
Glioblastoma Standard of Care: Effects on Tumor Evolution and Reverse Translation in Preclinical Models


Abstract
Simple Summary
Abstract
1. Introduction
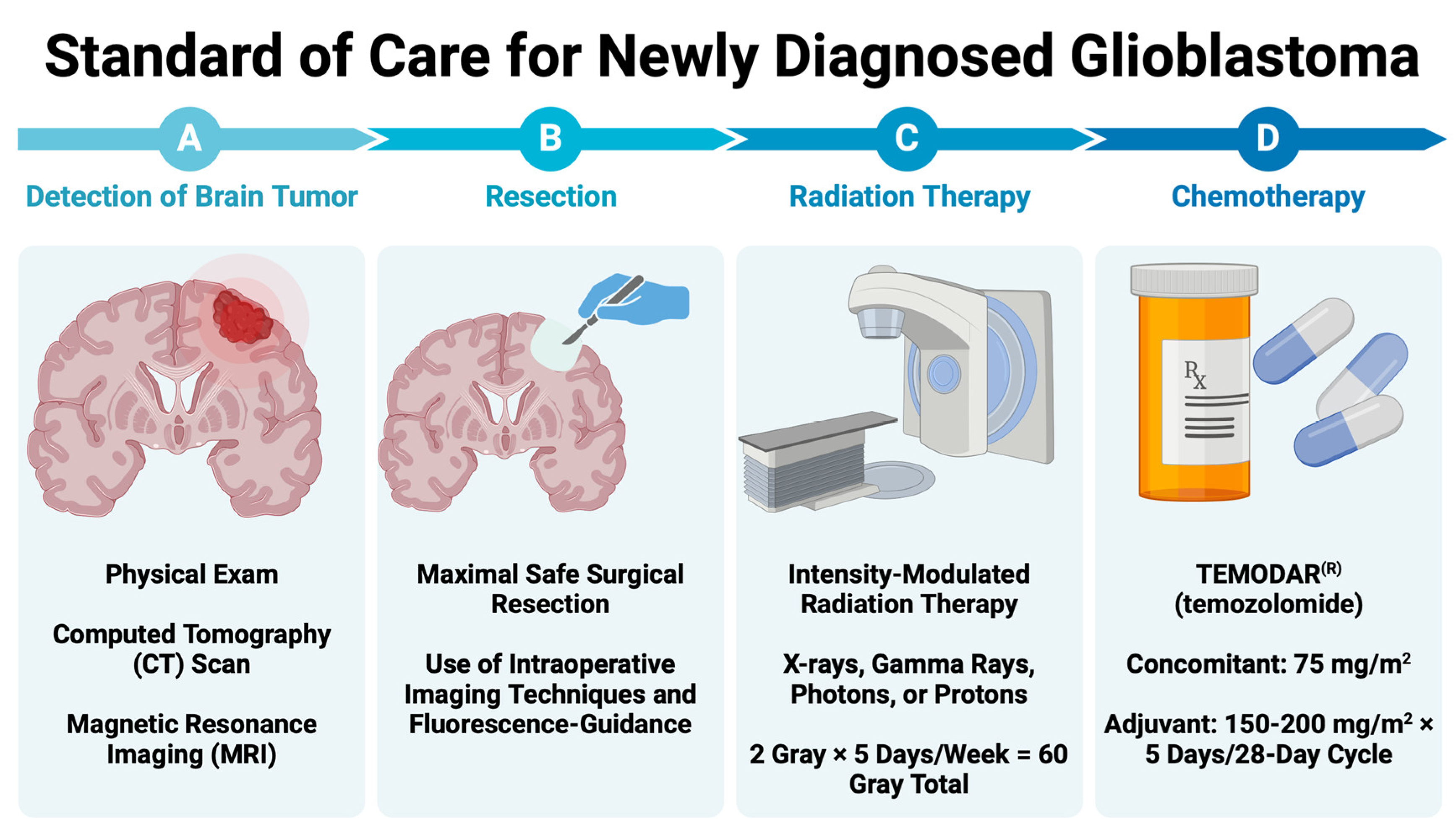
2. GBM Diagnosis and Standard of Care
2.1. Resection of Contrast-Enhancing Tissue
2.1.1. Resection of Contrast-Enhancing Tissue
2.1.2. Resection of Non-Contrast-Enhancing Tissue
2.1.3. Resection Using Fluorescent Imaging Agents
2.2. Radiation Therapy (RT)
RT Techniques
2.3. Chemotherapy
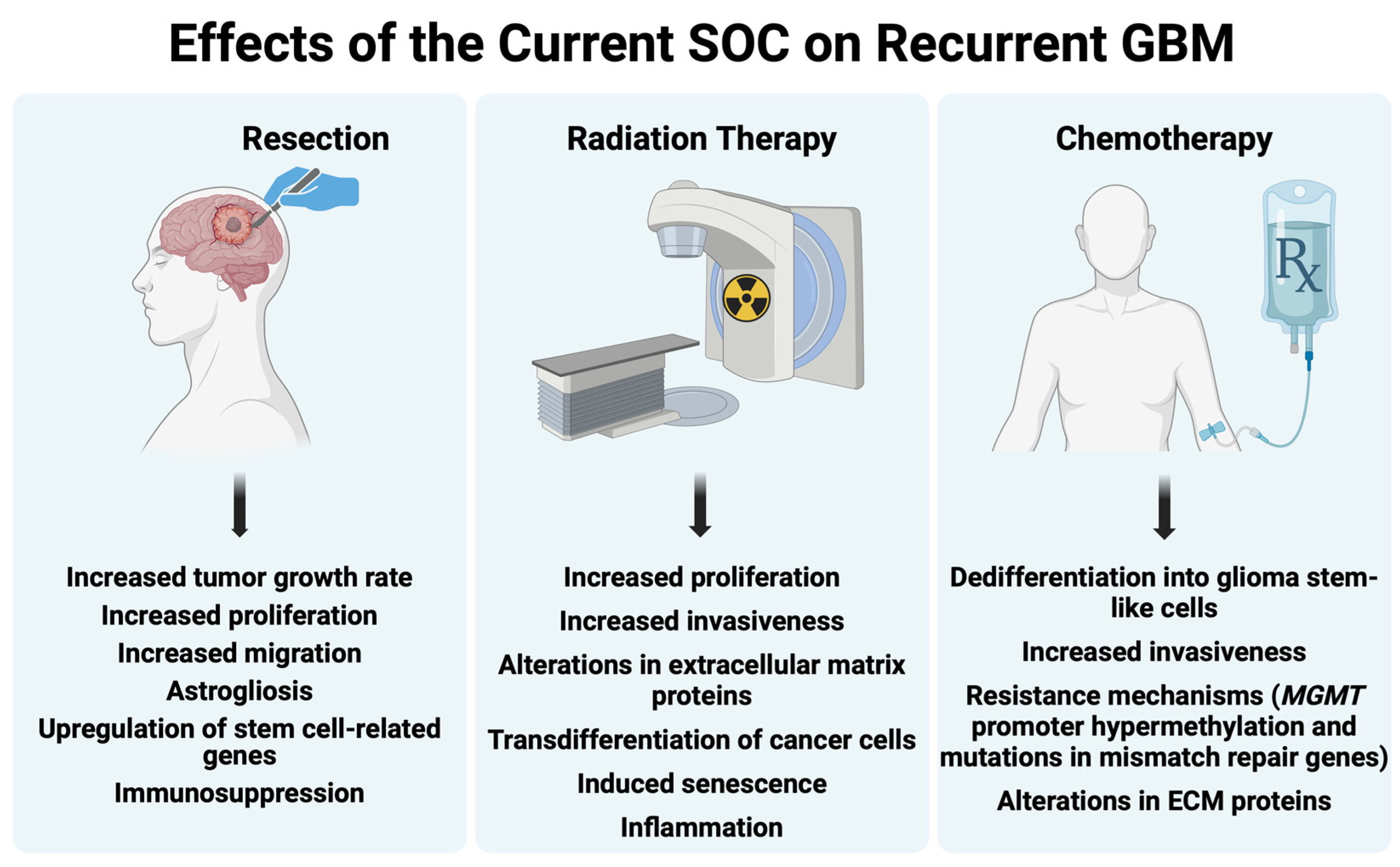
3. The Effect of the Standard of Care on GBM
3.1. Surgery—Effects on Recurrent GBM
3.1.1. Biopsy
3.1.2. Resection
3.1.3. Anesthesia
3.2. Radiation Therapy—Effects on Recurrent GBM
3.2.1. Effects on GBM Tumor
Genotypic and Phenotypic Alterations
Metabolic Alterations
3.2.2. Effects on Tumor Microenvironment
Extracellular Matrix
Vasculature
Immune System
3.3. Chemotherapy—Effects on GBM
3.3.1. Cellular Differentiation
3.3.2. Drug Resistance
3.3.3. Tumor Microenvironment
4. Preclinical Models Recapitulating GBM Standard of Care
4.1. Resection
4.1.1. White Light Resection
4.1.2. Punch Biopsy
4.1.3. Fluorescence-Guided Resection: Fluorescent-Labeled Cells
4.1.4. Fluorescence-Guided Resection: Fluorescent Imaging Agents
4.2. Radiation Therapy
4.3. Chemotherapy
4.3.1. Route of Administration
4.3.2. Dosing Schedule
4.4. Combination Therapies
4.4.1. Resection + TMZ (or Other Novel Therapies)
4.4.2. Radiation Therapy + TMZ
4.4.3. Radiation Therapy + TMZ + Novel Therapeutic Strategies
4.4.4. Resection + Radiation Therapy + Chemotherapy (Stupp Protocol)
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro-Oncology 2023, 25, iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Ostrom, Q.T.; Cioffi, G.; Neff, C.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. Primary brain and other central nervous system tumors in the United States (2014–2018): A summary of the CBTRUS statistical report for clinicians. Neurooncol Pract. 2022, 9, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.C.; Mercier, M.-C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef] [PubMed]
- Oster, C.; Lazaridis, L.; Feldheim, J.; Schmidt, T.; Kleinschnitz, C.; Kebir, S.; Glas, M. Systematic Review of Phase Iii Trials in Newly Diagnosed Glioblastoma 2005–2021. Neuro-Oncology 2022, 24, 77. [Google Scholar] [CrossRef]
- Gunjur, A.; Balasubramanian, A.; Hafeez, U.; Menon, S.; Cher, L.; Parakh, S.; Gan, H.K. Poor correlation between preclinical and patient efficacy data for tumor targeted monotherapies in glioblastoma: The results of a systematic review. J. Neurooncol. 2022, 159, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Simon, B.; Leonie de Boer, N.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. BBA—Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef] [PubMed]
