This feature paper describes the first-principles studies on designing polyatomic molecular systems with strong electron-acceptor (superhalogens) or electron-donor (superalkalis) properties and potential chemical applications in material chemistry. These research studies not only describe novel superatoms but also verify the usefulness of computational chemistry and solid-state physics methods to predict the stability and physicochemical properties of chemical compounds, supervised learning methods for classification and prediction with a particular focus on the use of these methods in assessing the application potential of superatoms. A concise description of selected research projects is presented in the following sections.
3.1. Prediction of the Electronic Stability of Superhalogen Anion from Topology
Negatively charged molecular systems play an important role in chemical reactions (both as the reagents and reaction products). Due to their potential applications in material chemistry, anions are the subject of extensive theoretical and experimental studies. One class of such compounds is superhalogen anions, whose large electronic stabilities may lead to the forming of unusual chemical materials. An unusual number of ligands in relation to the valency of the central atom has a stabilization effect on the ability of the superhalogen systems to bind an extra electron. To investigate the relationship between superhalogen’s properties of electron affinity and their structure and composition, the quantitative structure-property relationship (QSPR) approach can be used. The QSPR technique mathematically links physicochemical properties with the structure of a molecule (
Figure 2). Descriptors are the quantitates that represent attributes of the molecules and aid in establishing the mathematical correlation. The usefulness of this method has been confirmed for different compounds (such as nanoparticles) [
35].
In [
36], the performance of the QSPR approach was tested to suggest an effective method that could be used to predict the electronic stability of superhalogen anions. A QSPR model was developed to relate superhalogen structural features with vertical electron detachment energy (VDE) for a set of MX
4− (M = B, Al; X = F, Cl, Br) anions. The methodology applied is schematically presented in
Figure 2 and involves the following steps: (i) collecting data set; (ii) calculating molecular descriptors for all studied compounds; (iii) splitting the compounds into training and validation sets; (iv) calibrating a QSPR model, (v) internal and external validating the model with the use of a test and validation sets, respectively.
The Genetic Algorithm was applied, combined with the Multiple Linear Regression approach to select variables and develop models. Relying on the evolutionary operations of ‘crossover and mutation’, the optimal combinations of descriptors that explain property variation within the training set were chosen. The most significant QSPR model is provided below (Equation (1) [
36]):
where N = 20, R
2 = 0.975, Q
2CV = 0.968, Q
2EXT = 0.960, RMSE
C = 0.146, RMSE
CV = 0.167, RMSE
EXT = 0.190.
The descriptors (SpMax4_Bh(s), ATS1m) depend on topology and will be discussed later (Equations (2)–(4)). The values of the determination coefficient (R
2), the cross-validation determination coefficient (Q
2CV), and the external validation coefficient (Q
2EXT) are close to 1, while the corresponding root mean square error values (RMSE
C, RMSE
CV, and RMSE
EXT) are both low and similar. Hence, the developed QSPR model is well-fitted, robust, and has good predictive abilities. In addition, the visual correlation between the observed and the predicted VDE for the training and validation sets is also satisfactory (
Figure 3a).
The leverage approach [
37] was performed to verify the chemical applicability domain of the developed model [
36]. The plot of the standardized residuals versus the leverage values (the Williams plot,
Figure 3b) confirmed that most molecules from the calibration and validation sets are located inside a rectangular area within the leverage threshold h* = 0.45 and ±3 standard deviation units [
36]. In the Williams plot, one compound (AlF
4−) was found to be an X outlier characterized by a leverage threshold (red line in
Figure 3b), and it can be explained as a compound with peculiar features poorly represented in the training set. However, this X-outlier has a small residual, and hence, it can be regarded as a stabilizing point, increasing the model’s precision [
38]. Explicitly, if the h
i values of training compounds are higher than h* and, simultaneously, the predictions for those molecules have small residuals, such molecules are so-called ‘good influence points’, stabilizing the model and making it more precise.
The main advantage of the developed QSPR model is that both descriptors belong to topology molecular descriptors. The topology molecular descriptors are defined as quantifiable properties that can be estimated from the connection table representation of a molecule (e.g., elements, formal charges, and bonds) and do not require knowledge of the atomic coordinates. The topology descriptors are not dependent on the conformation of a molecule and are suitable for non-optimized chemical structure studies. The SpMax4_Bh descriptor is derived from the molecular graph and calculated from the Burden matrices,
Bh(
I). The Burden matrices are augmented adjacency matrices defined to account for heteroatoms and bond multiplicity as the following (Equation (2)):
The diagonal elements are the intrinsic state (I-state) of atoms, while the off-diagonal elements (corresponding to pairs of bonded atoms) are the square roots of conventional bond orders
* (i.e., 1 for single bonds), and all other matrix elements are set at 0.001 [
39]. The SpMax4_Bh(s) descriptor provides the fourth largest positive eigenvalue for each matrix weighted by intrinsic state (I-state). The intrinsic state (I-state) of the
ith atom was estimated with Equation (3):
where
ni is the principal quantum number of the last electronic shell,
is the number of valence electrons, and
is the number of sigma electrons of the ith atom in the molecular structure.
The SpMax4_Bh(s) descriptor strongly depends on the ligands which the superhalogen system consists of. The primary factor differentiating the intrinsic state values among halogen atoms is the principal quantum number of the last electronic shell (ni, Equation (3)). In fact, there is a strong relationship between the VDE value and the nature of the orbitals involved in the electron addition. The principal quantum number defines the energy of an electron and the size of the orbital (i.e., the distance from the nucleus of the peak in a radial probability distribution plot). As the principal quantum number increases, the orbital becomes larger, and the electron density is farther from the nucleus. As ni increases, the electron also has a higher potential energy and is, therefore, less tightly bound to the nucleus. Consequently, involving ligands with larger principal quantum numbers results in a VDE decrease.
The second descriptor in the developed QSPR model is the ATS (autocorrelation of topological structure) descriptor. The ATS descriptor is a measure of the probability of finding objects of defined properties within a distance of interest [
40]. The distance between atoms is estimated as the number of bonds between the respective atoms (topological distance, i.e., the length (number of involved bonds) of the shortest path between the two atoms). The ATS1m descriptor is weighted by the atomic mass of atom pairs at a topological distance equal to 1. In other words, the ATS1m value is obtained by summing all the products
mi ×
mj of all the pairs of atoms
i and
j, for which topological distance equals one as:
where
nAT is the total number of molecule atoms and
is equal to one if atoms
i and
j are connected via chemical bond, zero otherwise.
The ATS1m descriptor (Equation (4)) depends on atomic mass. Different central atoms and ligands affect the VDE of the superhalogen anion. The most striking difference I observed while comparing the BX
4− and AlX
4−, as the replacement of B central atom with Al leads to the electron binding energy (VDE) increase from 6.218 eV (BCl
4−) to 7.016 eV (AlCl
4−) [
9]. The same pattern I found for BF
3Cl
− (VDE = 6.428 eV) and AlF
3Cl
− (VDE = 7.245 eV) as well as for the BF
2Cl
2− (VDE = 6.089 eV) and AlF
2Cl
2− (VDE = 6.990 eV) anions [
9]. In the case of each BX
4−/AlX
4− pair considered (X = F, Cl, Br), the increase in the VDE is accompanied by the increase of the atomic mass of the central atom. Namely, the boron atom (atomic mass equal to 9 a.u.) as a central atom provides a considerably smaller VDE value than those obtained for the corresponding species in which the role of the central atom plays aluminum (atomic mass equal to 27 a.u.). This observation is consistent with the changes in the ATS1m that can be derived from (Table 1 of [
36]). As far as the influence of the ligands on the VDE values is concerned, in the BF
4−/BCl
4−/BBr
4− and AlF
4−/AlCl
4−/AlBr
4− series, the VDE decreases from 8.975 eV to 5.625 eV and from 9.789 eV to 6.346 eV, respectively [
9]. This significant decrease in the VDE value caused by replacing four F ligands with either four Cl or four Br atoms shows that a larger atomic mass of the ligand results in a smaller VDE of the resulting superhalogen anion.
The interpretation of the physical meaning of the topological descriptors used in the developed QSPR model suggests that the electron stability of the MX
4− anions (M = B, Al; X = F, Cl, Br) can be predicted from the atomic mass of the atoms comprising the superhalogen systems. The highest VDE values are expected for species containing, at the same time, a central atom with a large atomic mass and ligands characterized by a small atomic mass. Indeed, the highest VDE is observed for the AlF
4− (VDE = 9.789 eV) anion and the replacement of one fluorine atom with Cl or Br leading to AlF
3Cl
− (VDE = 7.245 eV) or AlF
3Br
− (VDE = 6.519 eV) reduces the VDE by 2.544 and 3.270 eV, respectively [
9]. This observation indicates that replacing only one small ligand in the superhalogen system with a larger atom is highly unfavorable because it reduces electronic stability. This issue will be discussed further in
Section 3.4.
The developed QSPR model predicts VDE directly from the topology molecular graphs and conforms with the general predictions formulated previously for the superhalogen anions [
9,
14]. Explicitly, the observation that a smaller ‘size’ of the halogen ligands usually leads to a larger vertical electron detachment energy is confirmed by the developed model. Based on the analysis of the obtained results [
36], it was demonstrated that the value of the VDE is closely related to the nature of the orbitals involved in electron addition, being the highest one within MF
4− (M = B, Al) and lower for ligands with higher principal quantum numbers. Moreover, the atomic mass of the central atom (B or Al in our case) and the atomic mass of ligands are strongly related to the VDE of the resulting anion.
In paper [
36], based on the two theoretical molecular descriptors calculated exclusively from the molecular structures, a QSPR model to estimate vertical electron detachment energies of MX
4− (M = B, Al; X = F, Cl, Br) superhalogen anions was developed. Both descriptors, as topology descriptors, do not depend on the conformation of a molecule and only use the atoms and connection information of the molecule for the calculation. The developed QSPR model, therefore, allows us to predict the electron binding energy of superhalogen anion based on the connection table representation of a molecule (
Figure 4). The advantage of this approach lies in the fact that it requires only the knowledge of the topology and does not require experimental quantities or quantum-mechanical computations. Hence, the developed QSPR model could provide reliable VDE values of superhalogen anions in the absence of theoretical characterization (e.g., due to insufficient computer resources). Moreover, the QSPR model reveals the mass of atoms contributing to the superhalogen anions and the number of principal quantum number (
ni) of ligands to be the most influential atomic properties in the structures of the superhalogen anions. The developed model and its interpretation described in detail in [
36] might be useful for theoretical and experimental chemists, especially those who design new materials with strong electron-acceptor properties.
3.2. Design of Superhalogen Anions Containing of Noble Gas Atoms
I designed and investigated theoretically superhalogen anions in which inert noble gas atoms play the role of central atoms. The previous theoretical studies [
8,
9,
17,
41,
42] and developed QSPR model [
36] allow us to assume that employing fluorine atoms could result in large electronic stability of the designed systems. Indeed, an ab initio calculation confirmed that mononuclear NgF
n− (n = 3, 5, 7) and binuclear Ng
2F
13− anions (Ng = Xe, Rn;
Figure 5) are geometrically, thermodynamically, and electronically stable systems in the gas phase [
43].
