Integrated Microfluidic Preconcentration and Nucleic Amplification System for Detection of Influenza A Virus H1N1 in Saliva

 , , , ,
, , , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Influenza A Virus (H1N1) and Its Titration
2.2. Preparation of Antibody-Conjugated Magnetic Nanoparticles (Ab-MNPs)
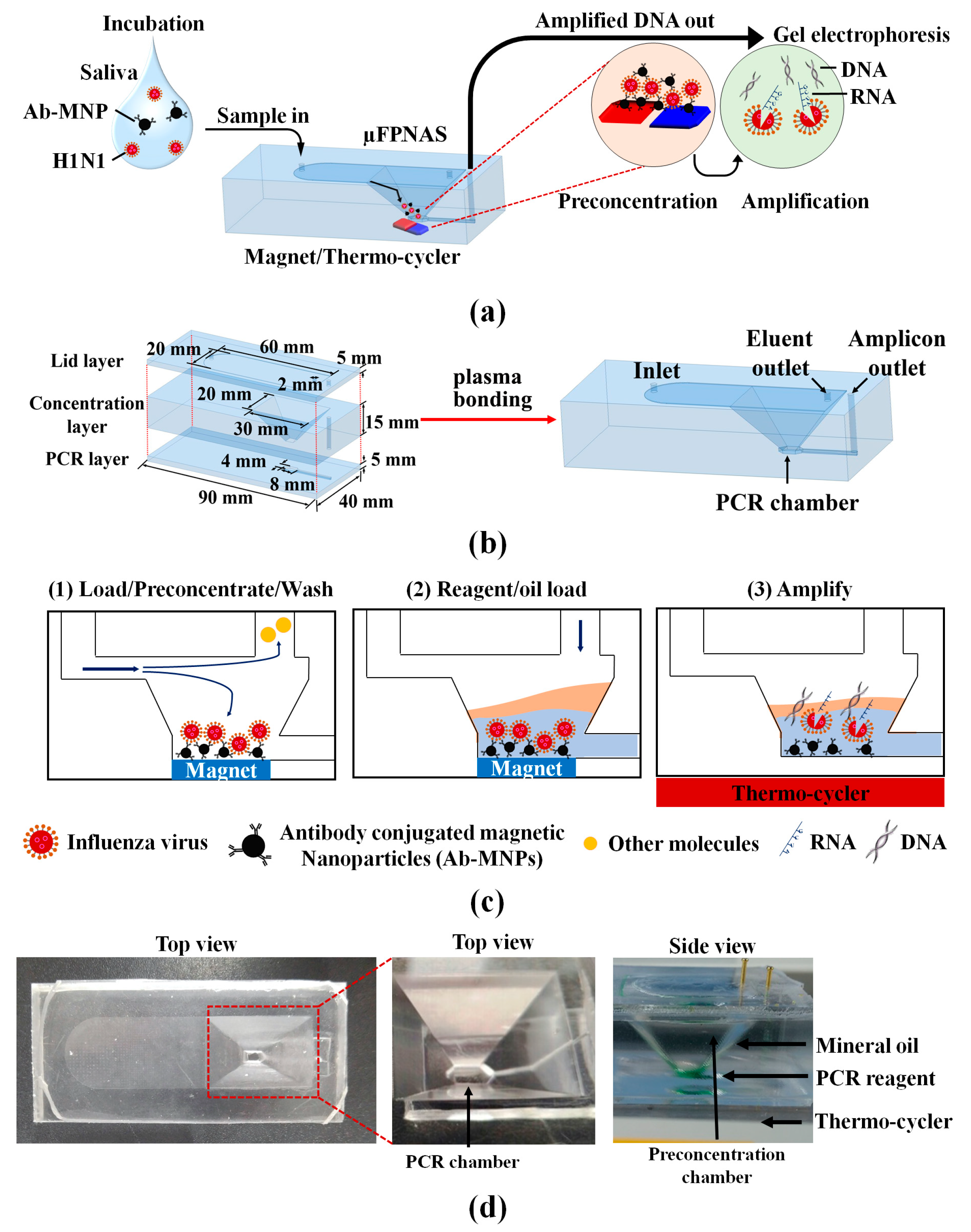
2.3. Microfabrication and Assembly of Microfluidic Preconcentration and Nucleic Amplification System (μFPNAS)
2.4. On-Chip Preconcentration of H1N1 in Phosphate-Buffered Saline (PBS) and Saliva
2.5. Virus-Capturing Efficiency
2.6. Numerical Analysis of Heat Transfer in μFPNAS during Thermocycling
2.7. On-Chip Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
2.8. Statistical Data Analysis
3. Results
3.1. Simulation of Heat Transfer Profiles at PCR Chamber
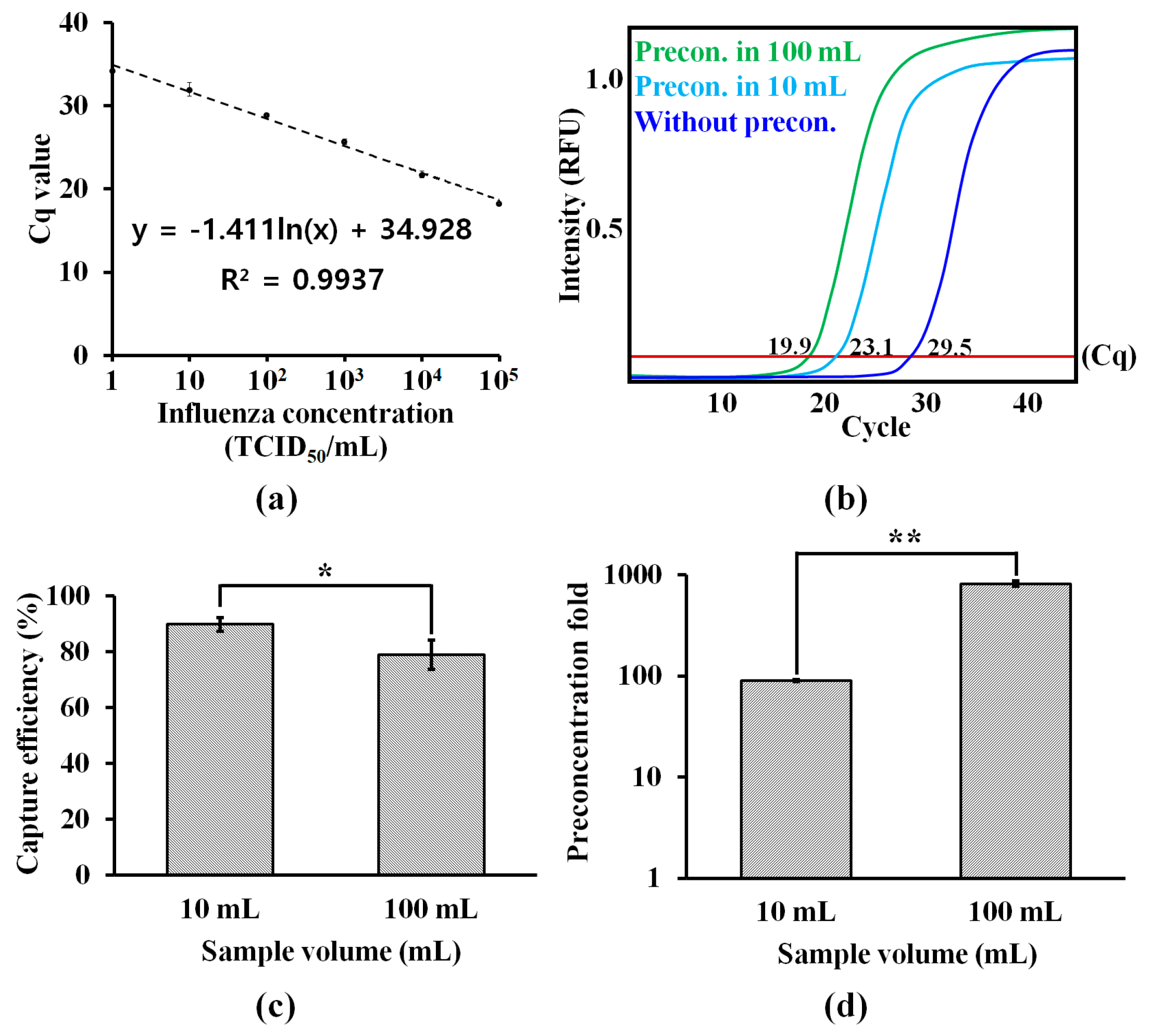
3.2. Effect of Sample Volume on Capturing Efficiency and Preconcentratin Fold
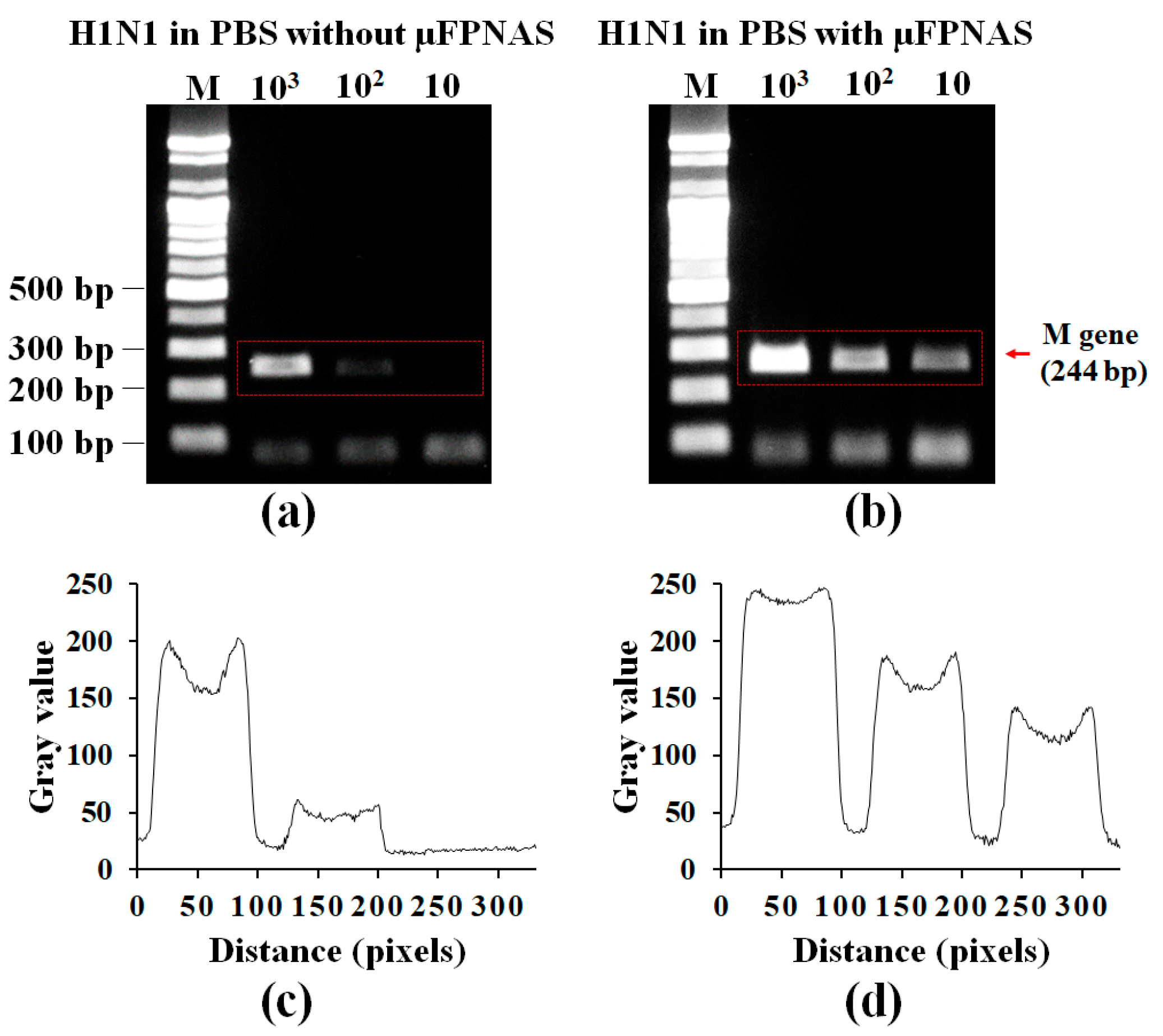
3.3. On-Chip RT-PCR for Preconcentration of H1N1 Virus and Amplification of Target RNA in PBS
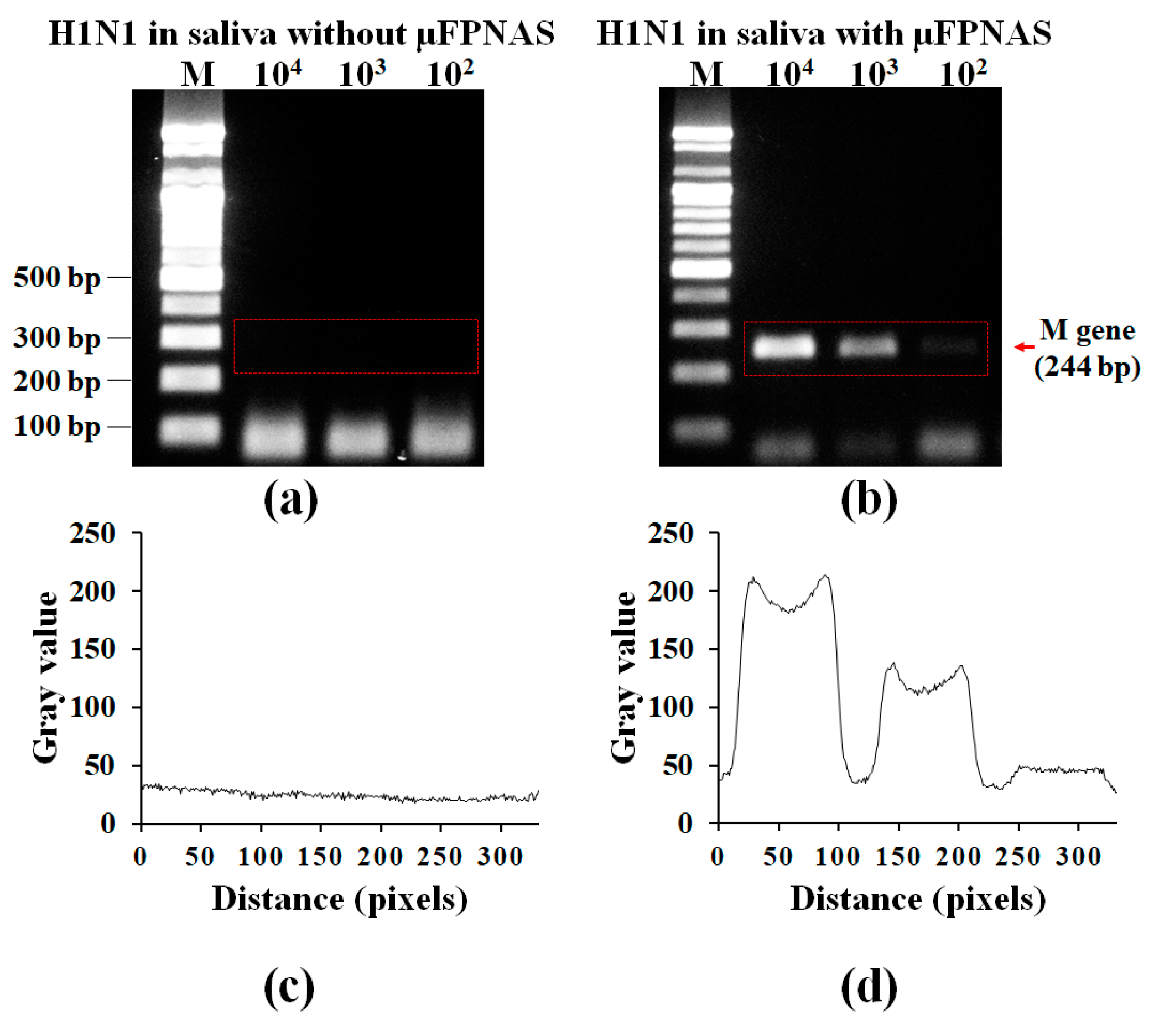
3.4. Spike Test in Saliva
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Taubenberger, J.K.; Morens, D.M. The pathology of influenza virus infections. Annu. Rev. Pathol.-Mech. 2008, 3, 499–522. [Google Scholar] [CrossRef]
- Camp, J.V.; Bagci, U.; Chu, Y.-K.; Squier, B.; Fraig, M.; Uriarte, S.M.; Guo, H.; Mollura, D.J.; Jonsson, C.B. Lower respiratory tract infection of the ferret by 2009 H1N1 pandemic influenza A virus triggers biphasic, systemic, and local recruitment of neutrophils. J. Virol. 2015, 89, 8733–8748. [Google Scholar] [CrossRef] [Green Version]