- Riva, M.; Bevers, S.; Wouters, R.; Thirion, G.; Vandenbrande, K.; Vankerckhoven, A.; Berckmans, Y.; Verbeeck, J.; Keersmaecker, K.D.; Coosemans, A. Towards more accurate preclinical glioblastoma modelling: Reverse translation of clinical standard of care in a glioblastoma mouse model. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, K.; Li, M.; Cui, Y.; Ren, X.; Yang, C.; Zhao, X.; Lin, S. Classification of Progression Patterns in Glioblastoma: Analysis of Predictive Factors and Clinical Implications. Front. Oncol. 2020, 10, 590648. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.S.; Holland, E.C. Standard of care therapy for malignant glioma and its effect on tumor and stromal cells. Oncogene 2012, 31, 1995–2006. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Burns, T.C. Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Z.; Li, J.; Huang, T.; Wang, Y.; Chang, L.; Zheng, W.; Ma, Y.; Chen, F.; Gong, X.; et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Sci. Rep. 2019, 9, 16058. [Google Scholar] [CrossRef]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef] [PubMed]
- Hoogstrate, Y.; Draaisma, K.; Ghisai, S.A.; van Hijfte, L.; Barin, N.; de Heer, I.; Coppieters, W.; van den Bosch, T.P.P.; Bolleboom, A.; Gao, Z.; et al. Transcriptome analysis reveals tumor microenvironment changes in glioblastoma. Cancer Cell 2023, 41, 678–692.e677. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Nandhabalan, M.; Murray, S.A.; Plaha, P. Glioblastoma: Clinical presentation, diagnosis, and management. BMJ 2021, 374, n1560. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. WHO Classification of Tumours: Central Nervous System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2021. [Google Scholar]
- Beiko, J.; Suki, D.; Hess, K.R.; Fox, B.D.; Cheung, V.; Cabral, M.; Shonka, N.; Gilbert, M.R.; Sawaya, R.; Prabhu, S.S.; et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro-Oncology 2014, 16, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Tomoszková, S.; Skarda, J.; Lipina, R. Potential Diagnostic and Clinical Significance of Selected Genetic Alterations in Glioblastoma. Int. J. Mol. Sci. 2024, 25, 4438. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- McGirt, M.J.; Chaichana, K.L.; Gathinji, M.; Attenello, F.J.; Than, K.; Olivi, A.; Weingart, J.D.; Brem, H.; Quiñones-Hinojosa, A. Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J. Neurosurg. 2009, 110, 156–162. [Google Scholar] [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Polley, M.-Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.M.; Hervey-Jumper, S.; Morshed, R.A.; Young, J.; Han, S.J.; Chunduru, P.; Zhang, Y.; Phillips, J.J.; Shai, A.; Lafontaine, M.; et al. Association of Maximal Extent of Resection of Contrast-Enhanced and Non–Contrast-Enhanced Tumor With Survival Within Molecular Subgroups of Patients With Newly Diagnosed Glioblastoma. JAMA Oncol. 