1. Cell Cycle and Intracellular Toxin Content
2.2. Transcriptome Sequencing and Assembly
2.3. Gene Functional Annotations
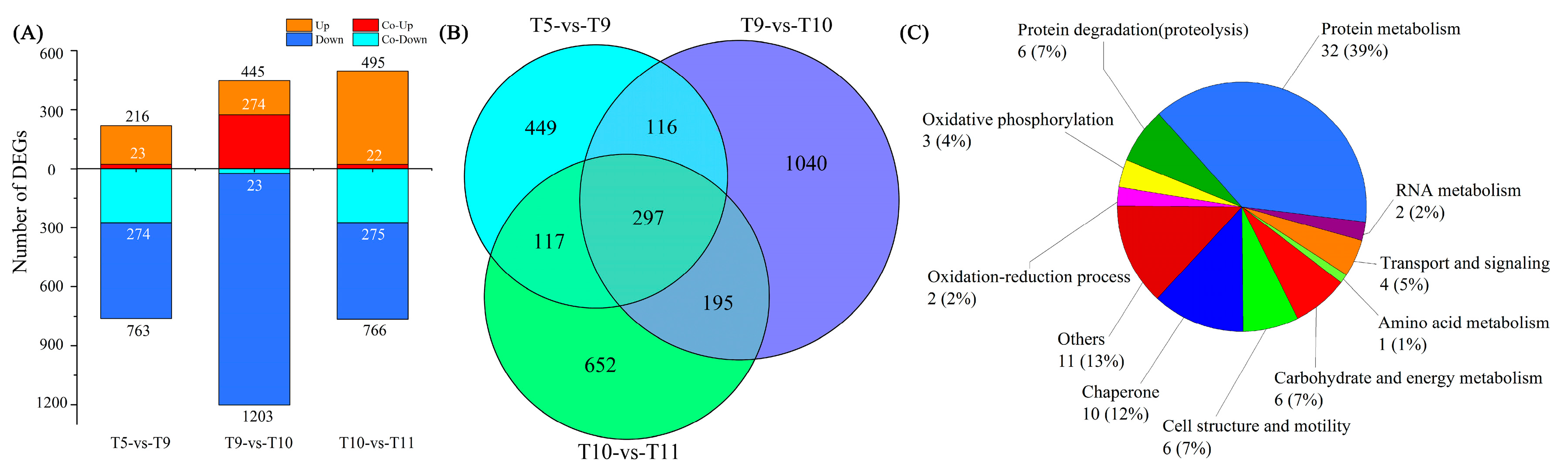
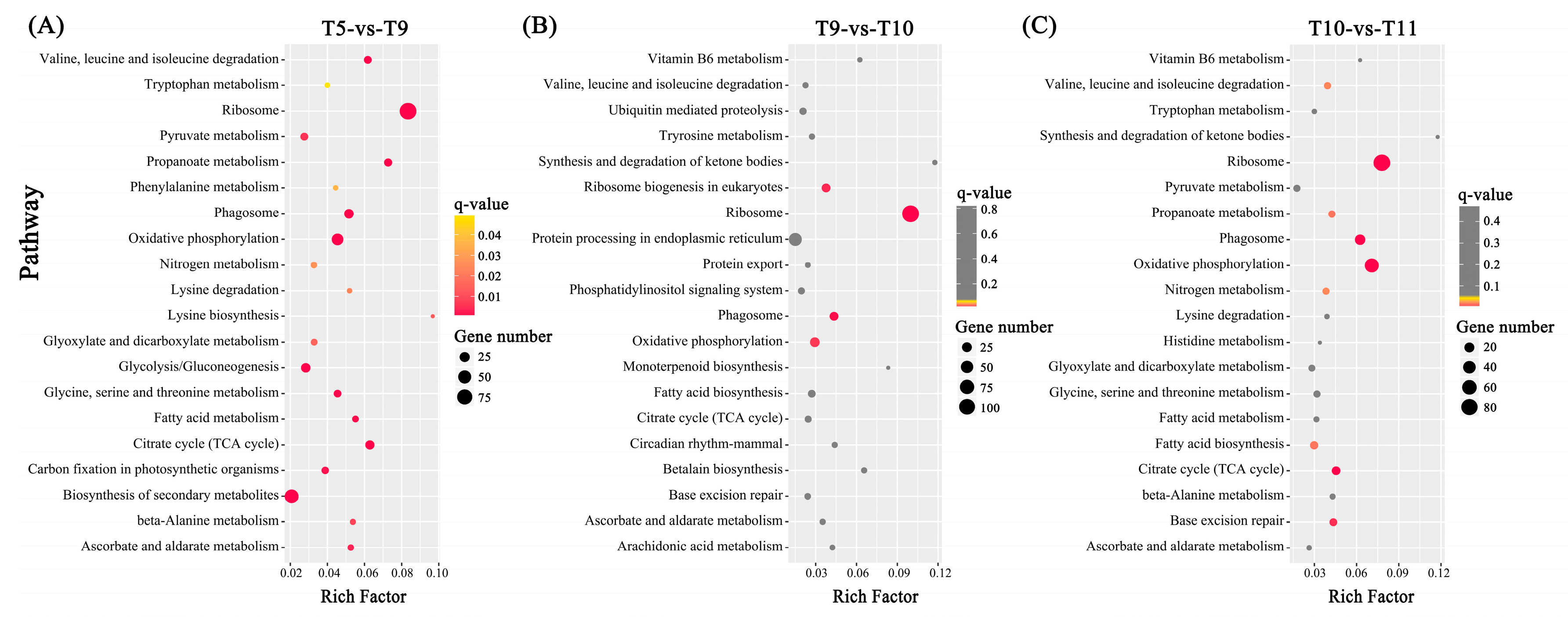
2.4. Differentially Expressed Genes (DEGs) among Four Samples
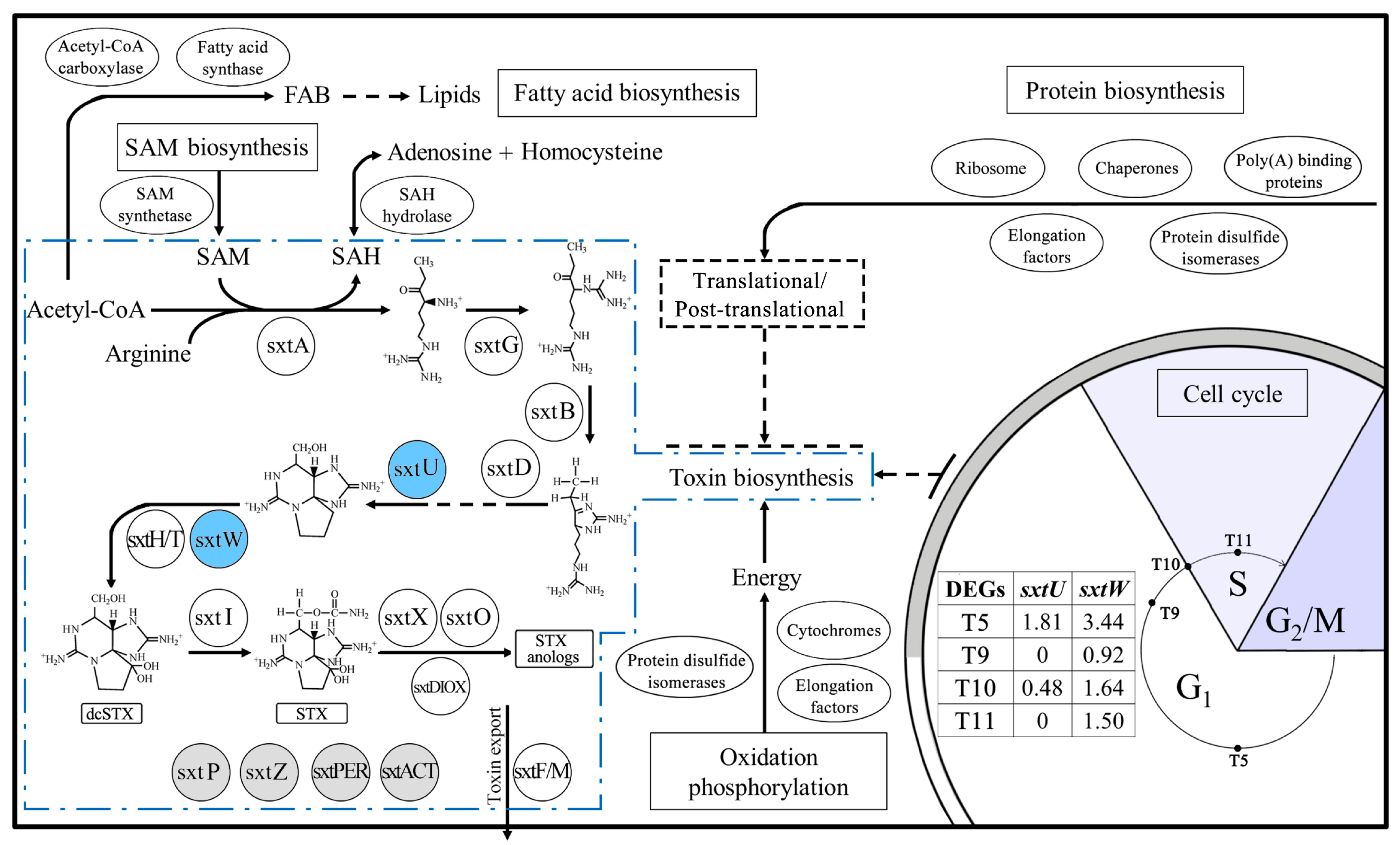
2.5. Genes Related to Toxin Biosynthesis
3. Discussion
3.1. Toxin Biosynthesis within the Cell Cycle
3.2. Variations of Toxin Related Genes at Different Toxin Biosynthesis Stages
3.3. Other Pathways Potentially Related to Toxin Biosynthesis
4. Conclusions
5. Materials and Methods
5.1. Culture Conditioning and Sample Collection
5.2. FCM and Intracellular Toxin Analysis
5.3. RNA Isolation
5.4. Transcriptome Sequencing
5.5. De Novo Assembly and Functional Annotations
5.6. DEGs Analysis
5.7. Identification of Sxt Genes
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Salcedo, T.; Upadhyay, R.J.; Nagasaki, K.; Bhattacharya, D. Dozens of toxin-related genes are expressed in a non-toxic strain of the dinoflagellate Heterocapsa circularisquama. Mol. Biol. Evol. 2012, 29, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y. Tetrodotoxin, saxitoxin and their significance in the study of excitation phenomena. Pharmacol. Rev. 1966, 18, 997–1049. [Google Scholar] [PubMed]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Hallegraeff, G.M. A review of harmful algal blooms and their apparent global increase. Phycologia 1993, 32, 79–99. [Google Scholar] [CrossRef]
- Kellmann, R.; Mihali, T.K.; Jeon, Y.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef] [PubMed]
- Mihali, T.K.; Kellmann, R.; Neilan, B.A. Characterisation of the paralytic shellfish toxin biosynthesis gene clusters in Anabaena circinalis AWQC131c and Aphanizomenon sp. NH-5. BMC Biochem. 2009, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Stucken, K.; John, U.; Cembella, A.; Murillo, A.A.; Soto-Liebe, K.; Fuentes-Valdés, J.J.; Friedel, M.; Plominsky, A.M.; Vásquez, M.; Glöckner, G. The smallest known genomes of multicellular and toxic cyanobacteria: Comparison, minimal gene sets for linked traits and the evolutionary implications. PLoS ONE 2010, 5, e9235. [Google Scholar] [CrossRef] [PubMed]
- Mihali, T.K.; Carmichael, W.W.; Neilan, B.A. A putative gene cluster from a Lyngbya wollei bloom that encodes paralytic shellfish toxin biosynthesis. PLoS ONE 2011, 6, e14657. [Google Scholar] [CrossRef] [PubMed]