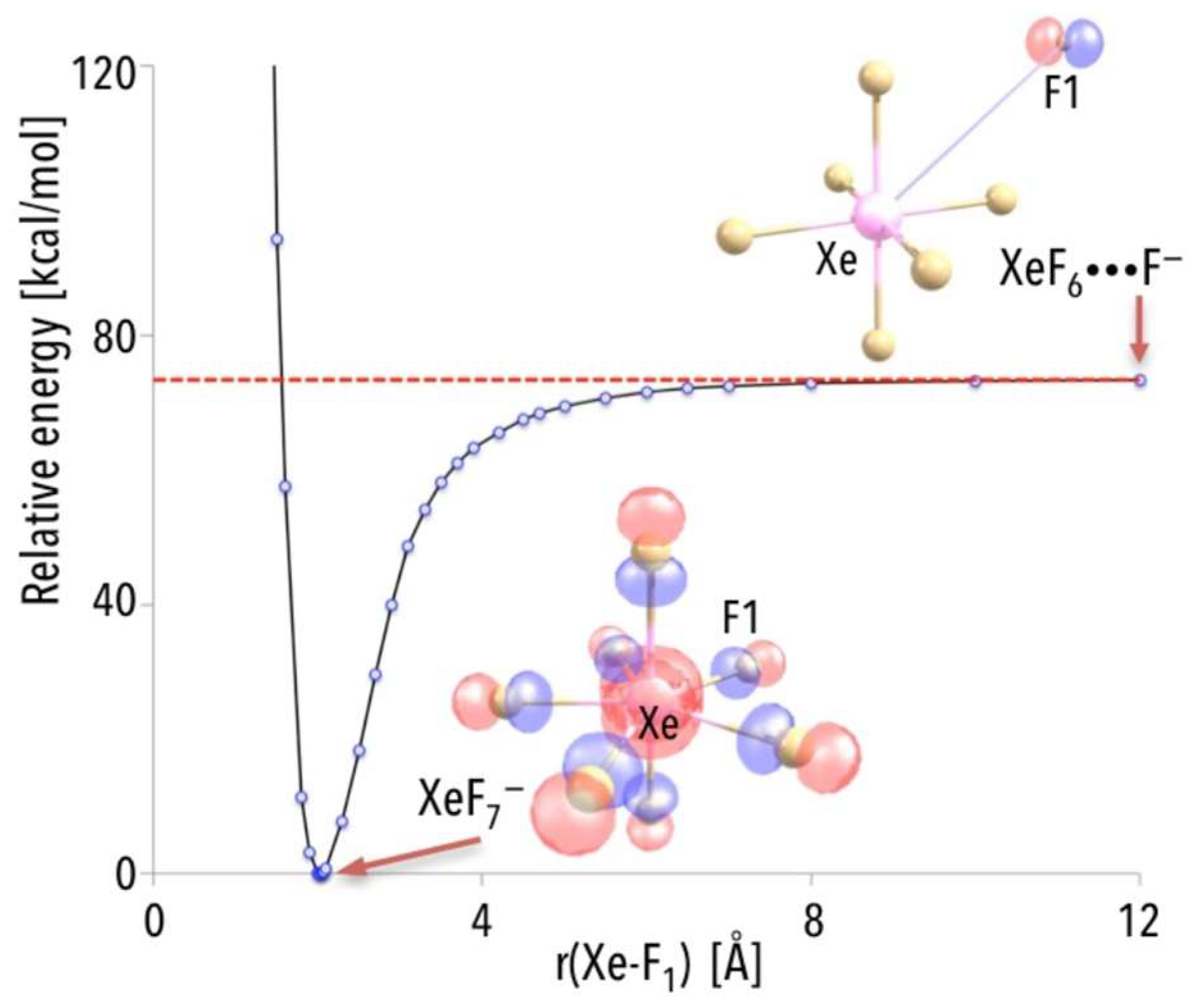
In [
43], the mechanism of the NgF
7− anion formation by assessing the energy change that accompanies the reaction of substrates (XeF
6 and F
−) leading to the creation of the XeF
7− anion was explained. While analyzing the energy profile for the XeF
6 + F
− → XeF
7− process, the excess electron must be assigned to F rather than to XeF
6 (for the separated F and XeF
6 systems, see
Figure 6) due to the considerably larger electron affinity of the fluorine atom. Indeed, as indicated by the localization of the highest energy molecular orbital (HOMO), the additional electron is in the vicinity of the F species (as depicted in
Figure 6). The part of the excess electron density gets transferred to the remaining F atoms as the originally distant F approaches XeF
6 to form the XeF
7− anion (see
Figure 6, where also the HOMO for the equilibrium XeF
7− structure is shown). Consequently, the additional electron is equally distributed (due to symmetry) among all F ligands in the XeF
7− anion. The energy smoothly decreases as the “extra” F atom approaches the neutral XeF
6 molecule, and there is no barrier that must be surmounted. Since the energy of the separated XeF
6 and F
− is much larger (by ca. 73 kcal/mol, see the asymptote in
Figure 6) than the energy of XeF
7− anion at its equilibrium structure, and having in mind that the XeF
6 + F
− → XeF
7− process is predicted to be barrier-free, one may expect the XeF
7− anion to be created spontaneously in the gas phase (whenever F
− ions find themselves in the vicinity of XeF
6 molecules) [
43].
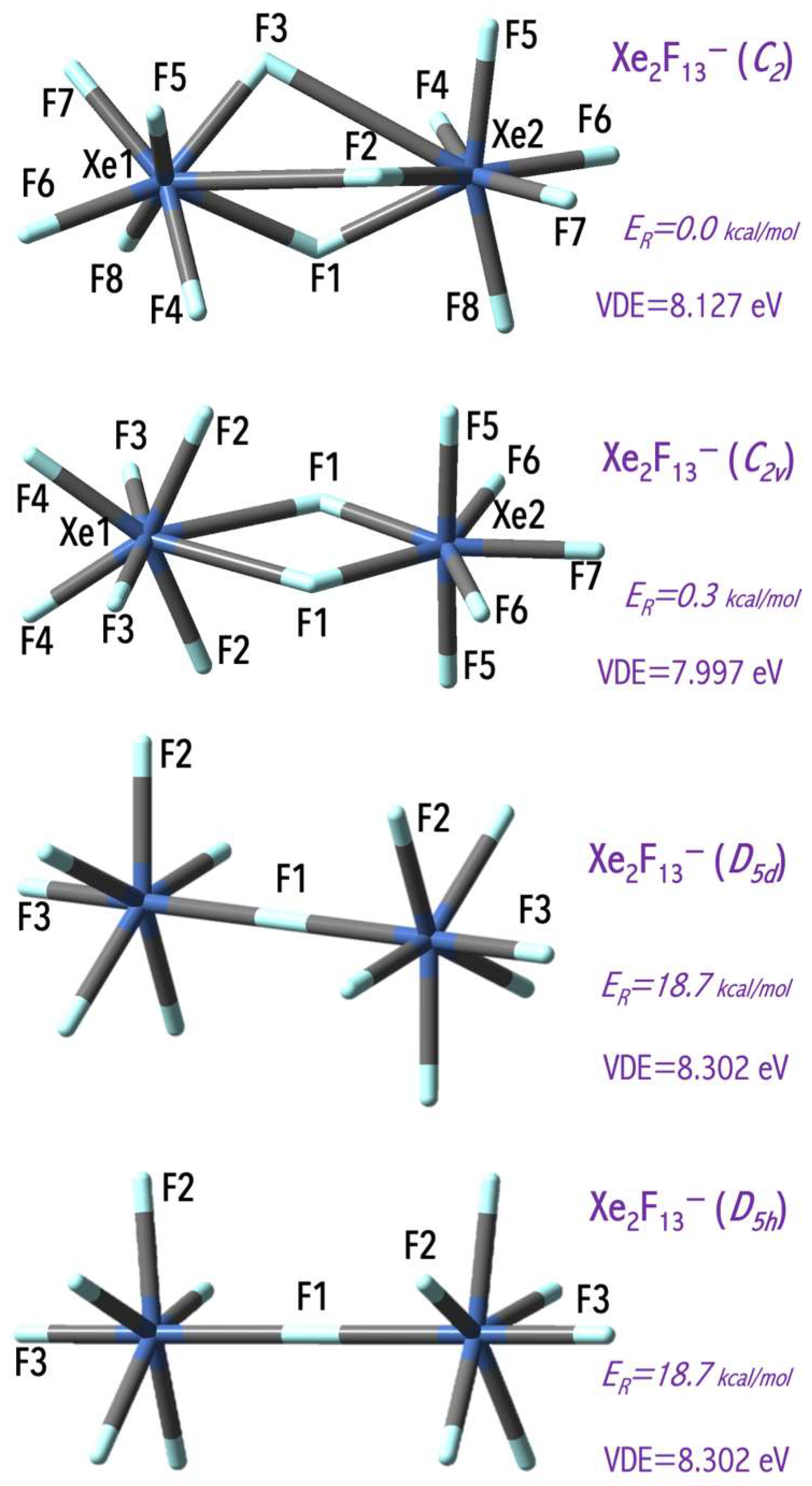
The values of the excess electron binding energies calculated for the Ng
nF
6n+1− anions (Ng = Xe, Rn) span the 5.2–9.6 eV [
43] range and exceed the electron affinity of the chlorine atom (3.62 eV [
44]). The estimated VDE values depend on central atom use and on the number of fluorine ligands (nF,
Figure 5). In general, a larger central atom (with a larger atomic mass) leads to a larger VDE value, and this observation agrees with the mathematical model for the VDE of superhalogen anion prediction [
36]. In addition, the VDE of the Rn
nF
6n+1− anions increases with the number of electronegative ligands (nF) increase due to the stabilization effect of the delocalization of excess negative charge over a larger number of ligands. Replacing mononuclear NgF
n− anions with binuclear Ng
2F
13− systems allow for binding more ligands without system destabilization (due to the reduction of ligand-ligand repulsion in a binuclear system in comparison to a mononuclear system). In addition, my results indicate that the VDE of the binuclear superhalogen Xe
2F
13− system depends on its geometrical structure and spans the 7.997–8.302 eV range, as shown in
Figure 7 [
43]. This observation conforms with the subsequent theoretical studies [
45,
46] indicating that VDE strongly depends on the superhalogen’s geometrical structure (if various isomeric structures are possible and stable).
3.3. Alternative Ligands in Superhalogen Design
A typical superhalogen has its central metal atom decorated with halogen ligands [
8,
9,
14]. However, both theoretical and experimental studies [
18] have shown that the presence of metal or halogen atoms is not mandatory for a cluster to exhibit superhalogen properties. Examples include the noble-gas-based superhalogen molecules [
43] and alternative superhalogen systems where fluoroxyl groups (OF) act as ligands [
45]. The
(M = Li, Na, K, Be, Mg, Ca, B, Al) ground states are highly symmetric structures and have large electronic stability, see
Figure 8. The estimated VDEs always exceed 5 eV, which suggests the superhalogen nature of the
anions. Despite large electronic stability, the
anions seem to be thermodynamically unstable toward
loss/formation [
45]. The thermodynamic instability of the
anions might be caused by the relatively large electron binding energy of the corresponding
fragmentation products. In
Figure 9, the comparison between VDE of
and
anions is presented. Replacing fluorine ligands with fluoroxyl groups always leads to a VDE decrease.
The above studies indicate that the electronegativity of the ligands determines the thermodynamic stability of superhalogen anions. In
Figure 9, the comparison between the VDE values of the
and
anions is depicted. Replacing fluorine ligands with fluoroxyl groups always leads to a decrease in the VDE value. The VDE of a superhalogen anion depends mainly on the electronegativity of the ligands with which this anion is decorated. In addition, the higher VDE of an anion is observed for a large number of such ligands due to the so-called ‘collective effects’ [
17]. The use of very strong electron acceptors such as fluorine ligands (EA = 3.401 eV [
47]) leads to an increase in VDE when compared to the system utilizing fluoroxyl groups (EA = 2.272 eV [
31]). Explicitly, in the Li(OF)
2−/Be(OF)
3−/B(OF)
4− and LiF
2−/BeF
3−/BF
4− series, the increase in VDE from 5.436 to 7.079 eV and from 6.510 to 8.975 eV [
9,
48], respectively (see
Figure 9) [
45]. Based on the ab initio computations, it was proved not only that electronegative substituents ensure large VDE but also that the difference in EA of ligands determines the thermodynamic stability of the resulting superhalogen anions [
45].