2020, 6, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.-J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Buitelaar, M.; Wagenaar, E.; van der Valk, M.A.; Scheffer, G.L.; Scheper, R.J.; Plösch, T.; Kuipers, F.; Elferink, R.P.J.O.; Rosing, H.; et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc. Natl. Acad. Sci. USA 2002, 99, 15649–15654. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chen, X.F.; Wang, L.G.; Yang, G.; Han, D.Y.; Teng, L.; Yang, M.C.; Wang, D.Y.; Shi, C.; Liu, Y.H.; et al. Increased expression of ABCB6 enhances protoporphyrin IX accumulation and photodynamic effect in human glioma. Ann. Surg. Oncol. 2013, 20, 4379–4388. [Google Scholar] [CrossRef]
- Mazurek, M.; Szczepanek, D.; Orzyłowska, A.; Rola, R. Analysis of Factors Affecting 5-ALA Fluorescence Intensity in Visualizing Glial Tumor Cells—Literature Review. Int. J. Mol. Sci. 2022, 23, 926. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Reulen, H.J.; Meinel, T.; Pichlmeier, U.; Schumacher, W.; Tonn, J.C.; Rohde, V.; Oppel, F.; Turowski, B.; Woiciechowsky, C.; et al. Extent of resection and survival in glioblastoma multiforme: Identification of and adjustment for bias. Neurosurgery 2008, 62, 564–576; discussion 564–576. [Google Scholar] [CrossRef] [PubMed]
- Schucht, P.; Knittel, S.; Slotboom, J.; Seidel, K.; Murek, M.; Jilch, A.; Raabe, A.; Beck, J. 5-ALA complete resections go beyond MR contrast enhancement: Shift corrected volumetric analysis of the extent of resection in surgery for glioblastoma. Acta Neurochir. 2014, 156, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Smith, E.J.; Barreau, A.; Nyaeme, M.; Cramer, S.W.; Najafali, D.; Krist, D.T.; Arnold, P.M.; Hassaneen, W. Comparison of fluorescein sodium, 5-ALA, and intraoperative MRI for resection of high-grade gliomas: A systematic review and network meta-analysis. J. Clin. Neurosci. 2022, 98, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.E.L.; Wright, J.; Sloan, A.E.; Brady-Kalnay, S.M. Fluorescent-Guided Surgical Resection of Glioma with Targeted Molecular Imaging Agents: A Literature Review. World Neurosurg. 2016, 90, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Buszek, S.M.; Al Feghali, K.A.; Elhalawani, H.; Chevli, N.; Allen, P.K.; Chung, C. Optimal Timing of Radiotherapy Following Gross Total or Subtotal Resection of Glioblastoma: A Real-World Assessment using the National Cancer Database. Sci. Rep. 2020, 10, 4926. [Google Scholar] [CrossRef] [PubMed]
