Cytochalasin-B-Inducible Nanovesicle Mimics of Natural Extracellular Vesicles That Are Capable of Nucleic Acid Transfer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mesenchymal Stem Cell (MSC) Isolation and Characterization
2.2. Extracellular Vesicle (EV) Isolation
2.3. Determination of EV, Cytochalasin-B-Inducible Nanovesicle (CINV), and Membrane-Derived Nanovesicle (MDNV) Concentrations and Size Estimation
2.4. Detergent-Free Generation of Membrane-Derived Nanovesicles (MDNVs)
2.5. Preparation of Cytochalasin-B-Inducible Nanovesicles (CINVs)
2.6. Loading of EVs, CINVs, and MDNVs with Fluorescein (FAM)-Labelled Oligodeoxyribonucleotide
2.7. Flow Cytometry Analysis of EVs, CINVs, and MDNVs
2.8. Cell Culture Experiments
2.9. Electron Microscopy Analysis
2.10. CINV Loading Capacity
2.11. Nuclease Protection Assay
2.12. Statistical Analysis
3. Results
3.1. Characterization of Human Endometrial MSCs
3.2. Isolation and Characterization of MSC-Derived Natural EVs and Their Artificial Mimics: CINVs and MDNVs
3.3. Loading of EVs, CINVs, and MDNVs with FAM-ON
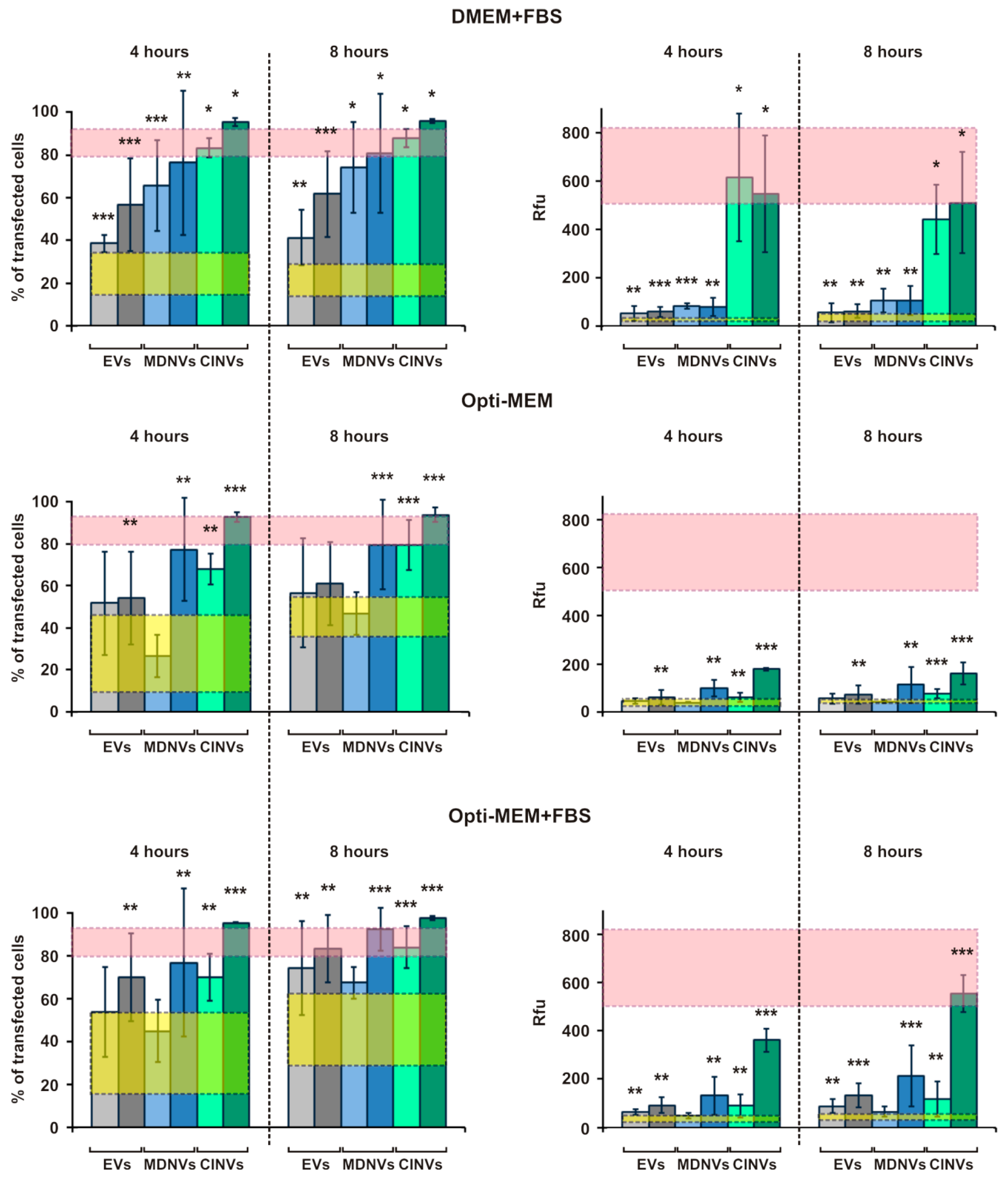
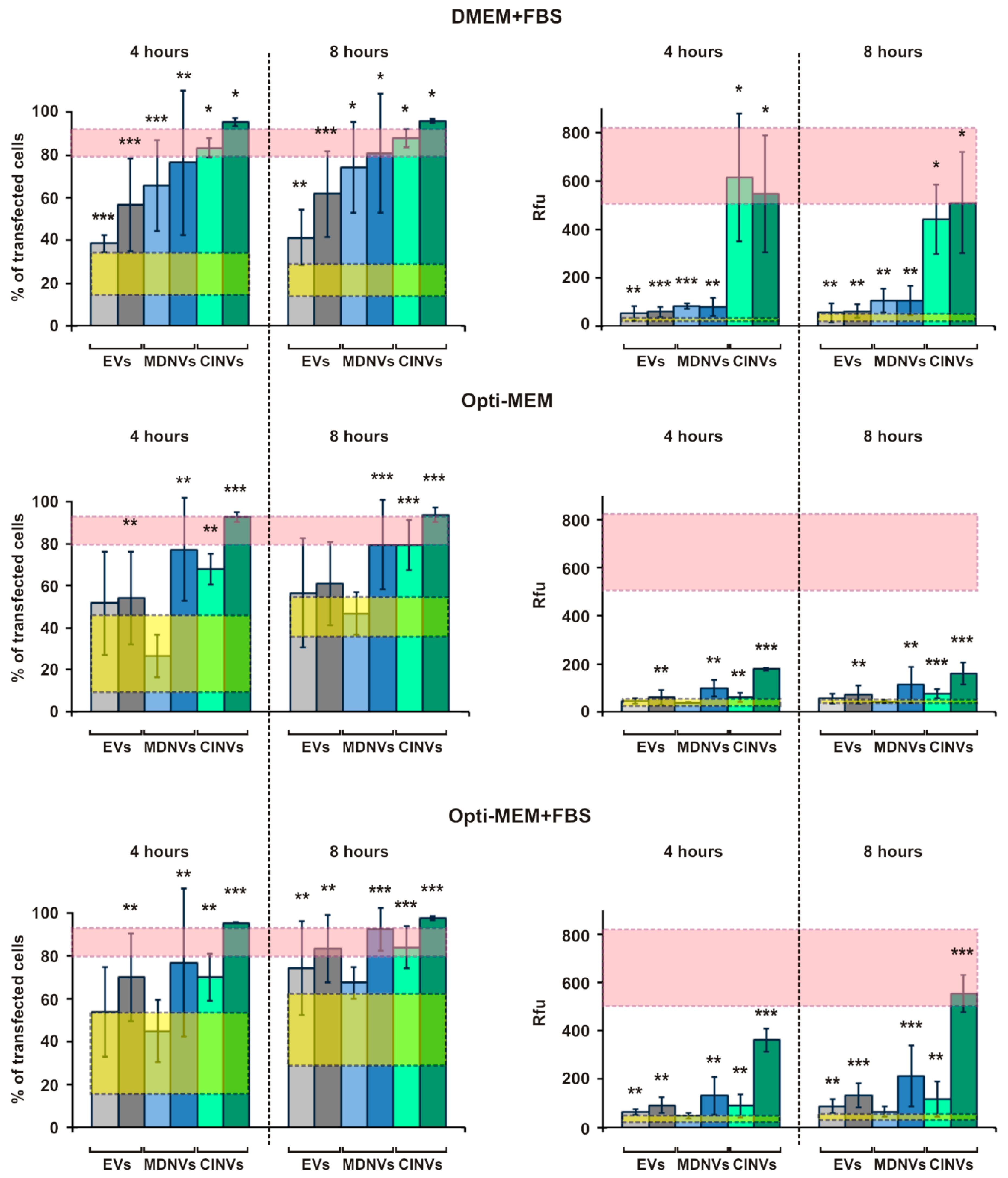
3.4. FAM-ON Delivery into HEK293 Cells Mediated by EVs, CINVs, and MDNVs
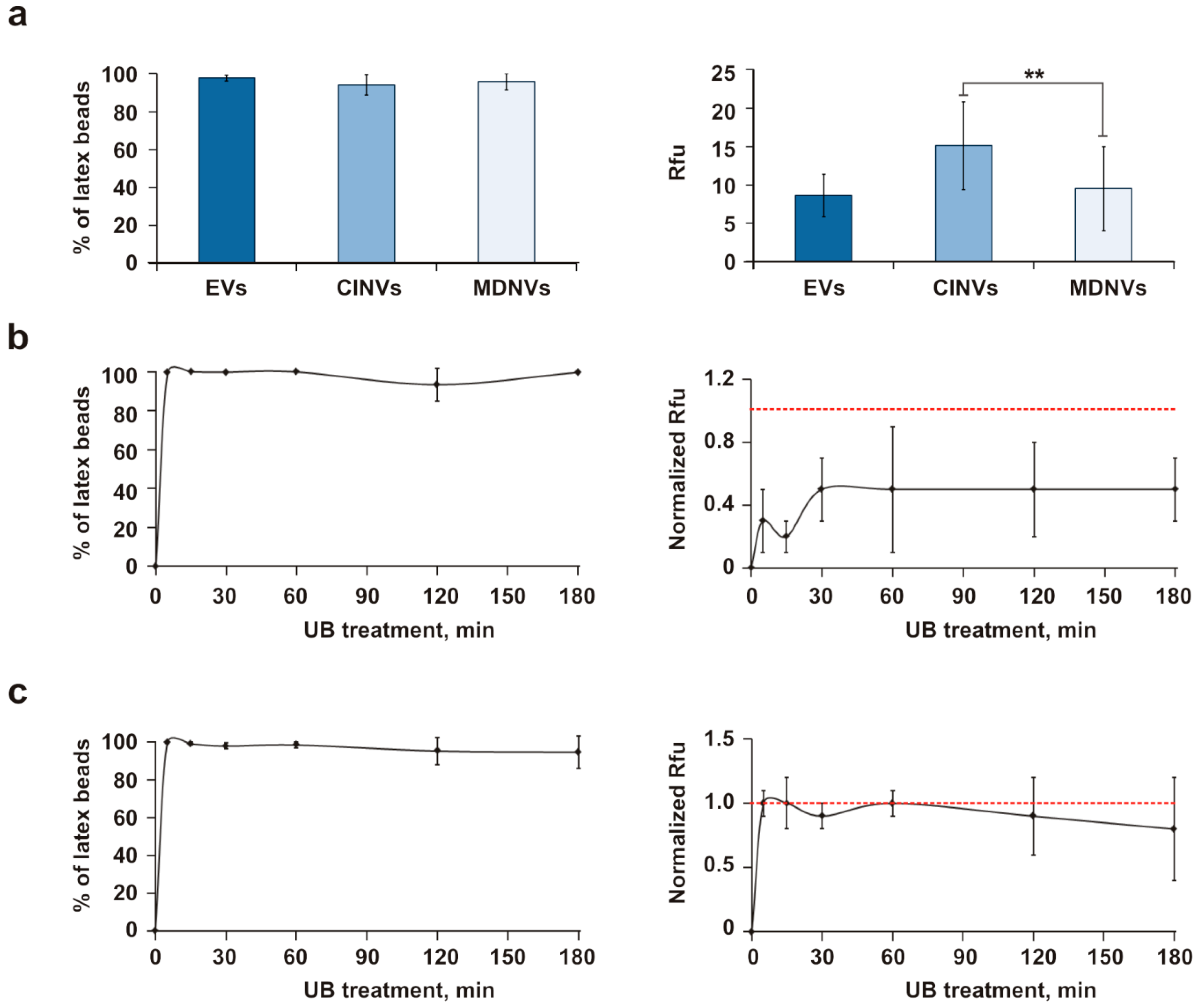
3.5. Evaluation of Fr/Th CINV Loading Capacity with FAM-ON
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Gudbergsson, J.M.; Jonsson, K.; Simonsen, J.B.; Johnsen, K.B. Systematic review of targeted extracellular vesicles for drug delivery—Considerations on methodological and biological heterogeneity. J. Controlled Release 2019, 306, 108–120. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Lakhal, S.; Mager, I.; Wood, M.J. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Delivery Rev. 2013, 65, 391–397. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Shim, G.; Kim, M.-G.; Park, J.Y.; Oh, Y.-K. Application of cationic liposomes for delivery of nucleic acids. Asian J. Pharm. Sci. 2013, 8, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Chernolovskaya, E.L.; Zenkova, M.A. Chemical modification of siRNA. Curr. Opin. Mol. Ther. 2010, 12, 158–167. [Google Scholar]