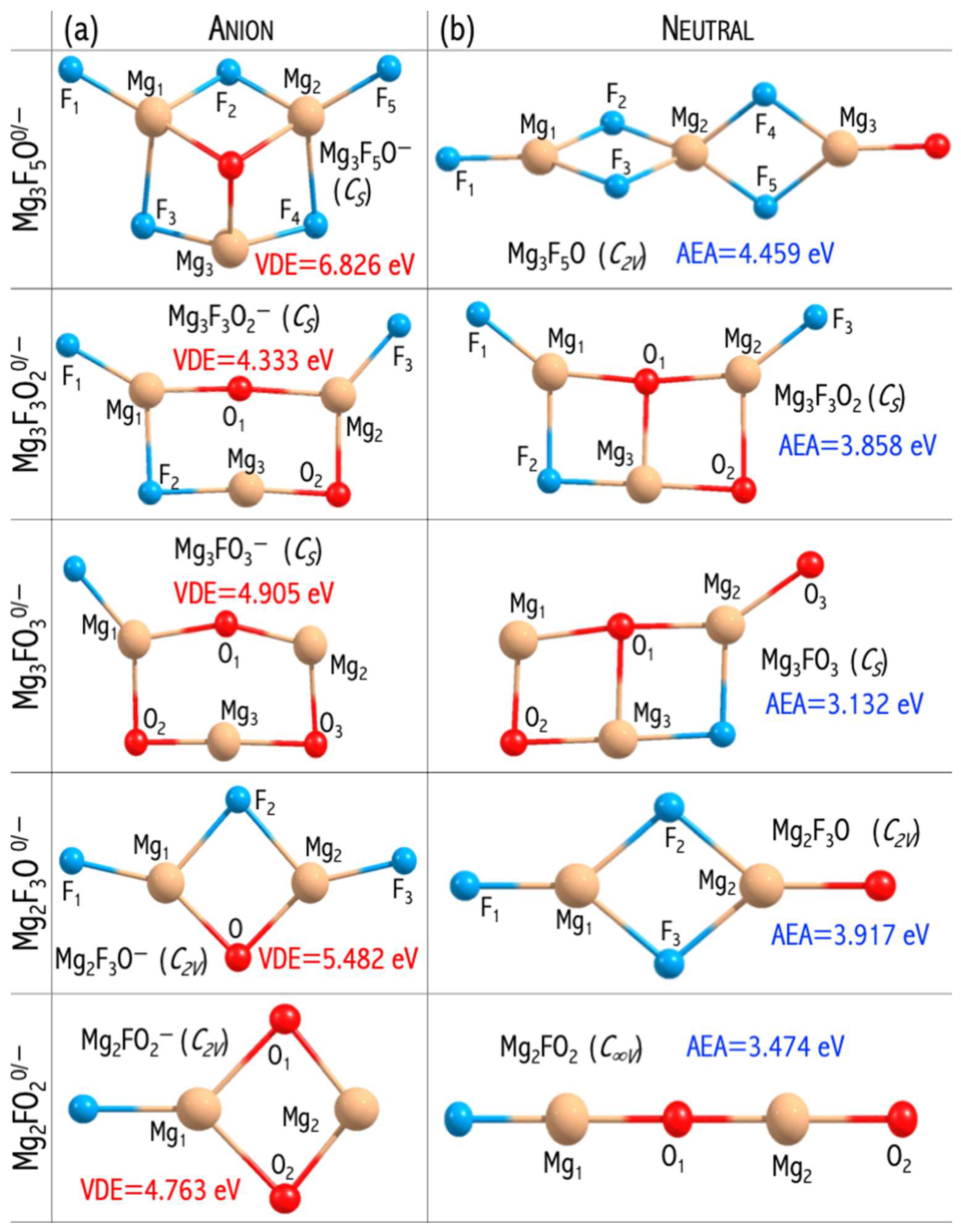
3.4. Polynuclear Superhalogens and Mixed Ligands
The design of polynuclear superhalogen allows to enhance the electron binging energy [
17,
20]. Inspired by the results obtained for binuclear superhalogen anions [
43], I decided to further investigate the influence of the number of central atoms (and ligands) on the electron binding energy in the resulting anions. In addition, I decided to check whether replacing fluoroxyl groups with oxygen and fluorine ligands would enhance the thermodynamic stability of the system. Based on the ab initio computations, I proved the ability to form stable superhalogen anions using mixed ligands (O, F) [
46]. Moreover, replacing fluoroxyl group [
45] with oxygen and fluorine ligands leads to thermodynamically stable
(n = 2, 3; m = 1−3) anions. The positive Gibbs free energy for the
process indicate that each superhalogen and its anion should be formed spontaneously. It means that the
or
species, once synthesized, should successively attach the MgF
2 molecules to develop polynuclear
systems [
46].
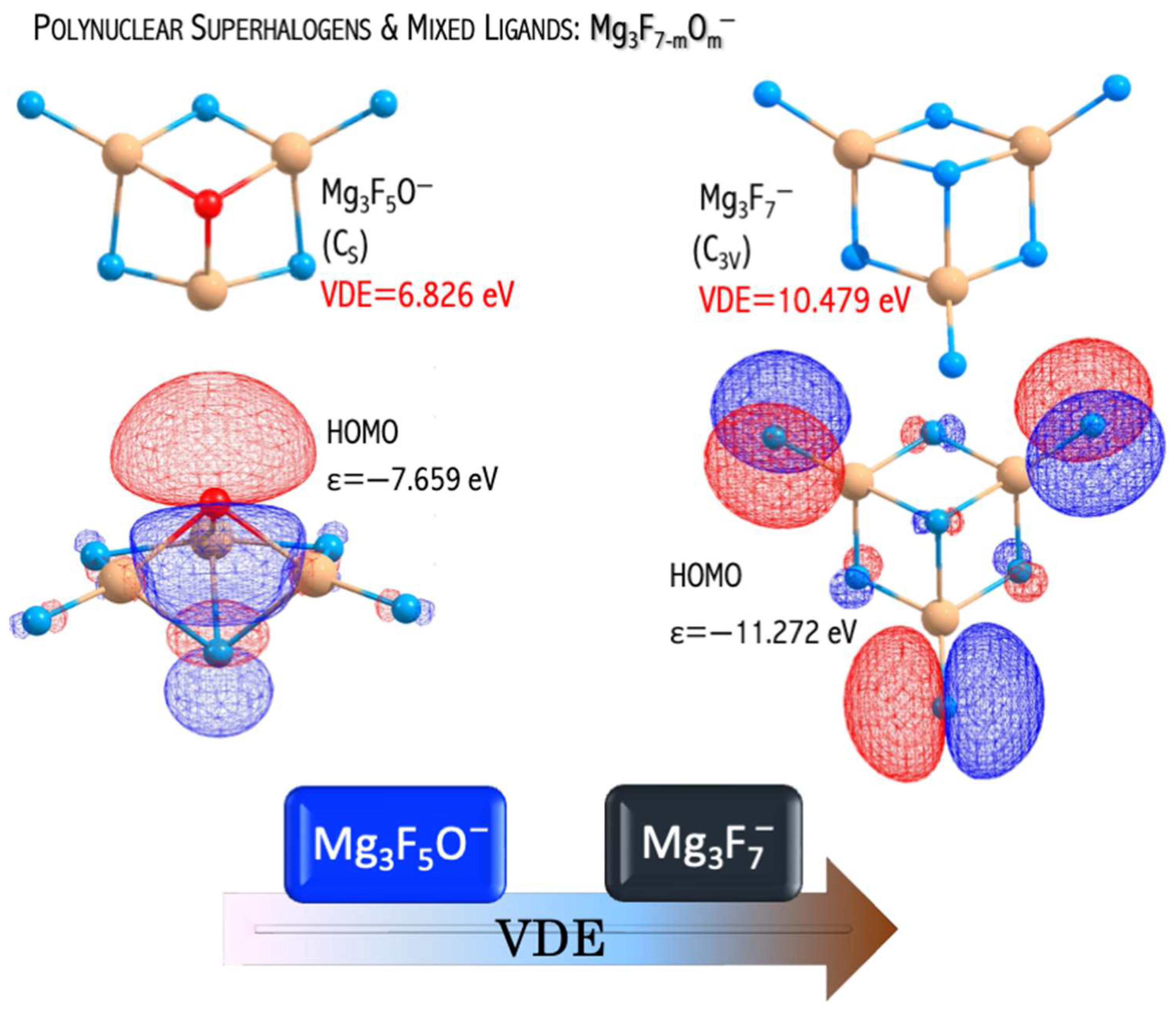
The
anions have large electronic stabilities. Their VDEs span the 4.763−6.826 eV range (
Figure 10) and considerably exceed the EA of the chlorine atom (3.62 eV [
44]) [
46]. The obtained VDE values, albeit significant, are lower than the VDE of the non-mixed
anions described in [
49]. These observations can be explained by the difference in their highest occupied molecular orbital (HOMO) nature. In the case of mixed
anions, the HOMO reveals the bonding and antibonding character with respect to the Mg−F and Mg−O interactions, respectively (
Figure 11). In turn, the non-mixed
anion has a non-bonding HOMO orbital consisting purely of fluorine atomic orbitals (
Figure 11). The described alteration in the HOMO nature was observed for the first time by Freza et al. [
7] and explains the electronic stability decrease with mixed ligand introduction. Therefore, the replacement of fluorine atoms with mixed ligands (F, O) leads to symmetry breaking in mixed superhalogen anions and subsequent decreases in electron stability (
Figure 11) [
46,
49].
Even though the VDEs of the designed oxyfluoride superhalogens are lower than those of the corresponding non-mixed fluoride systems (described in [
49]), still, the designed superatoms do have very large VDEs (approach 7 eV) [
46]. Hence, the possibility of utilizing mixed superhalogens in chemical applications should be promising and is yet to be discussed in the following
Section 3.6.
The enhanced high electronic stability of the designed superatoms [
43,
45,
46,
49] is associated with the following:
- (i)
large electronegativity of the ligands (F, O, OF) that efficiently supports strong extra electron binding,
- (ii)
the large number of electronegative ligands that ensure the efficient negative charge distribution in the anion,
- (iii)
HOMO nature (i.e., it is mainly contributed by atomic orbitals of electronegative ligands),
- (iv)
the positive charges of metal cores provide extra stabilization of an electron delocalized over ligands.
The above factors have a stabilization effect on the ability of the designed systems to bind an extra electron. Overall, the Ng
nF
6n+1− (Ng = Xe, Rn; n = 1–2) [
43],
((M = Li, Na, K, Be, Mg, Ca, B, Al) [
45],
(n = 2, 3; m = 0−3) [
46,
49] have VDE exceeding the electron affinity of Cl atom (3.62 eV [
44]) and can act as a strong oxidizing agent [
49,
50,
51].
3.5. Designed Superhalogens as Strong Oxidizing Agents
The superhalogens described in the above sections have a large electron affinity. Therefore, a natural continuation of the above research was to verify the hypothesis on the ability of the designed superhalogen systems to act as oxidizing agents in chemical processes. Explicitly, the stability and the charge flow in [
D][superhalogen] complexes, where
D is an atom (e.g., Li) or molecule (metal oxide, C
60 fullerene), were verified. The electron affinity of superhalogen determines its ability to ionize molecules effectively. In my studies, I considered the superhalogens whose electron binding energy (expressed by the VDE of the corresponding anion) covers the 4.8–10.5 eV range. I chose both mononuclear (LiCl
2, LiF
2, LiI
2, MgCl
3, MgF
3, AlCl
4, AlF
4) [
7,
8,
9] as well as polynuclear Mg
3F
7 [
49] superhalogens due to their simple structure and sufficient electron affinity.