- Dziąbowska, K.; Czaczyk, E.; Nidzworski, D. Detection methods of human and animal influenza virus—current trends. Biosensors 2018, 8, 94. [Google Scholar] [CrossRef] [Green Version]
- Gatherer, D. The 2009 H1N1 influenza outbreak in its historical context. J. Clin. Virol. 2009, 45, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, B.; Larsson, C.; Zweygberg, B.W. Simultaneous detection and typing of influenza viruses A and B by a nested reverse transcription-PCR: Comparison to virus isolation and antigen detection by immunofluorescence and optical immunoassay (FLU OIA). J. Clin. Microbiol. 2001, 39, 134–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachucki, C.T.; Creticos, C. Early detection of influenza virus by using a fluorometric assay of infected tissue culture. J. Clin. Microbiol. 1988, 26, 2664–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouchier, R.A.; Bestebroer, T.M.; Herfst, S.; Van Der Kemp, L.; Rimmelzwaan, G.F.; Osterhaus, A.D. Detection of influenza A viruses from different species by PCR amplification of conserved sequences in the matrix gene. J. Clin. Microbiol. 2000, 38, 4096–4101. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Goyal, S.M.; Kang, S.; Farnham, M.; Joo, H. Detection and subtyping of swine influenza H1N1, H1N2 and H3N2 viruses in clinical samples using two multiplex RT-PCR assays. J. Virol. Methods 2002, 102, 53–59. [Google Scholar] [CrossRef]