- D′Agostino, P.M.; Moffitt, M.C.; Neilan, B.A. Current knowledge of paralytic shellfish toxin biosynthesis, molecular detection and evolution. In Toxins and Biologically Active Compounds from Microalgae; CRC Press: Boca Raton, FL, USA, 2014; pp. 251–280. [Google Scholar]
- Stüken, A.; Orr, R.J.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of nuclear-encoded genes for the neurotoxin saxitoxin in dinoflagellates. PLoS ONE 2011, 6, e20096. [Google Scholar] [CrossRef] [PubMed]
- Orr, R.J.; Stuken, A.; Murray, S.A.; Jakobsen, K.S. Evolutionary acquisition and loss of saxitoxin biosynthesis in dinoflagellates: The second "core" gene, sxtG. Appl. Environ. Microbiol. 2013, 79, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.A.; Wiese, M.; Stuken, A.; Brett, S.; Kellmann, R.; Hallegraeff, G.; Neilan, B.A. SxtA-based quantitative molecular assay to identify saxitoxin-producing harmful algal blooms in marine waters. Appl. Environ. Microbiol. 2011, 77, 7050–7057. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.A.; Diwan, R.; Orr, R.J.; Kohli, G.S.; John, U. Gene duplication, loss and selection in the evolution of saxitoxin biosynthesis in alveolates. Mol. Phylogenet. Evolut. 2015, 92, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.F.; Lin, L.; Wang, D.Z. Comparative transcriptome analysis of a toxin-producing dinoflagellate Alexandrium catenella and its non-toxic mutant. Mar. Drugs 2014, 12, 5698–5718. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.D.; Wisecaver, J.H.; Brosnahan, M.L.; Kulis, D.M.; Anderson, D.M.; Bhattacharya, D.; Plumley, F.G.; Erdner, D.L. Evolution of saxitoxin synthesis in cyanobacteria and dinoflagellates. Mol. Biol. Evol. 2013, 30, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Moreno Diaz de la Espina, S.; Alverca, E.; Cuadrado, A.; Franca, S. Organization of the genome and gene expression in a nuclear environment lacking histones and nucleosomes: The amazing dinoflagellates. Eur. J. Cell Biol. 2005, 84, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Taroncher-Oldenburg, G.; Kulis, D.M.; Anderson, D.M. Toxin variability during the cell cycle of the dinoflagellate Alexandrium fundyense. Limnol. Oceanogr. 1997, 42, 1178–1188. [Google Scholar] [CrossRef]
- Taroncher-Oldenburg, G.; Kulis, D.M.; Anderson, D.M. Coupling of saxitoxin biosynthesis to the G1 phase of the cell cycle in the dinoflagellate Alexandrin fundyense: Temperature and nutrient effects. Nat. Toxins 1999, 7, 207–219. [Google Scholar] [CrossRef]