- Järver, P.; Coursindel, T.; Andaloussi, S.E.; Godfrey, C.; Wood, M.J.; Gait, M.J. Peptide-mediated Cell and In Vivo Delivery of Antisense Oligonucleotides and siRNA. Mol Ther Nucleic Acids 2012, 1, e27. [Google Scholar] [CrossRef] [PubMed]
- Alshaer, W.; Hillaireau, H.; Fattal, E. Aptamer-guided nanomedicines for anticancer drug delivery. Adv. Drug Delivery Rev. 2018, 134, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, M.K.; Khan, I.; Mani, P.; Chattopadhyay, P.; Sarkar, D.P.; Sinha, S. Combination of hepatocyte specific delivery and transformation dependent expression of shRNA inducing transcriptional gene silencing of c-Myc promoter in hepatocellular carcinoma cells. BMC Cancer 2014, 14, 582. [Google Scholar] [CrossRef] [PubMed]
- Voráčková, I.; Ulbrich, P.; Diehl, W.E.; Ruml, T. Engineered retroviral virus-like particles for receptor targeting. Arch. Virol. 2014, 159, 677–688. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, J.; Holtrop, M.; Wilschut, J.; Huckriede, A. Reconstituted influenza virus envelopes as an efficient carrier system for cellular delivery of small-interfering RNAs. Gene Ther. 2006, 13, 400–411. [Google Scholar] [CrossRef]
- Subramanian, N.; Mani, P.; Roy, S.; Gnanasundram, S.V.; Sarkar, D.P.; Das, S. Targeted delivery of hepatitis C virus-specific short hairpin RNA in mouse liver using Sendai virosomes. J. Gen. Virol. 2009, 90, 1812–1819. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Controlled Release 2013, 172, 229–238. [Google Scholar] [CrossRef]
- Wang, J.-H.; Forterre, A.V.; Zhao, J.; Frimannsson, D.O.; Delcayre, A.; Antes, T.J.; Efron, B.; Jeffrey, S.S.; Pegram, M.D.; Matin, A.C. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol. Cancer Ther. 2018, 17, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Shtam, T.A.; Kovalev, R.A.; Varfolomeeva, E.Y.; Makarov, E.M.; Kil, Y.V.; Filatov, M.V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun. Signal. 2013, 11, 88. [Google Scholar] [CrossRef]
- Wahlgren, J.; De L Karlson, T.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef]