- Ito, M.; Watanabe, M.; Nakagawa, N.; Ihara, T.; Okuno, Y. Rapid detection and typing of influenza A and B by loop-mediated isothermal amplification: Comparison with immunochromatography and virus isolation. J. Virol. Methods 2006, 135, 272–275. [Google Scholar] [CrossRef]
- Shan, S.; Ko, L.-S.; Collins, R.A.; Wu, Z.; Chen, J.; Chan, K.-Y.; Xing, J.; Lau, L.-T.; Yu, A.C.-H. Comparison of nucleic acid-based detection of avian influenza H5N1 with virus isolation. Biochem. Biophys. Res. Commun. 2003, 302, 377–383. [Google Scholar] [CrossRef]
- Lv, J.; Wei, B.; Chai, T.; Xia, X.; Miao, Z.; Yao, M.; Gao, Y.; Huang, R.; Yang, H.; Roesler, U. Development of a real-time RT-PCR method for rapid detection of H9 avian influenza virus in the air. Arch. Virol. 2011, 156, 1795. [Google Scholar] [CrossRef] [PubMed]
- Cuchacovich, R. Clinical applications of the polymerase chain reaction: An update. Infect. Dis. Clin. North Am. 2006, 20, 735–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Rothman, R.E. PCR-based diagnostics for infectious diseases: Uses, limitations, and future applications in acute-care settings. Lancet Infect. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef]
- Johnson, S.R.; Martin, D.H.; Cammarata, C.; Morse, S.A. Alterations in sample preparation increase sensitivity of PCR assay for diagnosis of chancroid. J. Clin. Microbiol. 1995, 33, 1036–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amicosante, M.; Richeldi, L.; Trenti, G.; Paone, G.; Campa, M.; Bisetti, A.; Saltini, C. Inactivation of polymerase inhibitors for Mycobacterium tuberculosis DNA amplification in sputum by using capture resin. J. Clin. Microbiol. 