- Barker II, F.G.; Prados, M.D.; Chang, S.M.; Gutin, P.H.; Lamborn, K.R.; Larson, D.A.; Malec, M.K.; McDermott, M.W.; Sneed, P.K.; Wara, W.M.; et al. Radiation response and survival time in patients with glioblastoma multiforme. J. Neurosurg. 1996, 84, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Samson, P.; Perkins, S.M.; Ansstas, G.; Chheda, M.G.; DeWees, T.A.; Tsien, C.I.; Robinson, C.G.; Campian, J.L. Impact of concurrent chemotherapy with radiation therapy for elderly patients with newly diagnosed glioblastoma: A review of the National Cancer Data Base. J. Neurooncol. 2017, 131, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.R.; Kirkpatrick, J.P.; Fiveash, J.B.; Shih, H.A.; Koay, E.J.; Lutz, S.; Petit, J.; Chao, S.T.; Brown, P.D.; Vogelbaum, M.; et al. Radiation therapy for glioblastoma: Executive summary of an American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. Pract. Radiat. Oncol. 2016, 6, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Gy, S.; Dracham, C.B.; Dey, T.; Madan, R.; Khosla, D.; Oinum, A.; Kapoor, R. Can 3D-CRT meet the desired dose distribution to target and OARs in glioblastoma? A tertiary cancer center experience. CNS Oncol. 2020, 9, CNS60. [Google Scholar] [CrossRef]
- Narayana, A.; Yamada, J.; Berry, S.; Shah, P.; Hunt, M.; Gutin, P.H.; Leibel, S.A. Intensity-modulated radiotherapy in high-grade gliomas: Clinical and dosimetric results. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Thibouw, D.; Truc, G.; Bertaut, A.; Chevalier, C.; Aubignac, L.; Mirjolet, C. Clinical and dosimetric study of radiotherapy for glioblastoma: Three-dimensional conformal radiotherapy versus intensity-modulated radiotherapy. J. Neurooncol. 2018, 137, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Barbarite, E.; Sick, J.T.; Berchmans, E.; Bregy, A.; Shah, A.H.; Elsayyad, N.; Komotar, R.J. The role of brachytherapy in the treatment of glioblastoma multiforme. Neurosurg. Rev. 2017, 40, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Miralbell, R.; Lomax, A.; Russo, M. Potential role of proton therapy in the treatment of pediatric medulloblastoma/primitive neuro-ectodermal tumors: Spinal theca irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1997, 38, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Chung, C.; Liu, D.D.; McAvoy, S.; Grosshans, D.; Al Feghali, K.; Mahajan, A.; Li, J.; McGovern, S.L.; McAleer, M.F.; et al. A prospective phase II randomized trial of proton radiotherapy vs. intensity-modulated radiotherapy for patients with newly diagnosed glioblastoma. Neuro-Oncology 2021, 23, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Bronk, J.K.; Amer, A.; Khose, S.; Flint, D.; Adair, A.; Yepes, P.; Grosshans, D.; Johnson, J.; Chung, C. Brain Radiation Necrosis Outside the Target Volume After Proton Radiation Therapy: Analyses of Multiparametric Imaging and Proton Biologic Effectiveness. Adv. Radiat. Oncol. 2022, 7, 101044. [Google Scholar] [CrossRef] [PubMed]