- Stremersch, S.; Vandenbroucke, R.E.; Van Wonterghem, E.; Hendrix, A.; De Smedt, S.C.; Raemdonck, K. Comparing exosome-like vesicles with liposomes for the functional cellular delivery of small RNAs. J. Controlled Release 2016, 232, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-p.; Lin, Z.-x.; Jiang, X.-y.; Yu, X.-y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharm. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pick, H.; Schmid, E.L.; Tairi, A.-P.; Ilegems, E.; Hovius, R.; Vogel, H. Investigating cellular signaling reactions in single attoliter vesicles. J. Am. Chem. Soc. 2005, 127, 2908–2912. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, S.B.; Bharti, D.; Jang, S.-J.; Hwang, S.-C.; Park, J.-K.; Shin, J.-K.; Byun, J.-H.; Park, B.-W.; Rho, G.-J. Cryopreservation of Human Wharton’s Jelly-derived Mesenchymal Stem Cells Following Controlled Rate Freezing Protocol Using Different Cryoprotectants; A Comparative Study. Int. J. Stem Cells 2015, 8, 155–169. [Google Scholar] [CrossRef]
- Lu, T.-J.; Chiu, F.-Y.; Chiu, H.-Y.; Chang, M.-C.; Hung, S.-C. Chondrogenic Differentiation of Mesenchymal Stem Cells in Three-Dimensional Chitosan Film Culture. Cell Transplant. 2017, 26, 417–427. [Google Scholar] [CrossRef]
- Reyes, M.; Lund, T.; Lenvik, T.; Aguiar, D.; Koodie, L.; Verfaillie, C.M. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood 2001, 98, 2615–2625. [Google Scholar] [CrossRef]
- D’Ippolito, G.; Diabira, S.; Howard, G.A.; Menei, P.; Roos, B.A.; Schiller, P.C. Marrow-isolated adult multilineage inducible (MIAMI) cells, a unique population of postnatal young and old human cells with extensive expansion and differentiation potential. J. Cell Sci. 2004, 117, 2971–2981. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.L.; Pike, L.J. A simplified method for the preparation of detergent-free lipid rafts. J. Lipid Res. 2005, 46, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Gomzikova, M.O.; Zhuravleva, M.N.; Miftakhova, R.R.; Arkhipova, S.S.; Evtugin, V.G.; Khaiboullina, S.F.; Kiyasov, A.P.; Persson, J.L.; Mongan, N.P.; Pestell, R.G.; et al. Cytochalasin B-induced membrane vesicles convey angiogenic activity of parental cells. Oncotarget 2017, 8, 70496–70507. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Controlled Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Dias, M.V.S.; Martins, V.R.; Hajj, G.N.M. Stress-Inducible Protein 1 (STI1): Extracellular Vesicle Analysis and Quantification. Methods Mol. Biol. 2016, 1459, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kossinova, O.A.; Gopanenko, A.V.; Tamkovich, S.N.; Krasheninina, O.A.; Tupikin, A.E.; Kiseleva, E.; Yanshina, D.D.; Malygin, A.A.; Ven’yaminova, A.G.; Kabilov, M.R.; et al. Cytosolic YB-1 and NSUN2 are the only proteins recognizing specific motifs present in mRNAs enriched in exosomes. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Charoenviriyakul, C.; Takahashi, Y.; Morishita, M.; Matsumoto, A.; Nishikawa, M.; Takakura, Y. Cell type-specific and common characteristics of exosomes derived from mouse cell lines: Yield, physicochemical properties, and pharmacokinetics. Eur. J. Pharm. Sci. 2017, 96, 316–322. [Google Scholar] [CrossRef]
- Stolk, M.; Seifert, M. Protein contaminations impact quantification and functional analysis of extracellular vesicle preparations from mesenchymal stromal cells. J. Stem Cells Regener. Med. 2015, 11, 44–47. [Google Scholar]
- Kesimer, M.; Gupta, R. Physical characterization and profiling of airway epithelial derived exosomes using light scattering. Methods 2015, 87, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Gomzikova, M.; Kletukhina, S.; Kurbangaleeva, S.; Rizvanov, A. Evaluation of Cytochalasin B-Induced Membrane Vesicles Fusion Specificity with Target Cells. Biomed. Res. Int. 2018, 2018, 7053623. [Google Scholar] [CrossRef]