1995, 33, 629–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lee, J.; Park, S. A 3D-printed millifluidic platform enabling bacterial preconcentration and DNA purification for molecular detection of pathogens in blood. Micromachines 2018, 9, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mättö, J.; Saarela, M.; Alaluusua, S.; Oja, V.; Jousimies-Somer, H.; Asikainen, S. Detection of Porphyromonas gingivalisfrom saliva by PCR by using a simple sample-processing method. J. Clin. Microbiol. 1998, 36, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Ochert, A.; Boulter, A.; Birnbaum, W.; Johnson, N.; Teo, C. Inhibitory effect of salivary fluids on PCR: Potency and removal. PCR Methods Appl. 1994, 3, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Heuvelink, A.E.; Talukder, K.A.; Zwietering, M.H.; De Boer, E. Evaluation of immunomagnetic separation and PCR for the detection of Escherichia coli O157 in animal feces and meats. J. Food Prot. 2006, 69, 2865–2869. [Google Scholar] [CrossRef]
- Ganesh, I.; Tran, B.M.; Kim, Y.; Kim, J.; Cheng, H.; Lee, N.Y.; Park, S. An integrated microfluidic PCR system with immunomagnetic nanoparticles for the detection of bacterial pathogens. Biomed. Microdevices 2016, 18, 116. [Google Scholar] [CrossRef]
- Zhu, P.; Shelton, D.R.; Li, S.; Adams, D.L.; Karns, J.S.; Amstutz, P.; Tang, C.-M. Detection of E. coli O157: H7 by immunomagnetic separation coupled with fluorescence immunoassay. Biosens. Bioelectron. 