I studied the influence of superhalogen type (mononuclear and polynuclear) on its ability to serve as an effective oxidizing agent for fullerene C
60 ionization [
49,
51]. The C
60 ionization (IE = 7.58 eV [
52]) is, however, difficult to occur and requires a strong oxidizer. It was demonstrated that utilizing very strong oxidizing agents (such as neutral superhalogens) leads to complete or partial C
60 nanoparticle ionization [
49,
51]. The fullerene, when combined with a properly chosen superhalogen (which plays the role of oxidizer), should form stable and strongly bound ionic (C
60)
•+(superhalogen)
− compounds, see
Figure 12a,b. The above conclusion was formulated based on the ab initio calculations on (i) the structural deformation of superhalogens and fullerene upon the ionization process, (ii) predicted charge flow between C
60 and each superhalogen (which allows for the estimation of the electron density flow from the nanoparticle to a superhalogen during the ionization process) and (iii) the interaction energy between the superhalogen and fullerene in the (C
60)
•+(superhalogen)
− system.
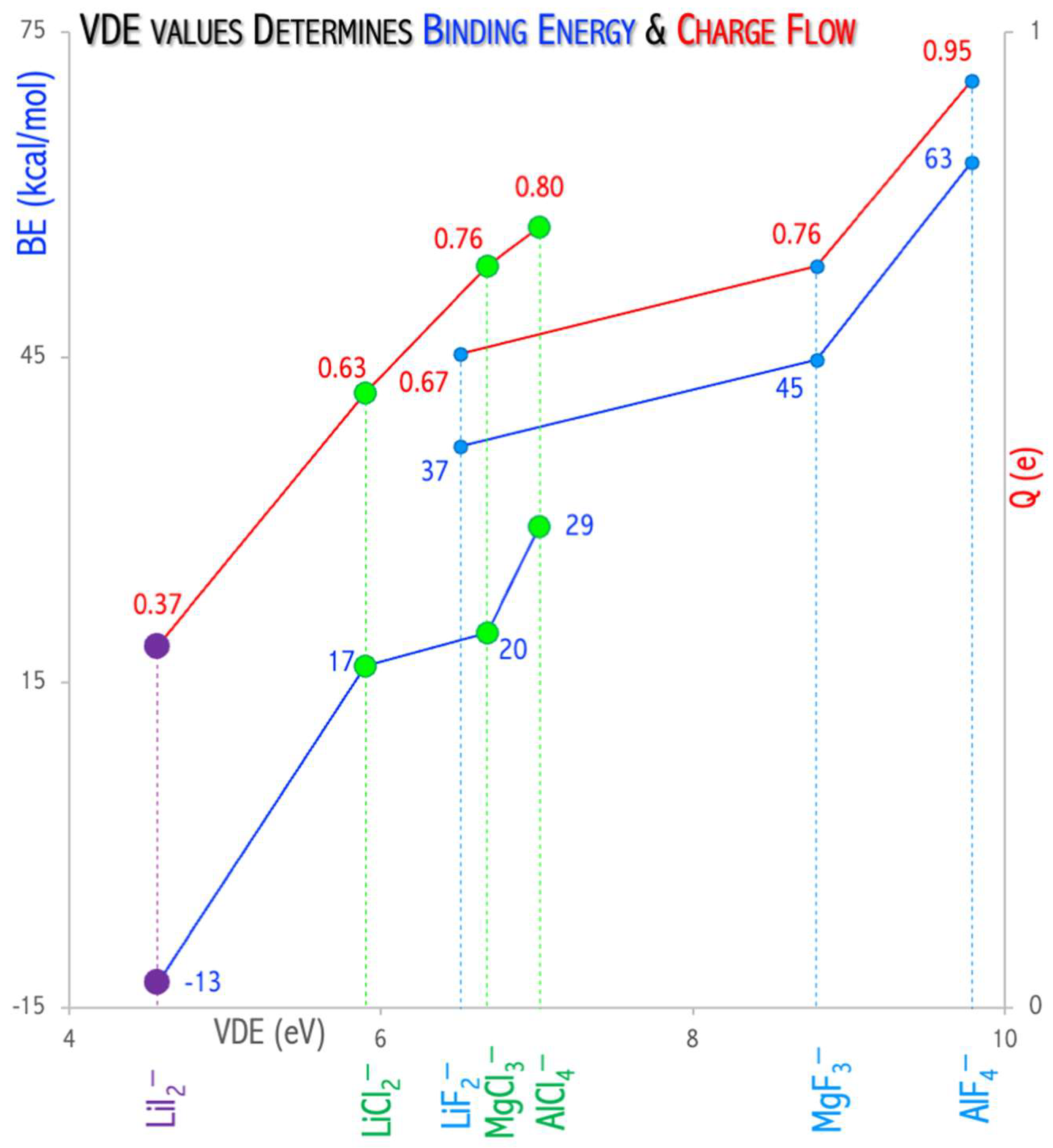
The value of vertical electron detachment energy in the superhalogen anion determines the stability and charge flow between C
60 and the superhalogen [
49,
51].
Figure 13 shows the increase in the interaction energy and charge flow with VDE increase. The process of fullerene binding by superhalogens (LiCl
2, LiF
2, LiI
2, MgCl
3, MgF
3, AlCl
4, AlF
4, Mg
3F
7) with a VDE in the range of 5.9–10.5 eV is exothermic (the interaction energy is 19.5–62.9 kcal/mol in the gas phase, 7.9–87.4 kcal/mol for acetonitrile, 8.3–83.8 for ortho-dichlorobenzene). The (C
60)···(LiI
2)
• complex formation is an endothermic process (the interaction energy is −12.7 kcal/mol (gas phase,
Figure 13), −6.9 kcal/mol (acetonitrile), −10.3 (ortho-dichlorobenzene,
Figure 12c). This means that the VDE (4.8 eV) of the LiI
2− anion is insufficient to lead to the effective oxidation of the fullerene molecule, and the LiI
2 superhalogen only forms a weakly bound complex (C
60)···(LiI
2)
•, see
Figure 12c [
51]. On the other hand, the use of a properly selected superhalogen (having a VDE above 5.9 eV, see
Figure 12a,b) leads to a strongly bonded ionic (C
60)
•+(superhalogen)
− compound [
49,
51].
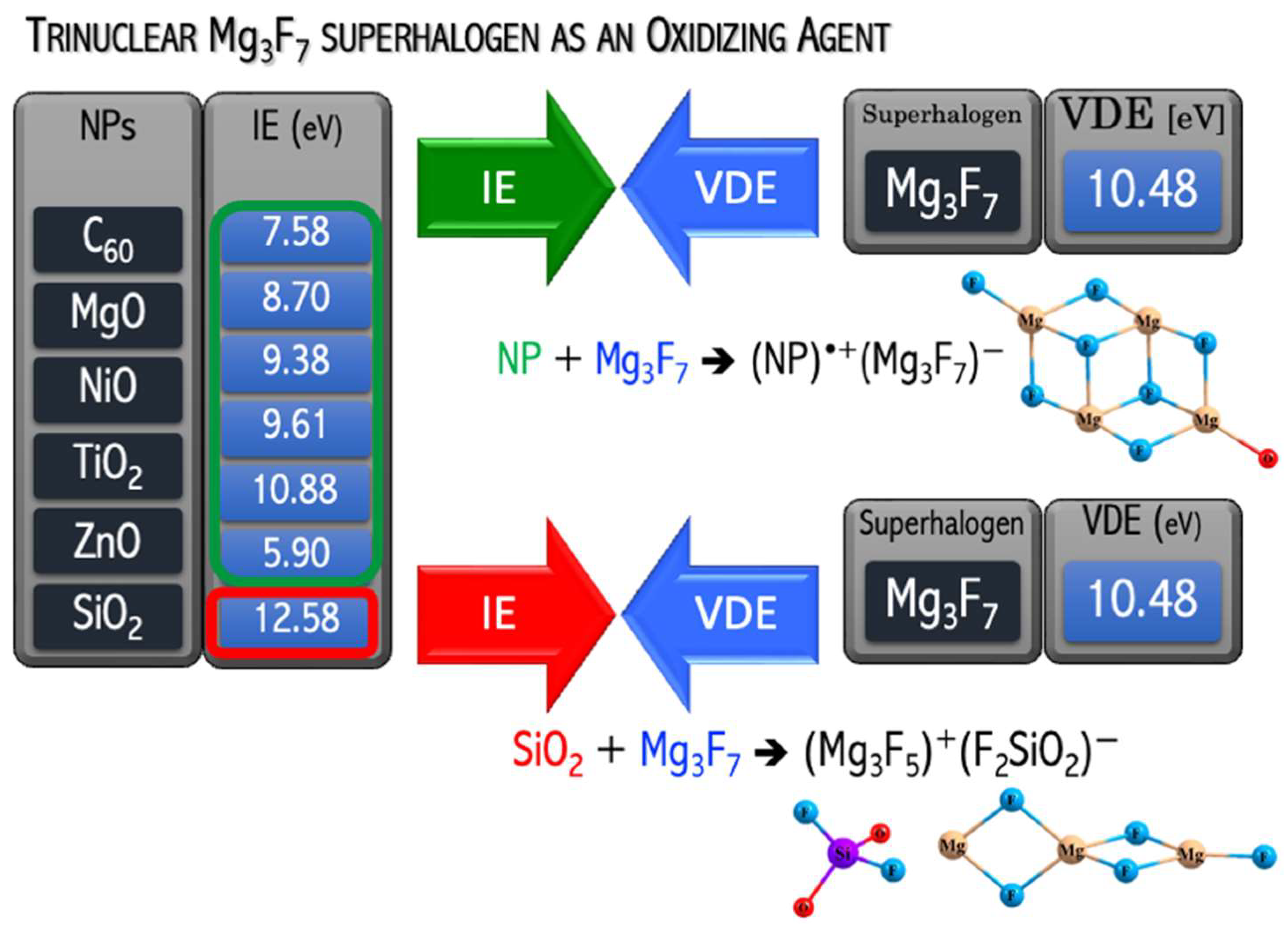
A natural continuation of the above research was a project in which the usefulness of the hypothesis about the possibility of binding and removing potentially toxic metal oxide nanoparticles from the environment was tested. In [
49], I demonstrated that the replacement of mononuclear superhalogens by the polynuclear Mg
3F
7 system leads to strongly bonded (C
60)
•+(superhalogen)
− compounds, see
Figure 14. In [
50], I verified the ability of the polynuclear Mg
3F
7 superhalogen to oxidize mono- and metal dioxides (CoO, CuO, MgO, NiO, ZnO, MnO
2, SiO
2, TiO
2). The verification performed for many [MeO
n][Mg
3F
7] systems was positive (
Figure 15). In particular, the Mg
3F
7 superhalogen can bind and form stable chemical compounds when interacting with metal oxides, whose ionization energy approaches 11.4 eV (
Figure 15). In contrast, the Mg
3F
7 superhalogen is not able to effectively ionize the silica oxide SiO
2, whose ionization energy exceeds 12 eV (
Figure 15). Based on the ab initio results [
50], the key role of the relationship between the ionization energy of the metal oxide and the binding energy of the excess electron of the superhalogen with which MeO
n interacts was indicated (
Figure 15).
The ab initio studies on [
D][superhalogen] systems [
49,
50,
51] not only explain the stability (or lack thereof) of the [
D][
A] chemical compounds (
D = C
60, CoO, CuO, MgO, MnO
2, NiO, SiO
2, TiO
2, ZnO;
A = LiF
2, LiCl
2, LiI
2, MgF
3, MgCl
3, AlF
4, AlCl
4, Mg
3F
7), but also they allow to predict the stability of almost any chemical systems built according to the donor-acceptor (
D–
A) scheme (
Figure 15) and can be useful in the design of new nanomaterials (e.g., binary nano-salts, which is yet to be discussed in
Section 3.10).