- Gleave, M.E.; Monia, B.P. Antisense therapy for cancer. Nat. Rev. Cancer 2005, 5, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.Y.; Sung, C.W.; Chen, C.; Cheng, C.M.; Lin, D.P.; Huang, C.T.; Hsu, M.Y. Advances in exosomes technology. Clin. Chim. Acta 2019, 493, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.L.; Zhu, J.; Liu, J.X.; Jiang, F.; Ni, W.K.; Qu, L.S.; Ni, R.Z.; Lu, C.H.; Xiao, M.B. A Comparison of Traditional and Novel Methods for the Separation of Exosomes from Human Samples. Biomed. Res. Int. 2018, 2018, 3634563. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.d.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef]
- Wang, K.; Jiang, Z.; Webster, K.A.; Chen, J.; Hu, H.; Zhou, Y.; Zhao, J.; Wang, L.; Wang, Y.; Zhong, Z.; et al. Enhanced Cardioprotection by Human Endometrium Mesenchymal Stem Cells Driven by Exosomal MicroRNA-21. Stem Cells Transl. Med. 2017, 6, 209–222. [Google Scholar] [CrossRef]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Hall, B.; Andreeff, M.; Marini, F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Bone Marrow-Deriv. Progenit. 2007, 180, 263–283. [Google Scholar] [CrossRef]
- Yeh Yeo, R.W. Efficiency of Exosome Production Correlates Inversely with the Developmental Maturity of MSC Donor. J. Stem Cell Res. Ther. 2013, 3, 1000145. [Google Scholar] [CrossRef]
- Grigor’eva, A.E.; Dyrkheeva, N.S.; Bryzgunova, O.E.; Tamkovich, S.N.; Chelobanov, B.P.; Ryabchikova, E.I. Contamination of exosome preparations, isolated from biological fluids. Biochem. Moscow Suppl. Ser. B 2017, 63, 91–96. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.; Sharma, S.; Gimzewski, J. The Role of Isolation Methods on a Nanoscale Surface Structure and its Effect on the Size of Exosomes. J. Circ. Biomark. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xing, H.; Xun, Z.; Yang, T.; Ding, P.; Cai, C.; Wang, D.; Zhao, X. Exosome-based small RNA delivery: Progress and prospects. Asian J. Pharm. Sci. 2018, 13, 1–11. [Google Scholar] [CrossRef]
- Jiang, L.; Vader, P.; Schiffelers, R.M. Extracellular vesicles for nucleic acid delivery: Progress and prospects for safe RNA-based gene therapy. Gene Ther. 2017, 24, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef]