2011, 30, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Kabir, S. Detection of Helicobacter pylori DNA in feces and saliva by polymerase chain reaction: A review. Helicobacter 2004, 9, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Mauk, M.G.; Hackett, B.A.; Cherry, S.; Bau, H.H.; Liu, C. Instrument-free point-of-care molecular detection of Zika virus. Anal. Chem. 2016, 88, 7289–7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemz, A.; Ferguson, T.M.; Boyle, D.S. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011, 29, 240–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enroth, H.; Engstrand, L. Immunomagnetic separation and PCR for detection of Helicobacter pylori in water and stool specimens. J. Clin. Microbiol. 1995, 33, 2162–2165. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shi, X.; Gehring, A.G.; Paoli, G.C. Automated immunomagnetic separation for the detection of Escherichia coli O157: H7 from spinach. Int. J. Food Microbiol. 2014, 179, 33–37. [Google Scholar] [CrossRef]
- Luppa, P.B.; Müller, C.; Schlichtiger, A.; Schlebusch, H. Point-of-care testing (POCT): Current techniques and future perspectives. Trends Anal. Chem. 2011, 30, 887–898. [Google Scholar] [CrossRef]
- Bu, M.; Christensen, T.B.; Smistrup, K.; Wolff, A.; Hansen, M.F. Characterization of a microfluidic magnetic bead separator for high-throughput applications. Sens. Actuators A-Phys. 2008, 145, 430–436. [Google Scholar] [CrossRef]
- Fedio, W.M.; Jinneman, K.C.; Yoshitomi, K.J.; Zapata, R.; Wendakoon, C.N.; Browning, P.; Weagant, S.D. Detection of E. coli O157: H7 in raw ground beef by Pathatrix™ immunomagnetic-separation, real-time PCR and cultural methods. Int. J. Food Microbiol. 2011, 148, 87–92. [Google Scholar] [CrossRef]