3.6. Superhalogens as Electrolytes of Lithium-Ion Batteries
Lithium-ion batteries (LIBs) have many applications—from portable electronic devices to electric vehicles. The LIBs, however, suffer from limited recycling life (<1000 cycles) as well as durability and safety issues, much of which is attributed to the current use of organic electrolytes. In this regard, the replacement of organic liquid electrolytes with inorganic electrolytes seems to be very appealing. Examples include LIBs consisting of LiBF
4, LiAsF
6, LiPF
6, LiFePO
4, LiClO
4, or LiN(SO
2F)
2 inorganic electrolytes, see
Figure 16b. The anionic subunits of these electrolytes are all superhalogen anions. However, superhalogen electrolytes have some limitations. LiBF
4 has the inferior ability to form solid electrolyte interphases (SEIs), LiClO
4 is explosive, while LiAsF
6 is poisonous [
53]. LiPF
6 decomposes to PF
5 and LiF, whereas PF
5 is readily hydrolyzing to form PF
3O and HF. PF
3O and HF are very reactive on the cathode and anode surfaces, which negatively impacts the electrode performance [
54]. LiFePO
4 suffers from the so-called ‘memory effect’ resulting in losing the usable capacity if recharged repeatedly after being only partially discharged [
55]. Besides, LIBs have limited performance at elevated temperatures and limited life cycles (due to surface phenomena on both electrodes). In this frame, the performance of the polynuclear magnesium-based superhalogen anions (1st column of
Figure 17) [
46,
49] as potential alternatives for electrolytes of lithium-ion batteries was tested.
For this purpose, I designed and examined the stability of the Li
+/Mg
nF
2n+1−2mO
m− (n = 2, 3; m = 0–3) compounds and corresponding ground states are provided in
Figure 17. Based on the ab initio calculations performed both in the gas phase and water solution, I demonstrated that polynuclear superhalogen anions can act as novel electrolytes in lithium-ion batteries. This conclusion is supported by the following:
- (i)
The geometrical stability of Li
+/Mg
nF
2n+1−2mO
m− equilibrium structures (
Figure 17).
- (ii)
Hardly any charge transfer (below 0.01 e) between lithium ion and each of superhalogen MgnF2n+1−2mOm− anions confirming the superhalogen anion stability in lithium salt due to negligible electronic density transfer from Li+ upon its interaction with superhalogen anion.
- (iii)
Vertical ionization potential (VIP) increasing upon Li
+ counterion introduction. The Li
+ counterion introduction results in more stable Li
+/Mg
nF
2n+1−2mO
m− compounds (VIP
CCSD(T) = 7.8–12.2 eV [
56]) in comparison to corresponding anions (VDE
CCSD(T) = 3.471–9.515 eV [
46,
49]).
- (iv)
The interaction energy between the Li
+ cation and a superhalogen anion (
) covering the 5.884–7.456 eV range (in a vacuum,
Figure 16a) and 0.365–0.750 eV range (in water solution) [
56]. The lower
lithium ion bonding the higher Li
+ ion conductivity.
- (v)
The low binding energy of H
2O to the electrolyte (
= 0.616–0.969 eV,
Figure 16a) as a criterium of chemical stability of electrolyte toward interaction with water molecules.
- (vi)
The large electrolyte stability window (EW of 9.03–14.70 V [
56]) ensures maximal potential difference between electrodes. The high operating voltage of the cell is achieved if the LUMO and HOMO levels of the electrolyte are separated by a large band gap.
Based on the ab initio computations and detailed analysis, a general recommendation on polynuclear superhalogen anion performance as an electrolyte in lithium-ion batteries was formulated. From the ab initio calculation result (performed both in a vacuum and with the PCM solvation model) analysis, the magnesium-based superhalogens Mg
nF
2n+1−2mO
m (n = 2, 3; m = 0–3) with high electron binding energies (manifested by the vertical electron detachment energies (VDEs) of their corresponding daughter anions of the 4.76–10.48 eV range, see
Figure 17) might serve as building blocks of lithium salts. Since the Li-ion binding energy of the Li
+/Mg
3F
7− salt (
= 5.884 eV,
Figure 16c) and sensitivity to water (
= 0.618 eV,
Figure 16c) were found to be lower compared to those of several currently used electrolytes in commercial Li-ion batteries (e.g., LiFePO
4), the Mg
3F
7-based electrolyte is recommended for lithium-ion battery application [
56]. The analysis of potential ion battery electrolytes from the superhalogen perspective [
56] opens a path for the design and synthesis of desirable superatomic systems as promising building blocks for new functional materials.
3.7. Superatoms as Building Blocks of Materials
Designing novel superatoms is aimed at proposing the building blocks for functional materials. The main benefit of replacing conventional bulk species with superatom-based materials is the possibility of tuning the properties of superatom. The resulting superatomic materials with unique and tailored properties extend the scope of materials science with desirable features (following the safe-by-design idea). Recently, Roy et al. [
57] synthesized the binary assembly of metal chalcogenide clusters (i.e., Co
6Se
8(P(C
2H
5)
3)
6 (
1A), Cr
6Te
8(P(C
2H
5)
3)
6 (
2A), and Ni
9Te
6(P(C
2H
5)
3)
8 (
3A)) with C
60 carbon clusters as building blocks. The [Co
6Se
8(P(C
2H
5)
3)
6][C
60]
2 and [Cr
6Te
8(P(C
2H
5)
3)
6][C
60]
2 solids resemble conventional [M
2+][X
−]
2 solids of the CdI
2 structure type. In turn, the Ni
9Te
6(P(C
2H
5)
3)
8, as a more metal-rich system, possesses a larger reducing ability than Co
6Se
8(P(C
2H
5)
3)
6 and Cr
6Te
8(P(C
2H
5)
3)
6 clusters and forms cubic crystals analogous to M
+X
− salts (like the natural rock salt does). Different electronic properties accompany crystallographic dissimilarity among these solids, as the
1A•2C
60 and
2B•2C
60 are gapped semiconductors, whereas
3A•C
60 has ferromagnetic characteristics. Even though the experimental results confirmed the ability of molecular clusters to act as building blocks of solid-state materials, they did not explain the atom-like phenomena of molecular clusters.
The obtained DFT results proved the effectiveness of first-principles calculations to access the atom-like nature of transition metal clusters and to evaluate the influence of the superatomic nature on the crystal structure and physicochemical properties of cluster-assembled materials. In particular, the superalkali nature of the
3A cluster was proved. A superalkali is a cluster of atoms exhibiting enormously low ionization energy (IE lower than the IEs of alkali metal atoms). The superalkali nature of the
3A superatom (Ni
9Te
6(P(C
2H
5)
3)
8) has been proved by low IE (of 2.910 eV that is lower than the IE of cesium atom (IE = 3.9 eV [
34]) and antibonding HOMO orbital in relevance to Ni–Te interactions, see
Figure 18b [
58]. The antibonding character of the HOMO orbital makes the molecule more susceptible to the ionization process (by lowering its IE value). In turn, the remaining metal clusters have larger electronic stabilities due to IE(
1A) = 4.307 eV and IE(
2A) = 4.543 eV and the bonding HOMO orbital with respect to Co–Se (
1A) i Cr–Te (
2A) interactions, see
Figure 18a [
58]. In the
1A and
2A clusters, the outermost electrons are more tightly bound, making these superatoms less electron-donating than chemically reactive
3A superalkali. Consequently,
1A and
2A form [M
2+][X
−]
2 salts of CdI
2-type crystallographic structure, while strong electron donor
3A agent transfers an electron to C
60 fullerene to form [Ni
9Te
6(P(C
2H
5)
3)
8]
+[C
60]
− salt (in analogy to the natural rock salt, see
Figure 19).
In [
58], we proved the effectiveness of the first-principles approach to assessing the activation energy of cluster-assembled materials. The energy gap between the HOMO and LUMO orbitals (
= ε
LUMO − ε
HOMO) is a measure of kinetic stability. A large
implies high kinetic stability and low chemical reactivity due to the energetic disadvantage of adding electrons to a high-lying LUMO or removing electrons from a low-lying HOMO and so to create the activated complex of any potential reaction. The
values of isolated components (1.13–3.00 eV, see
Figure 18a–c) explain both the relatively high reactivity of the
3A superatom (
= 1.13 eV) and low BE of C
60/C
60 dimer (BE = 0.31 eV,
= 3.00 eV). The
is closely correlated to band gaps in solid states. The
1A•2C
60 and
2A•2C
60 systems have been proven to possess good electrical conductance with a semiconducting behavior, their Arrhenius activation energies (E
a) being of about 150 meV (
1A•2C
60) and 100 meV (
2A•2C
60) [
57]. Considering the obtained
(68–89 meV [
58]),
1–
2AB systems are small-gap semiconductors. The applied B3LYP-D3 functional is expected to underestimate the
, which then correlates nicely with the experimental activation energies.
The
1–
3AB systems show a monotonic decrease in
values with a decrease in the ionization energy of metal clusters. All systems (
1–
3AB) show a reduction in
values as compared to pure
1–
3A species. This
reduction is the largest for the
3AB system (to 0.48 eV) and gives evidence for the generation of a new HOMO orbital between the original HOMO and LUMO orbitals; consequently, the original HOMO of
3A cluster becomes HOMO-1 and the
becomes significantly smaller than in the pure
3A metal cluster [
58]. This means that the
3A superalkali shifts the valence electron to C
60, and a new energy level is generated into the occupied orbital site. A superalkali, as a strong electron donor, enables the
reduction of their complexes, which enhances their conductance properties [
59]. In addition,
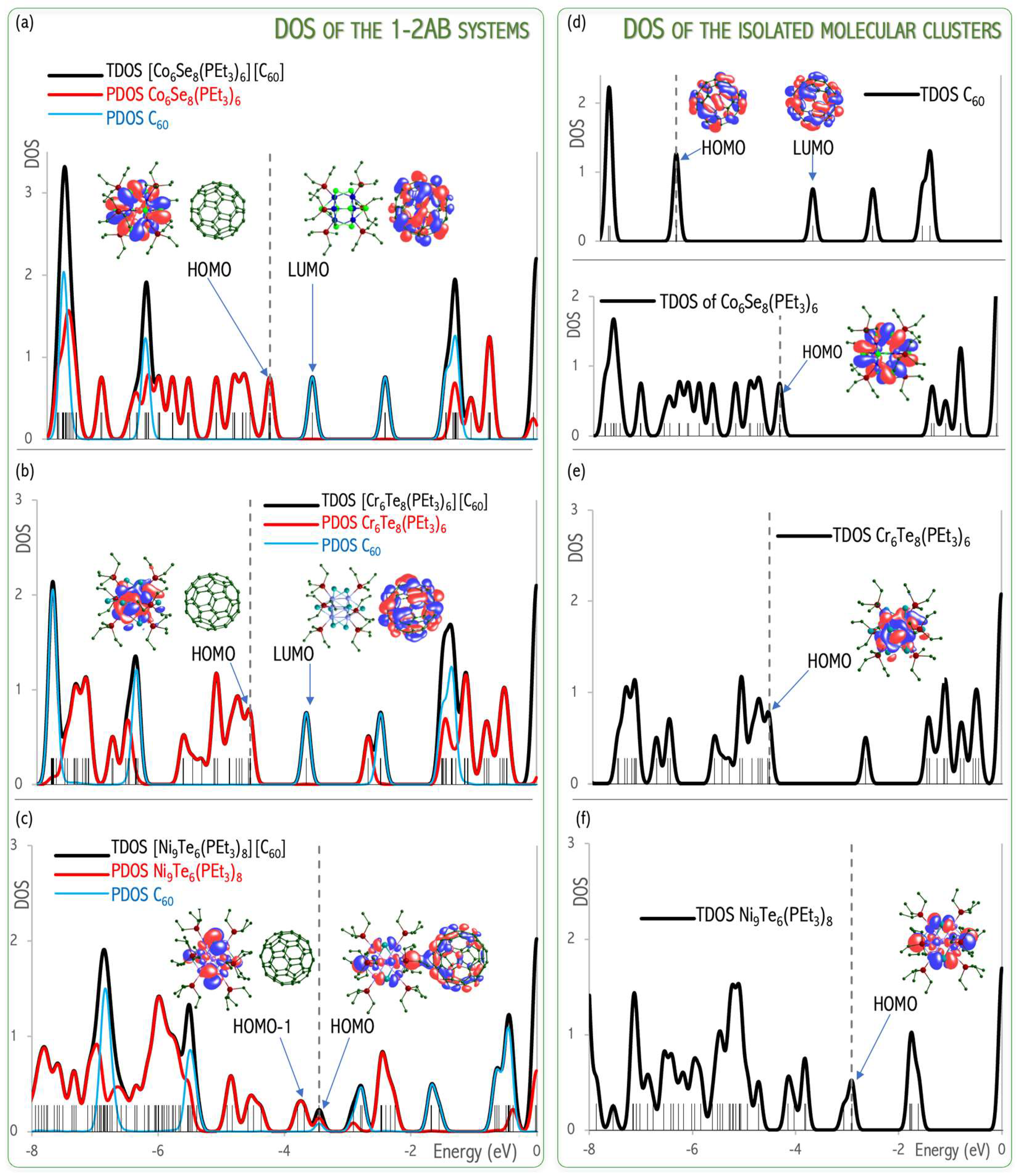
Figure 20a,b of DOS shows that the states of the
1–
2AB systems exhibit a very localized nature. Their HOMOs are contributed atomic orbitals of the metal cluster and thus not by the anion (C
60), as would be expected by an ionic solid. This observation implies a minor ionic character of the
1–
2AB chemical systems. On the contrary, the
3AB reveals that the HOMO consists of hybridized
3A superalkali and C
60 orbitals forming covalent-like
3A-C
60 interaction, see
Figure 20c [
58,
60]. These results proved the effectiveness of first-principles calculations to assess the influence of superatomic nature on the crystallographic structure and physicochemical properties of cluster-assembled materials.