- Maroto, R.; Zhao, Y.; Jamaluddin, M.; Popov, V.L.; Wang, H.; Kalubowilage, M.; Zhang, Y.; Luisi, J.; Sun, H.; Culbertson, C.T.; et al. Effects of storage temperature on airway exosome integrity for diagnostic and functional analyses. J. Extracell. Vesicles 2017, 6, 1359478. [Google Scholar] [CrossRef]
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noel, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410. [Google Scholar] [CrossRef]
- Lim, J.H.; Park, J.; Oh, E.H.; Ko, H.J.; Hong, S.; Park, T.H. Nanovesicle-based bioelectronic nose for the diagnosis of lung cancer from human blood. Adv. Healthcare Mater. 2014, 3, 360–366. [Google Scholar] [CrossRef]
- Lim, J.H.; Oh, E.H.; Park, J.; Hong, S.; Park, T.H. Ion-channel-coupled receptor-based platform for a real-time measurement of G-protein-coupled receptor activities. ACS Nano 2015, 9, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Gomzikova, M.O.; Zhuravleva, M.N.; Vorobev, V.V.; Salafutdinov, I.I.; Laikov, A.V.; Kletukhina, S.K.; Martynova, E.V.; Tazetdinova, L.G.; Ntekim, A.I.; Khaiboullina, S.F.; et al. Angiogenic activity of cytochalasin B-induced membrane vesicles of human mesenchymal stem cells. Cell Biol. 2019, in press. [Google Scholar]
- Mao, Z.; Cartier, R.; Hohl, A.; Farinacci, M.; Dorhoi, A.; Nguyen, T.-L.; Mulvaney, P.; Ralston, J.; Kaufmann, S.H.E.; Möhwald, H.; et al. Cells as factories for humanized encapsulation. Nano Lett. 2011, 11, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
- Wakatsuki, T.; Schwab, B.; Thompson, N.C.; Elson, E.L. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J. Cell Sci. 2001, 114, 1025–1036. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Nanovesicles | Ø ≤ 50 nm (Mean ± SD, nm) | 50 < Ø < 300 nm (Mean ± SD, nm) | Ø ≥ 300 nm (Mean ± SD, nm) | PDI |
|---|---|---|---|---|
| EVs | 0–1% (40 ± 10 nm) | 99–100% (210 ± 90 nm) | - | 0.2 |
| CINVs | - | 10–25% (130 ± 40 nm, 65 ± 10 nm) | 75–90% (500 ± 220 nm) | 0.5 |
| MDNVs (Fr/Th) | 3–4% (30 ± 5 nm) | ≈70% (140 ± 60 nm) | ≈25% (510 ± 170 nm) | 0.4 |
| MDNVs (5 min UB) | - | 100% (190 ± 130 nm) | - | 0.4 |
| MDNVs (15 min UB) | 0–6% (30 ± 10 nm) | 94–100% (180 ± 90 nm) | - | 0.3 |
| MDNVs (30 min UB) | 0–0.2% (15 ± 2 nm) | ≈90% (180 ± 120 nm) | ≈10% (1040 ± 400 nm) | 0.5 |