- Pamme, N. Magnetism and microfluidics. Lab Chip 2006, 6, 24–38. [Google Scholar] [CrossRef]
- Roda, A.; Mirasoli, M.; Roda, B.; Bonvicini, F.; Colliva, C.; Reschiglian, P. Recent developments in rapid multiplexed bioanalytical methods for foodborne pathogenic bacteria detection. Mikrochim. Acta 2012, 178, 7–28. [Google Scholar] [CrossRef]
- Xia, N.; Hunt, T.P.; Mayers, B.T.; Alsberg, E.; Whitesides, G.M.; Westervelt, R.M.; Ingber, D.E. Combined microfluidic-micromagnetic separation of living cells in continuous flow. Biomed. Microdevices 2006, 8, 299. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Banada, P.P.; Chatni, M.R.; Lim, K.S.; Bhunia, A.K.; Ladisch, M.; Bashir, R. A multifunctional micro-fluidic system for dielectrophoretic concentration coupled with immuno-capture of low numbers of Listeria monocytogenes. Lab Chip 2006, 6, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Fujitsuka, A.; Tsukagoshi, H.; Arakawa, M.; Goto-Sugai, K.; Ryo, A.; Okayama, Y.; Mizuta, K.; Nishina, A.; Yoshizumi, M.; Kaburagi, Y. A molecular epidemiological study of respiratory viruses detected in Japanese children with acute wheezing illness. BMC Infect. Dis. 2011, 11, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besecker, J.; Cornell, K.A.; Hampikian, G. Dynamic passivation with BSA overcomes LTCC mediated inhibition of PCR. Sens. Actuators B Chem. 2013, 176, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Lien, K.