3.8. Superalkali Substitution in Perovskite
The ABX
3-type perovskites (where A and B are cations, while X is an anion) receive attention as efficacious light absorbers for solar cells due to their high-power conversion efficiency (up to 24%). The photoelectric conversion efficiency is determined by a suitable band structure. Cation (A) substitution can be an efficient approach to adjust the electronic band structure of lead halide CsPbBr
3 perovskites. The examples include introducing superalkali cations to replace the Cs
+ cation in the CsPbBr
3 material [
61]. The bimetallic superalkalis (LiMg, NaMg, LiCa, and NaCa) were chosen since they are structurally simple compounds and have a strong tendency to detach one electron (IE = 4.57–4.92 eV [
62]) to achieve a closed-shell cation. Based on the first-principles calculations, it was evaluated whether the A-site cation may fit within the cavities in the BX
3 framework based on the Goldschmidt tolerance factor (
α, Equation (5)):
where
is the ionic radius of the A-cation,
is the ionic radius of the B-cation,
is the ionic radius of the anion.
The perovskite structure is favored by the following tolerance factors:
α = 0.90–1.0 for cubic perovskites and
α > 1.00 for tetragonal structures [
63]. The obtained tolerance factors are 0.94 (MgLi–PbBr
3), 1.03 (NaMg–PbBr
3), 1.04 (CaLi–PbBr
3), and 1.07 (CaNa–PbBr
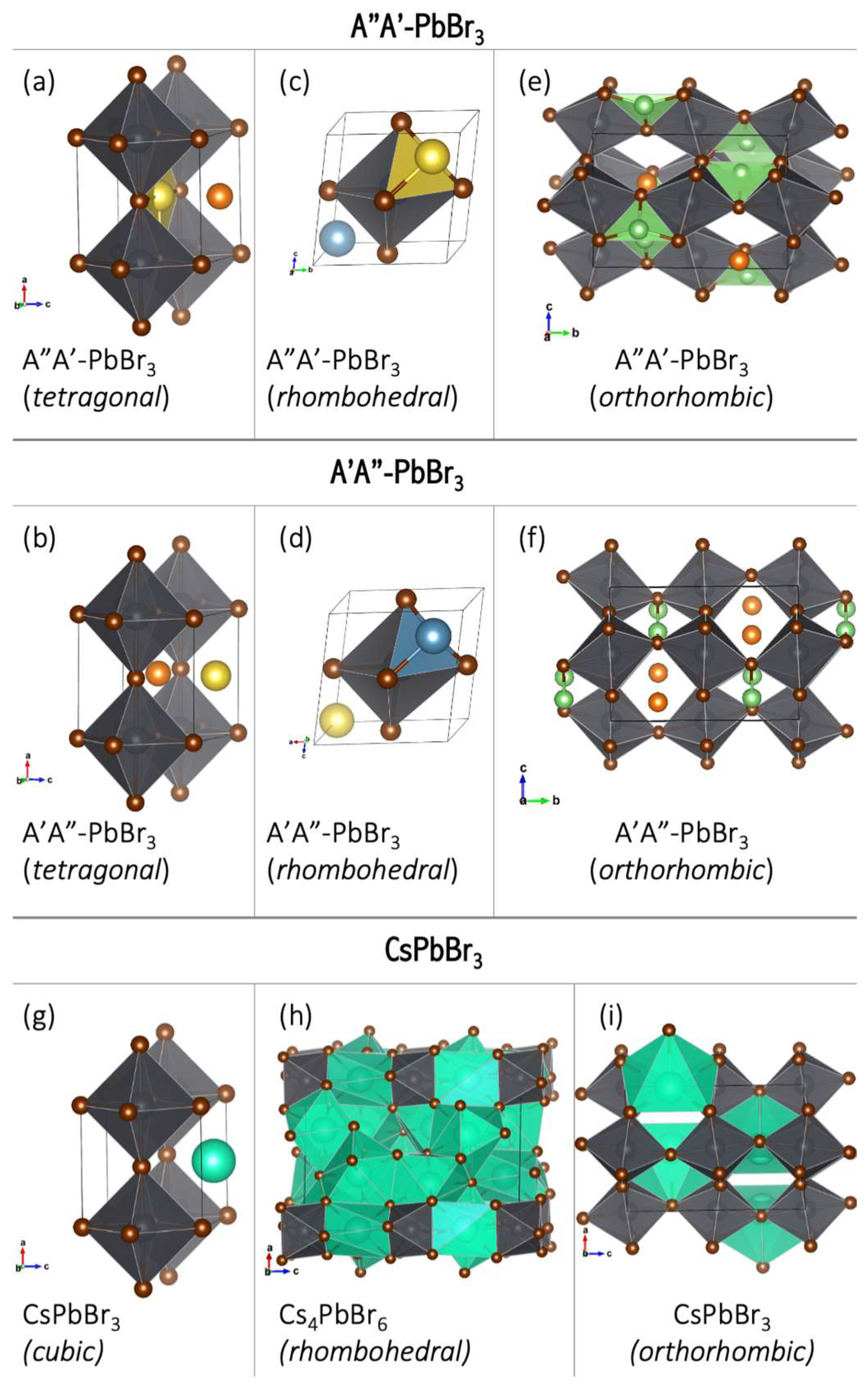
3), respectively, implying that the bimetallic superalkalis are a viable substitute for cesium in stable perovskites. The perovskite structure is, however, distorted from a cubic structure to a tetragonal, rhombohedral, or orthorhombic structure (
Figure 21). This distortion arises because of the modified size of the A ion, which causes a tilting of the BX
6 octahedra to optimize A–X bonding [
33]. Because all superalkali cations considered in [
61] are significantly less electropositive (the electron binding energy in the 4.57–4.92 eV range [
14]) than cesium (IE = 3.9 eV) [
34], the E
g values of superalkali–PBBr
3 are much lower than that of CsPbBr
3 (E
g of 2.48 eV) [
28]. Based on DFT calculations, LiMg
+ and NaMg
+ superalkalis with smaller ionic radii (2.21 and 2.61 Å, respectively) form semiconductors with a band gap from 0.25 to 1.54 eV. The superalkalis with larger ionic radii (i.e., LiCa
+ and NaCa
+) create systems with a metallic nature. Hence, bimetallic superalkalis with ionic radii up to 2.6 Å can form stable perovskites with low band gaps, whereas larger superalkalis (i.e., ionic radii approaching 2.7 Å) form metallic systems.
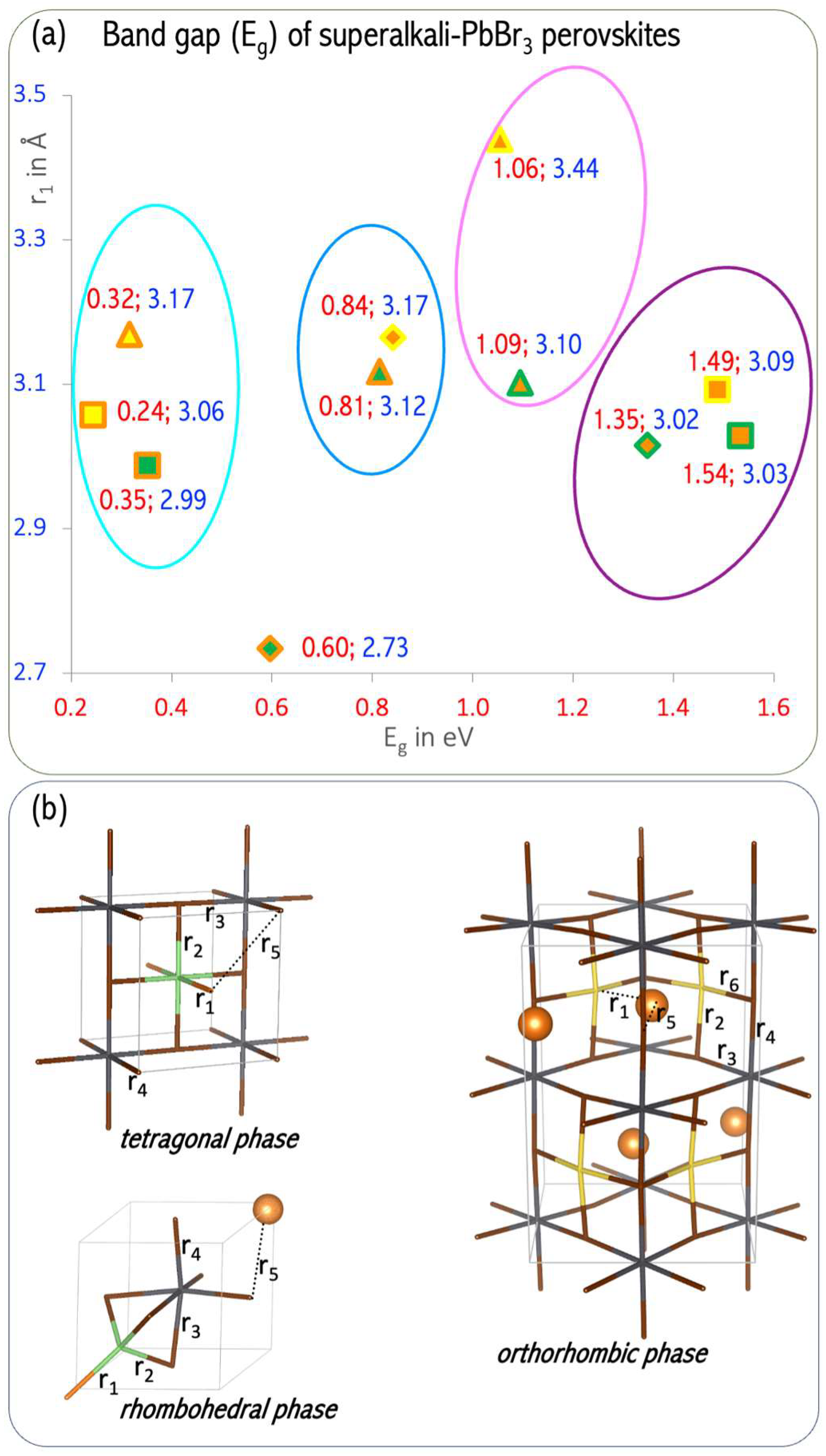
Based on a detailed analysis of the relationship between the length of the alkali metal–alkaline-earth metal (A′–A″) bond and the band gap (E
g), we formulated recommendations for the modification of the electronic structure and the design of perovskites with desirable properties [
61]. The lowest E
g value (0.24–0.35 eV) can be obtained for A′Mg–PbBr
3 perovskites with A′–A″ bond lengths in the range of 2.99–3.70 Å (marked in cyan in
Figure 22). In turn, the highest value of E
g (1.35–1.54 eV, marked in violet in
Figure 22) can be expected for MgA′–PbBr
3 systems with a very narrow A′–A″ bond range (3.02–3.09 Å [
61]). The band gap width of 0.8 eV corresponds to the narrow A′–A″ bond range of 3.12–3.17 Å (circled in blue in
Figure 22) corresponding to the orthorhombic structure of LiMg–PbBr
3 and the rhombohedral structure of MgNa–PbBr
3. The orthorhombic MgA′–PbBr
3 structures correspond to an E
g in the range of 1.06–1.09 eV (circled in magenta in
Figure 22), for which the A′–A″ distances differ by 0.34 Å. The analysis of the structure–band gap width relationship demonstrated the usefulness of superalkalis for designing perovskites with the desired photoelectric properties.