- Siu, G.Y.; Young, M.C.; Chan, D.K.O. Environmental and nutritional factors which regulate population dynamics and toxin production in the dinoflagellate Alexandrium catenella. In Asia-pacific Conference on Science and Management of Coastal Environment; Wong, Y.S., Tam, N.Y., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 1997; Volume 123, pp. 117–140. [Google Scholar]
- Harlow, L.D.; Negri, A.; Hallegraeff, G.M.; Koutoulis, A. Sam, Sahh and Map gene expression during cell division and paralytic shellfish toxin production of Alexandrium catenella (Dinophyceae). Phycologia 2007, 46, 666–674. [Google Scholar] [CrossRef]
- Ogata, T.; Kodama, M.; Ishimaru, T. Toxin production in the dinoflagellate Protogonyaulax tamarensis. Toxicon 1987, 25, 923–928. [Google Scholar] [CrossRef]
- John, E.H.; Flynn, K.J. Modelling changes in paralytic shellfish toxin content of dinoflagellates in response to nitrogen and phosphorus supply. Mar. Ecol. Prog. Ser. 2002, 225, 147–160. [Google Scholar] [CrossRef]
- Cho, Y.; Ogawa, M.; Hirota, M.; Oshima, Y. Effects of mitomycin C and colchicine on toxin production and cell cycle regulation in the dinoflagellate Alexandrium tamarense. Harmful Algae 2011, 10, 235–244. [Google Scholar] [CrossRef]
- Cho, Y.; Ogawa, M.; Yotsu-Yamashita, M.; Oshima, Y. Effect of 5-fluoro-2′-deoxyuridine on toxin production and cell cycle regulation in marine dinoflagellate, Alexandrium tamarense. Harmful Algae 2014, 32, 64–72. [Google Scholar] [CrossRef]
- Taroncher-Oldenburg, G.; Anderson, D.M. Identification and characterization of three differentially expressed genes, encoding S-adenosylhomocysteine hydrolase, methionine aminopeptidase, and a histone-like protein, in the toxic dinoflagellate Alexandrium fundyense. Appl. Environ. Microbiol. 2000, 66, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Gao, Y.; Lin, L.; Hong, H.S. Comparative proteomic analysis reveals proteins putatively involved in toxin biosynthesis in the marine dinoflagellate Alexandrium catenella. Mar. Drugs 2013, 11, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sui, Z.; Chang, L.; Kang, K.; Ma, J.; Kong, F.; Zhou, W.; Wang, J.; Guo, L.; Geng, H.; et al. Transcriptome de novo assembly sequencing and analysis of the toxic dinoflagellate Alexandrium catenella using the illumina platform. Gene 2014, 537, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Carmichael, W.W.; Evans, W.R. Factors influencing growth and toxin production by cultures of the freshwater cyanobacterium Lyngbya wollei farlow ex gomont. J. Appl. Phycol. 1997, 9, 55–63. [Google Scholar] [CrossRef]