-Y.; Lin, J.-L.; Liu, C.-Y.; Lei, H.-Y.; Lee, G.-B. Purification and enrichment of virus samples utilizing magnetic beads on a microfluidic system. Lab Chip 2007, 7, 868–875. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Abafogi, A.T.; Tran, B.M.; Kim, J.; Lee, J.; Chen, Z.; Bae, P.K.; Park, K.; Shin, Y.-B.; van Noort, D.; et al. Integrated Microfluidic Preconcentration and Nucleic Amplification System for Detection of Influenza A Virus H1N1 in Saliva. Micromachines 2020, 11, 203. https://doi.org/10.3390/mi11020203
Kim Y, Abafogi AT, Tran BM, Kim J, Lee J, Chen Z, Bae PK, Park K, Shin Y-B, van Noort D, et al. Integrated Microfluidic Preconcentration and Nucleic Amplification System for Detection of Influenza A Virus H1N1 in Saliva. Micromachines. 2020; 11(2):203. https://doi.org/10.3390/mi11020203
Chicago/Turabian StyleKim, Yonghee, Abdurhaman Teyib Abafogi, Buu Minh Tran, Jaewon Kim, Jinyeop Lee, Zhenzhong Chen, Pan Kee Bae, Kyoungsook Park, Yong-Beom Shin, Danny van Noort, and et al. 2020. "Integrated Microfluidic Preconcentration and Nucleic Amplification System for Detection of Influenza A Virus H1N1 in Saliva" Micromachines 11, no. 2: 203. https://doi.org/10.3390/mi11020203
APA StyleKim, Y., Abafogi, A. T., Tran, B. M., Kim, J., Lee, J., Chen, Z., Bae, P. K., Park, K., Shin, Y.-B., van Noort, D., Lee, N. Y., & Park, S. (2020). Integrated Microfluidic Preconcentration and Nucleic Amplification System for Detection of Influenza A Virus H1N1 in Saliva. Micromachines, 11(2), 203. https://doi.org/10.3390/mi11020203