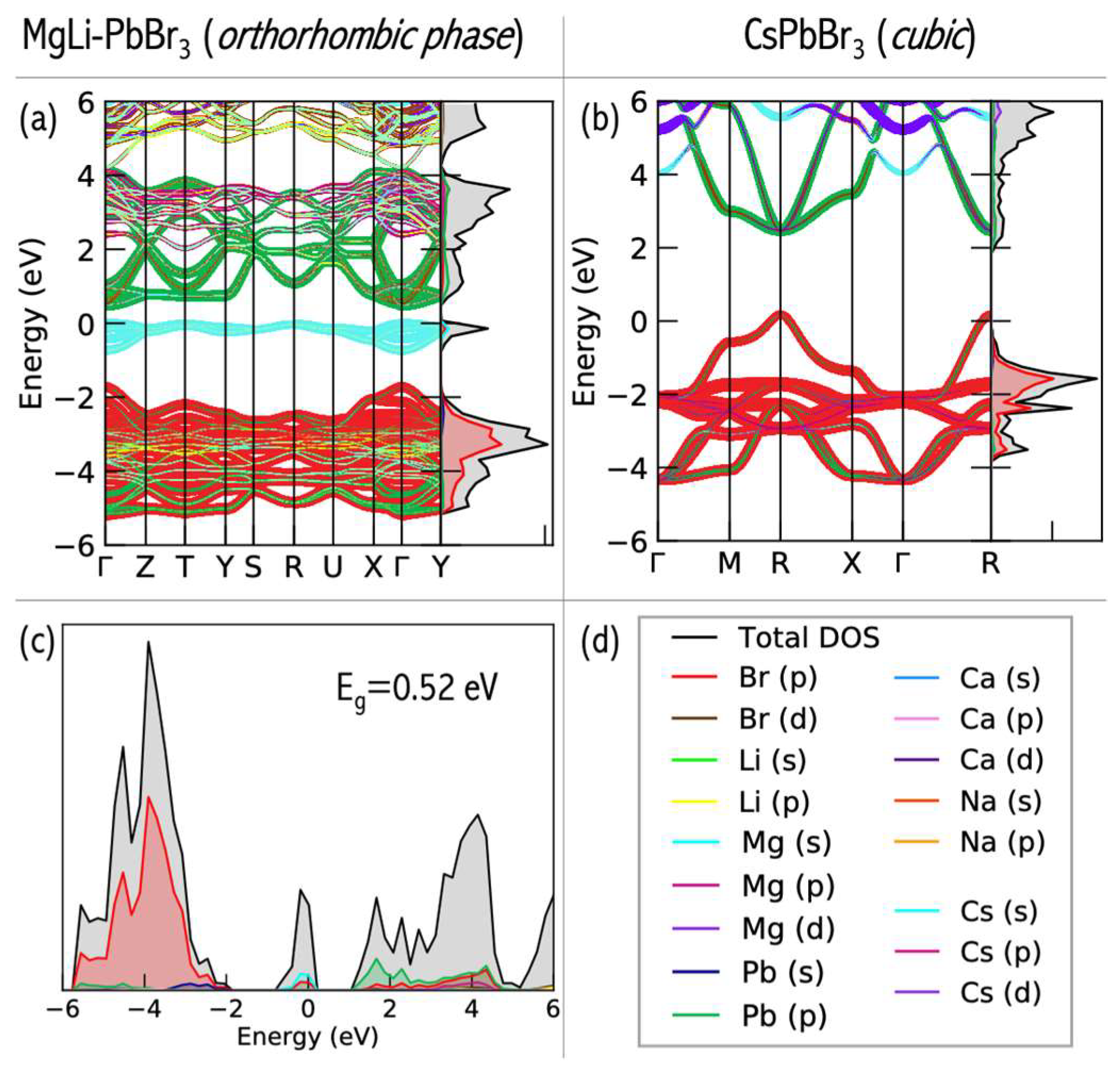
A significant consequence of superalkali insertion is band structure modification in the resulting perovskites [
61]. The CsPbBr
3 perovskite has the cesium states very deep inside the conduction band (more than 2 eV above the band edge, see
Figure 23b) and is not expected to play any significant role in the light adsorptions and to succeed in electronic processes. The introduction of the diatomic superalkali replacing the Cs atom in CsPbBr
3 leads to the formation of a new band in the energy range of −2 to 0 eV in all A′A″–PbBr
3 species (see
Figure 23a), as compared to CsPbBr
3. Within 1 eV below the valence band edge, these are ascribed to the alkali earth metal states. The A-site atom offers the main contributions for the valence band maximum (VBM) states of superalkali–PbBr
3 compounds, which can prevent the degradation of the PbBr
3 basic frame. Consequently, the transfer of electrons occurs at the A-site cation rather than halogen site atoms, which implies that the latter is not activated under light illumination [
32]. Since halogen site atoms are not involved in the VBM states and are not activated under UV light illumination, the superalkali–PbBr
3 compounds can be stable under these conditions.
The DFT results allowed us to formulate recommendations regarding the tunning of electronic properties of CsPbBr
3 perovskites upon superalkali substitution [
61]. The obtained band gap values strongly depend on the superalkali used and the structure formed. The electronic states of the A-site cations can be extended and directly contribute to the frontier orbitals of the lead bromide perovskite via A-site superalkali substitution, which provides electronic tunability to the band edge electronic configuration. Inserting diatomic superatomic species at the cationic A-sites of CsPbBr
3 reduces the band gap of lead halide perovskites. More specifically, the use of LiMg and NaMg superalkalis leads to band gaps in the 0.24–1.54 eV range. The band gaps of MgLi–PbBr
3 (1.35 eV) and MgNa–PbBr
3 (1.06 eV) are lower than the E
g of CsPbBr
3 (2.48 eV) and within the optimal E
g for single-junction solar cells. The E
g of 1.35 eV makes MgLi–PbBr
3 a promising candidate as a light absorber for solar cell applications. The above results [
61] constitute the first step for the development of novel perovskite-like materials with desirable electronic properties.
3.9. Novel Polynuclear Superalaklis
In 1982, Gutsev and Boldyrev introduced the simple formula
MLk+1 to describe one class of superalkalis, where
M is a k-valent electronegative central atom ligated with k + 1 alkali-metal atoms (
L) [
25]. The first superalkali system described in the literature superalkali system was Li
3O, whose experimental ionization energy (IE = 3.54 ± 0.3 eV [
64]) is lower than the IE of the cesium atom (3.89 eV). Its geometrical structure and ionization energy were investigated by quantum-mechanics calculations (adiabatic and vertical ionization energies were estimated to be in the 3.45–3.60 eV range [
65,
66]). Typical examples of superalkalis are FLi
2 [
67] OLi
3, ONa
3, OK
3 [
5], NLi
4 [
66], and CLi
5 [
6]. More recently, a lot of effort has been devoted to proposing alternative superalkali species, including lithium-based polynuclear superalkalis [
68], aromatic superalkali species [
32], and organo-zintl clusters (such as P
7R
4) [
33]. Despite these achievements, it is still desirable to obtain novel superalkali species to seek even lower ionization energy. The M
nL
n·k+1 polynuclear superalkali design seems to be the most promising direction to obtain efficient reducers. The examples include the N
4Mg
6M (M = Li, Na, K) polynuclear superalkalis, where four electronegative nitrogen central atoms are decorated with six electropositive magnesium atoms and one alkali metal atom (see
Figure 24).
The adiabatic ionization energies obtained at the CCSD(T)/6-311+G(3df) level of theory read 4.710 (N
4Mg
6Li), 4.564 (N
4Mg
6Na), and 4.414 eV (N
4Mg
6K) and are smaller than those of the sodium atom (5.14 eV at the same theory level), which emphasize their superalkali nature. This conclusion is also confirmed by their large HOMO–LUMO gaps (3.662–3.958 eV), binding energies per atom (3.05–3.12 eV), and the strong ionic interactions between electronegative nitrogen atoms and electropositive metal atoms. The HOMOs of N
4Mg
6M superalkalis are highly delocalized over the whole clusters to reduce the repulsion among the electrons, see
Figure 24. The highly diffuse HOMO also implies that the extra electron is loosely bound by the nuclei, giving rise to the low ionization energy. Also, the high thermodynamic stability of the designed superalkalis is due to the strong bonding character between metal atoms and the antibonding nature of the nitrogen-metal atoms interaction (lowering the ionization energy). Consequently, the N
4Mg
6M clusters have excellent reducing potential and can be used in the synthesis of charge-transfer salts and to activate moderately reactive molecules.
Carbon dioxide (CO
2), a greenhouse gas, is a cheap, nontoxic, and abundant carbon feedstock. So, developing efficient catalysts that convert CO
2 into fuel and value-added chemicals is vital to reduce the growing energy crisis and global warming. CO
2 can also be transformed into one-carbon (C
1) molecules (e.g., formic acid, formaldehyde, methanol, methane) that are the building blocks of the fuel and chemical industry. Unfortunately, the high thermodynamic stability of CO
2 makes the conversion process difficult. Converting CO
2 (a covalently bonded linear molecule) into fuel requires catalysts of high activity to add an electron (
Figure 25). This is a challenging transformation as CO
2− has a bent geometry, and the activation energy is high [
70]. In [
69], it was demonstrated that the N
4Mg
6M (M = Li, Na, K) superalkalis can reduce CO
2 and form superalkali/CO
2 complexes in which CO
2 is negatively charged.
Since my goal was to design superatoms for chemical applications, the most important part of [
69] was the assessment of the practicality and stability of superalkali/CO
2 systems. The possibility of reducing carbon dioxide by N
4Mg
6M superalkalis was studied. Based on the ab initio calculations, it was demonstrated that N
4Mg
6M/CO
2 systems represent two interacting ionic fragments (i.e., N
4Mg
6M
+ and CO
2−,
Figure 26). In superalkali/CO
2 complexes, the O–C–O bond angle was smaller (by 9%) than that in the isolated CO
2− anion and the amount of charge transfer was equal to 0.8
e. The charge transfer made possible by the low ionization energy of the superalkali seems to play a role in carbon dioxide activation, with a smaller ionization energy promoting the transfer of an electron to CO
2. The bonding interaction between carbon and the electronegative atoms of the reductant also bends the linear CO
2 structure. The above observations and their interpretations allowed us to consider N
4Mg
6M superalkalis as strong reducing agents.
Superalkalis can serve as building blocks of cluster-assembled materials or catalysts for CO
2 activation. I indicated the superatom compounds that can be formed to serve as building blocks for cluster-assembled materials (
Figure 26). I confirmed their structural stability. The high stability and donor-acceptor nature of the superalkali-superhalogen salts motivated me to design and evaluate a crystal structure of cluster-assembled superatomic compounds [
71].