- Moustafa, A.; Loram, J.E.; Hackett, J.D.; Anderson, D.M.; Plumley, F.G.; Bhattacharya, D. Origin of saxitoxin biosynthetic genes in cyanobacteria. PLoS ONE 2009, 4, e5758. [Google Scholar] [CrossRef] [PubMed]
- D′Agostino, P.M.; Song, X.; Neilan, B.A.; Moffitt, M.C. Comparative proteomics reveals that a saxitoxin-producing and a nontoxic strain of Anabaena circinalis are two different ecotypes. J. Proteome Res. 2014, 13, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- D′Agostino, P.M.; Song, X.; Neilan, B.A.; Moffitt, M.C. Proteogenomics of a saxitoxin-producing and non-toxic strain of Anabaena circinalis (cyanobacteria) in response to extracellular NaCl and phosphate depletion. Environ. Microbiol. 2016, 18, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Lidie, K.B.; Ryan, J.C.; Barbier, M.; Van Dolah, F.M. Gene expression in florida red tide dinoflagellate Karenia brevis: Analysis of an expressed sequence tag library and development of DNA microarray. Mar. Biotechnol. 2005, 7, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Erdner, D.L.; Anderson, D.M. Global transcriptional profiling of the toxic dinoflagellate Alexandrium fundyense using massively parallel signature sequencing. BMC Genom. 2006, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Lin, S. Genomic understanding of dinoflagellates. Res. Microbiol. 2011, 162, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, S.; Bayer, T.; Aranda, M.; Liew, Y.J.; Carr, A.; Micklem, G.; Voolstra, C.R. Integrating microRNA and mRNA expression profiling in Symbiodinium microadriaticum, a dinoflagellate symbiont of reef-building corals. BMC Genom. 2013, 14, 704. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z.; et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Mittag, M.; Li, L.; Hastings, J.W. The mRNA level of the circadian regulated Gonyaulax luciferase remains constant over the cycle. Chronobiol. Int. 1998, 15, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Fagan, T.; Morse, D.; Hastings, J.W. Circadian synthesis of a nuclear-encoded chloroplast glyceraldehyde-3-phosphate dehydrogenase in the dinoflagellate Gonyaulax polyedra is translationally controlled. Biochemistry 1999, 38, 7689–7695. [Google Scholar] [CrossRef] [PubMed]