| MDNVs (60 min UB) | - | 80% (230 ± 200 nm) | 20% (1060 ± 400 nm) | 0.5 |
| MDNVs (2 h UB) | 3–6% (40 ± 10 nm) | ≈70% (160 ± 80 nm) | ≈25–30% (1070 ± 400 nm) | 0.5 |
| MDNVs (3 h UB) | 0–2% (30 ± 10 nm) | 98–100% (210 ± 150 nm) | - | 0.4 |
| MDNVs (5 min UB + Fr/Th) | 0–1% (20 ± 5 nm) | ≈75% (150 ± 80 nm) | ≈25% (890 ± 380 nm) | 0.6 |
| MDNVs (15 min UB + Fr/Th) | - | 100% (190 ± 130 nm) | - | 0.4 |
| MDNVs (30 min UB + Fr/Th) | 2–3% (20 ± 5 nm) | 97–98% (230 ± 210 nm) | - | 0.6 |
| MDNVs (60 min UB + Fr/Th) | - | 50% (90 ± 30 nm) | 50% (300 ± 150 nm) | 0.5 |
| MDNVs (2 h UB + Fr/Th) | 10% (50 ± 10 nm) | 50% (150 ± 50 nm) | 40% (1870 ± 750 nm) | 0.7 |
| MDNVs (3 h UB + Fr/Th) | - | 30% (90 ± 30 nm) | 70% (810 ± 270 nm) | 0.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oshchepkova, A.; Neumestova, A.; Matveeva, V.; Artemyeva, L.; Morozova, K.; Kiseleva, E.; Zenkova, M.; Vlassov, V. Cytochalasin-B-Inducible Nanovesicle Mimics of Natural Extracellular Vesicles That Are Capable of Nucleic Acid Transfer. Micromachines 2019, 10, 750. https://doi.org/10.3390/mi10110750
Oshchepkova A, Neumestova A, Matveeva V, Artemyeva L, Morozova K, Kiseleva E, Zenkova M, Vlassov V. Cytochalasin-B-Inducible Nanovesicle Mimics of Natural Extracellular Vesicles That Are Capable of Nucleic Acid Transfer. Micromachines. 2019; 10(11):750. https://doi.org/10.3390/mi10110750
Chicago/Turabian StyleOshchepkova, Anastasiya, Alexandra Neumestova, Vera Matveeva, Lyudmila Artemyeva, Ksenia Morozova, Elena Kiseleva, Marina Zenkova, and Valentin Vlassov. 2019. "Cytochalasin-B-Inducible Nanovesicle Mimics of Natural Extracellular Vesicles That Are Capable of Nucleic Acid Transfer" Micromachines 10, no. 11: 750. https://doi.org/10.3390/mi10110750
APA StyleOshchepkova, A., Neumestova, A., Matveeva, V., Artemyeva, L., Morozova, K., Kiseleva, E., Zenkova, M., & Vlassov, V. (2019). Cytochalasin-B-Inducible Nanovesicle Mimics of Natural Extracellular Vesicles That Are Capable of Nucleic Acid Transfer. Micromachines, 10(11), 750. https://doi.org/10.3390/mi10110750