3.10. Molecular Crystals vs. Superatomic Lattice
In [
69], I demonstrated that the designed N
4Mg
6M (M = Li, Na, K) superalkali systems form stable and strongly bound ionic compounds with superhalogens (AlCl
4 and AlF
4,
Figure 26). The resulting [superalkali]
+[superhalogen]
− supersalts reveal large cohesive energy (130–201 kcal/mol [
69]), which increases with the increase in the ionization energy of superalkali. Hence, it is expected that [N
4Mg
6Li][AlCl
4] systems might form stable solids [
69]. Indeed, using a first-principles approach, we demonstrated that binary assemblies of superatomic compounds can be created through charge transfer between non-charged molecular clusters and utilizing intercluster electrostatic attraction as a driving force for co-assembly [
71]. We combined pairs of complementary molecular clusters in which one cluster is electron-accepting (superhalogen) and the other is electron-donating (superalkali),
Figure 27.
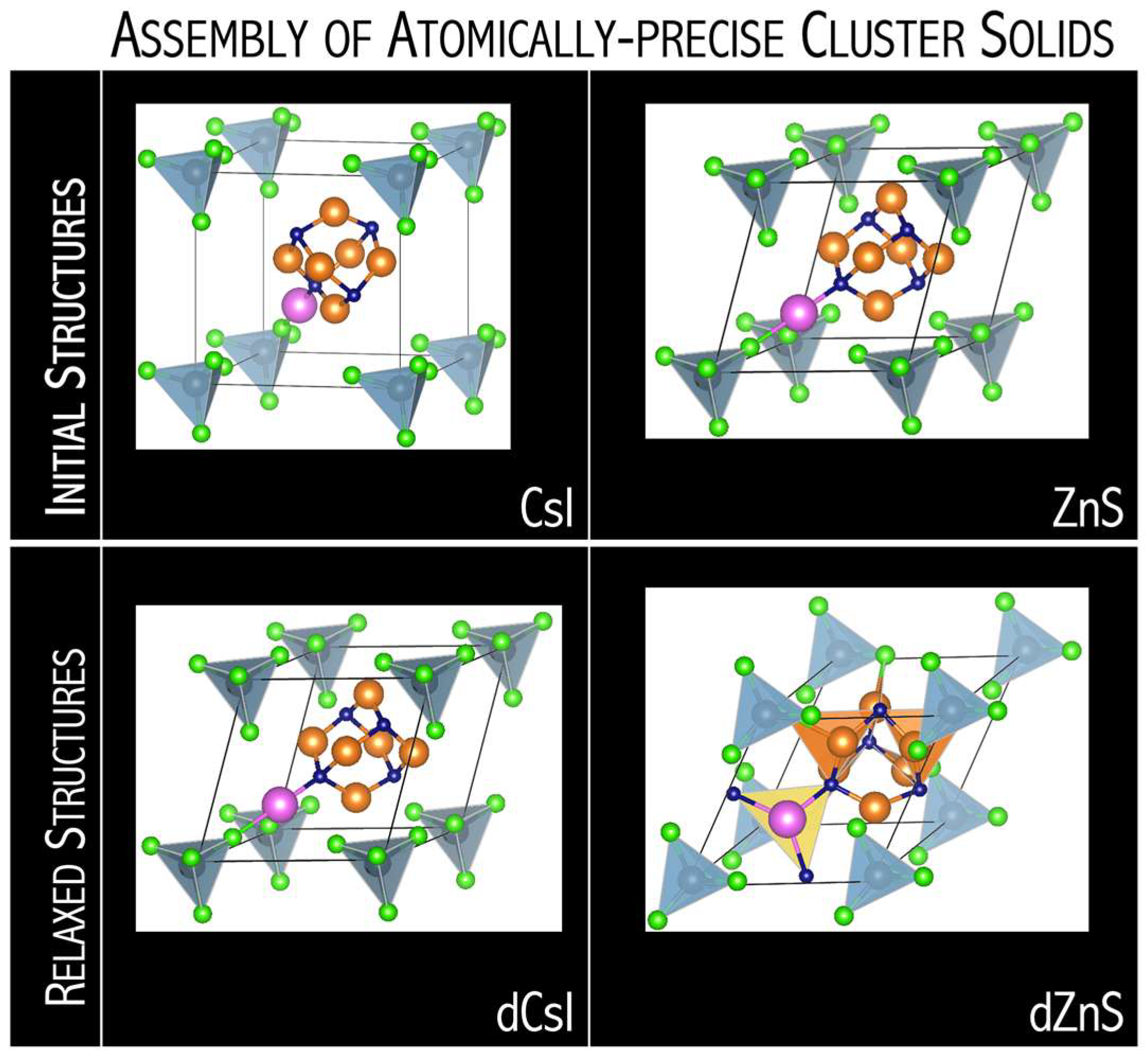
Analysis of the first-principles results indicates that the [N
4Mg
6M]
+[AlCl
4]
− and [N
4Mg
6M]
+[AlF
4]
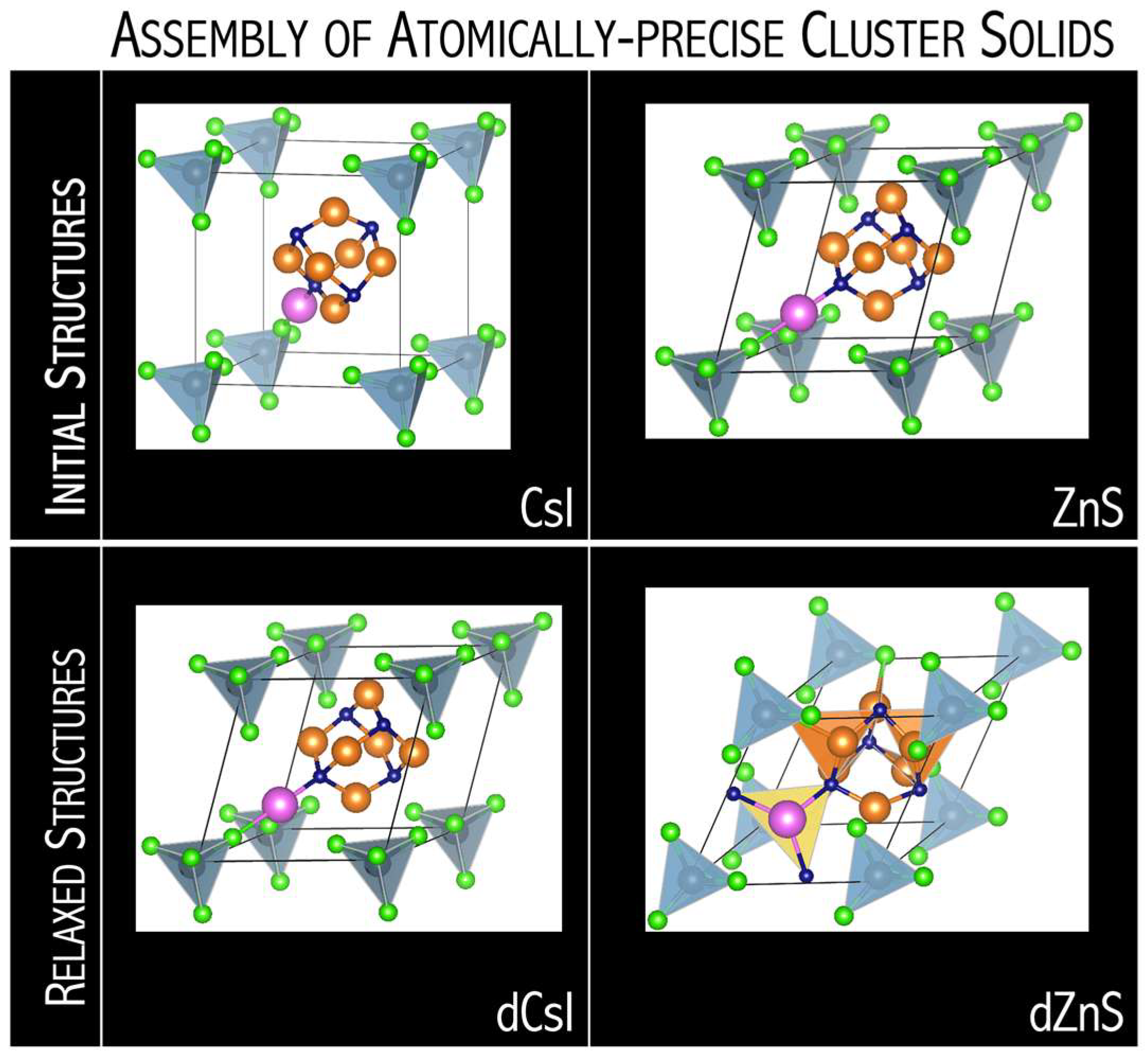
−revealed a nanoscale atom nature by crystallizing in a similar packing as disordered zinc blende (dZnS,
Figure 28), as well as also by resembling the empirical oxidation states of their atomic cation and anion analogs [
71]. The N
4Mg
6M–AlX
4 superatomic systems can also form molecular clusters where clusters are held to one another by electrostatic interactions (dCsI in
Figure 28). The DFT results emphasize how the structure of superatomic solids can be tuned upon single-atom substitution. The competition between superhalogen size (
dSH) and superalkali diameter (
dSA) is a key factor for predicting the stability and crystalline form of superalkali-superhalogen supersalts. The N
4Mg
6M superalkalis form solids when combined with superhalogen oxidizing agents whose
dSA/
dSH ratio is within the 1.32–1.78 range [
71]. A larger
dSA/
dSH ratio results in the close-packed superatomic lattice, whereas a smaller
dSA/
dSH ratio favors the formation of larger intermolecular distances in molecular crystals. The lowest
dSA/
dSH ratio (of 1.32) was found for N
4Mg
6Li–AlCl
4, which forms molecular solids for dCsI and dNaCl phases. In turn, N
4Mg
6Na–AlCl
4 and N
4Mg
6K–AlCl
4 with larger
dSA/
dSH ratios (1.44 and 1.46, respectively) favors close-packed superatomic phases forming [
71].
The [superalkali][superhalogen] solids reveal the electronic structure and the band gap tunability. The ground states of N
4Mg
6Li–AlCl
4, N
4Mg
6K–AlCl
4, and N
4Mg
6K-AlF
4 solids are semiconductors with indirect band gap (0.122, 0.151, and 0.103 eV) [
71]. In turn, in N
4Mg
6Na–AlCl
4, N
4Mg
6Li–AlF
4, and N
4Mg
6Na–AlF
4 solids, the Fermi level cuts across the valence band, revealing a metallic nature. The substitution of Cl by F reduces the band gap from 0.122 eV (N
4Mg
6Li–AlCl
4) to 0.000 eV (N
4Mg
6Li–AlF
4) and from 0.151 eV (N
4Mg
6K–AlCl
4) to 0.103 eV (N
4Mg
6K–AlF
4) [
71]. The band gap changes on halogen atom substitution are accompanied by valence band dispersion increase and are influenced by electronegativity (larger electron affinity). Based on our calculations, the electronic structures of the superalkali-superhalogen solids can be modified via alkali metal/halogen substitution, which provides GAP tunability. Overall, the first-principles results described in [
71] demonstrate how modification of the superatoms allows for tuning of the superstructure and electronic configuration and can be used to establish desirable functional materials.
The analysis of the first-principles computations proved the tunability of electronic and crystal structures of supersalts upon individual superatom modification. The DFT computations not only conform to the ability of superatoms to serve as building blocks of chemical materials but also allow us to predict the influence of superatomic structure modification on the electronic structure of the resulting materials. The new knowledge obtained within [
71] can be used to form functional materials with desirable properties. In this way, superatoms can replace elements of the periodic table, especially those with increasing scarcity. Nowadays, we risk seeing many of the natural elements that make up the world around us run out due to limited supplies, their location in conflict areas, or the incapacity to fully recycle them. Substitutes for scarce elements can be obtained by replacing them with superatoms. Superatoms can be made of combinations of plentiful supply atoms. Thus, the ability to design superatoms offers a new concept for materials design and the opportunity for their electronic structure to be tuned through minor, targeted modifications of their composition that take advantage of their predictable electronic structure.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}