- Van Le, Q.U.Y.; Samson, G.U.Y.; Desjardins, Y. Opposite effects of exogenous sucrose on growth, photosynthesis and carbon metabolism of in vitro plantlets of tomato (L. esculentum Mill.) grown under two levels of irradiances and CO2 concentration. J. Plant Physiol. 2001, 158, 599–605. [Google Scholar] [CrossRef]
- Wiese, M.; Murray, S.A.; Alvin, A.; Neilan, B.A. Gene expression and molecular evolution of sxtA4 in a saxitoxin producing dinoflagellate Aexandrium catenella. Toxicon 2014, 92, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Perini, F.; Galluzzi, L.; Dell′Aversano, C.; Iacovo, E.D.; Tartaglione, L.; Ricci, F.; Forino, M.; Ciminiello, P.; Penna, A. SxtA and sxtG gene expression and toxin production in the mediterranean Alexandrium minutum (Dinophyceae). Mar. Drugs 2014, 12, 5258–5276. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Neilan, B.A. Biochemical characterization of paralytic shellfish toxin biosynthesis in vitro. J. Phycol. 2007, 43, 497–508. [Google Scholar] [CrossRef]
- Zhang, S.-F.; Zhang, Y.; Xie, Z.-X.; Zhang, H.; Lin, L.; Wang, D.-Z. iTRAQ-based quantitative proteomic analysis of a toxigenic dinoflagellate Alexandrium catenella and its non-toxic mutant. Proteomics 2015, 15, 4041–4050. [Google Scholar] [CrossRef] [PubMed]
- Galasinski, W. Eukaryotic polypeptide elongation system and its sensitivity to the inhibitory substances of plant origin. Proc. Soc. Exp. Biol. Med. 1996, 212, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Gile, G.H.; Patron, N.J.; Keeling, P.J. EFL GTPase in cryptomonads and the distribution of EFL and EF-1α in chromalveolates. Protist 2006, 157, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, M.; Burd, C.G.; Dreyfuss, G. The mRNA poly(A)-binding protein: Localization, abundance, and RNA-binding specificity. Exp. Cell Res. 1994, 211, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.; Gilbert, H.F. Protein disulfide isomerase. Biochim. Biophys. Acta 2004, 1699, 35–44. [Google Scholar] [CrossRef]
- Kim, K.H.; Lopez-Casillas, F.; Bai, D.H.; Luo, X.; Pape, M.E. Role of reversible phosphorylation of acetyl-CoA carboxylase in long-chain fatty acid synthesis. FASEB J. 1989, 3, 2250–2256. [Google Scholar] [PubMed]
- Wang, L.H.; Lee, H.H.; Fang, L.S.; Mayfield, A.B.; Chen, C.S. Fatty acid and phospholipid syntheses are prerequisites for the cell cycle of Symbiodinium and their endosymbiosis within sea anemones. PLoS ONE 2013, 8, e72486. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.C.; Wong, J.T. Lipid biosynthesis and its coordination with cell cycle progression. Plant Cell Physiol. 2005, 46, 1973–1986. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 1987, 23, 633–638. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, H.; Lin, S. Tandem repeats, high copy number and remarkable diel expression rhythm of form II RubisCO in Prorocentrum donghaiense (Dinophyceae). PLoS ONE 2013, 8, e71232. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Li, C.; Zhang, Y.; Wang, Y.Y.; He, Z.P.; Lin, L.; Hong, H.S. Quantitative proteomic analysis of differentially expressed proteins in the toxicity-lost mutant of Alexandrium catenella (Dinophyceae) in the exponential phase. J. Proteom. 2012, 75, 5564–5577. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, H.; Zhuang, Y.; Tran, B.; Gill, J. Spliced leader-based metatranscriptomic analyses lead to recognition of hidden genomic features in dinoflagellates. Proc. Natl. Acad. Sci. USA 2010, 107, 20033–20038. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wohlrab, S.; Glöckner, G.; Guillou, L.; John, U. Genomic insights into processes driving the infection of Alexandrium tamarense by the parasitoid Amoebophrya sp. Eukaryot. Cell 2014, 13, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Furmanek, T.; Kryvi, H.; Krossoy, C.; Totland, G.K.; Grotmol, S.; Wargelius, A. Transcriptome sequencing of atlantic salmon (Salmo salar L.) notochord prior to development of the vertebrae provides clues to regulation of positional fate, chordoblast lineage and mineralisation. BMC Genom. 2014, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, F.J.; Blanca, J.; Ziarsolo, P.; Gramazio, P.; Plazas, M.; Anderson, G.J.; Prohens, J.; Vilanova, S. The first de novo transcriptome of pepino (Solanum muricatum): Assembly, comprehensive analysis and comparison with the closely related species S. caripense, potato and tomato. BMC Genom. 2016, 17, 321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-F.; Yuan, C.-J.; Chen, Y.; Chen, X.-H.; Li, D.-X.; Liu, J.-L.; Lin, L.; Wang, D.-Z. Comparative transcriptomic analysis reveals novel insights into the adaptive response of Skeletonema costatum to changing ambient phosphorus. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR gene indices clustering tools (TGICL): A software system for fast clustering of large est datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 1165–1188. [Google Scholar]
- Li, G.; Jia, Q.; Zhao, J.; Li, X.; Yu, M.; Samuel, M.S.; Zhao, S.; Prather, R.S.; Li, C. Dysregulation of genome-wide gene expression and DNA methylation in abnormal cloned piglets. BMC Genom. 2014, 15, 811. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO: TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number of Unigenes |
|---|---|
| NR | 72,936 |
| NT | 8091 |
| KEGG | 54,818 |
| Swiss-Prot | 49,749 |
| GO | 15,432 |
| COG | 45,162 |
| At least in one database | 74,261 |
| STX Gene | Putative Function | A. catenella Unigenes |
|---|---|---|
| sxtA | Aspartate aminotransferase | 17 |
| sxtB | Cytidine deaminase | 2 |
| sxtD | Sterole desaturase | 1 |
| sxtF/M | Toxic compound efflux protein | 2 |
| sxtG | Amidinotransferase | 3 |
| sxtH/T/DIOX | Phenylpropionate dioxygenase | 15 |
| sxtI | O-carbamoyltransferase | 4 |
| sxtO | Adenylylsulfate kinase | 1 |
| sxtP | STX-binding protein | 2 |
| sxtU | Short-chain alcohol dehydrogenase | 66 |
| sxtW | Ferredoxin | 5 |
| sxtX | Cephalosporin hydroxylase | 1 |
| sxtZ | Two-component sensor histidine kinase | 14 |
| sxtPER | Permease | 1 |
| sxtACT | Acyl-CoA dependent acyltransferase | 4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, S.-F.; Lin, L.; Wang, D.-Z. Whole Transcriptomic Analysis Provides Insights into Molecular Mechanisms for Toxin Biosynthesis in a Toxic Dinoflagellate Alexandrium catenella (ACHK-T). Toxins 2017, 9, 213. https://doi.org/10.3390/toxins9070213
Zhang Y, Zhang S-F, Lin L, Wang D-Z. Whole Transcriptomic Analysis Provides Insights into Molecular Mechanisms for Toxin Biosynthesis in a Toxic Dinoflagellate Alexandrium catenella (ACHK-T). Toxins. 2017; 9(7):213. https://doi.org/10.3390/toxins9070213
Chicago/Turabian StyleZhang, Yong, Shu-Fei Zhang, Lin Lin, and Da-Zhi Wang. 2017. "Whole Transcriptomic Analysis Provides Insights into Molecular Mechanisms for Toxin Biosynthesis in a Toxic Dinoflagellate Alexandrium catenella (ACHK-T)" Toxins 9, no. 7: 213. https://doi.org/10.3390/toxins9070213
APA StyleZhang, Y., Zhang, S.-F., Lin, L., & Wang, D.-Z. (2017). Whole Transcriptomic Analysis Provides Insights into Molecular Mechanisms for Toxin Biosynthesis in a Toxic Dinoflagellate Alexandrium catenella (ACHK-T). Toxins, 9(7), 213. https://doi.org/10.3390/toxins9070213