- Harrabi, S.B.; von Nettelbladt, B.; Gudden, C.; Adeberg, S.; Seidensaal, K.; Bauer, J.; Bahn, E.; Mairani, A.; Alber, M.; Haberer, T.; et al. Radiation induced contrast enhancement after proton beam therapy in patients with low grade glioma—How safe are protons? Radiother. Oncol. 2022, 167, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Mizumoto, M.; Kohzuki, H.; Sugii, N.; Sakurai, H.; Ishikawa, E. High-dose proton beam therapy versus conventional fractionated radiation therapy for newly diagnosed glioblastoma: A propensity score matching analysis. Radiat. Oncol. 2023, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Dose-Escalated Photon IMRT or Proton Beam Radiation Therapy Versus Standard-Dose Radiation Therapy and Temozolomide in Treating Patients With Newly Diagnosed Glioblastoma. NCT02179086. Available online: https://clinicaltrials.gov/study/NCT02179086 (accessed on 21 February 2024).
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Chambers, A.; Spithoff, K.; Laperriere, N. Gliadel wafers in the treatment of malignant glioma: A systematic review. Curr. Oncol. 2007, 14, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Agosti, E.; Zeppieri, M.; De Maria, L.; Tedeschi, C.; Fontanella, M.M.; Panciani, P.P.; Ius, T. Glioblastoma Immunotherapy: A Systematic Review of the Present Strategies and Prospects for Advancements. Int. J. Mol. Sci. 2023, 24, 15037. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Muscat, A.M.; Wong, N.C.; Drummond, K.J.; Algar, E.M.; Khasraw, M.; Verhaak, R.; Field, K.; Rosenthal, M.A.; Ashley, D.M. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 2018, 9, 7844–7858. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro-Oncology 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Alieva, M.; Margarido, A.S.; Wieles, T.; Abels, E.R.; Colak, B.; Boquetale, C.; Jan Noordmans, H.; Snijders, T.J.; Broekman, M.L.; van Rheenen, J. Preventing inflammation inhibits biopsy-mediated changes in tumor cell behavior. Sci. Rep. 2017, 7, 7529. [Google Scholar] [CrossRef]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-Oncology 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, J.; Ying, Z.; Chen, B.; Han, A.; Liang, Y.; Song, L.; Yuan, J.; Li, J.; Li, M. Astrocyte elevated gene-1 upregulates matrix metalloproteinase-9 and induces human glioma invasion. Cancer Res. 2010, 70, 3750–3759. [Google Scholar] [CrossRef] [PubMed]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of astrocytes by glioma cells to facilitate invasiveness: A mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J. Neurosci. 2003, 23, 4034–4043. [Google Scholar] [CrossRef] [PubMed]
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 2007, 99, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, A.M.; Halle, B.; Cedile, O.; Burton, M.; Baun, C.; Thisgaard, H.; Anand, A.; Hubert, C.; Thomassen, M.; Michaelsen, S.R.; et al. Surgical resection of glioblastomas induces pleiotrophin-mediated self-renewal of glioblastoma stem cells in recurrent tumors. Neuro-Oncology 2022, 24, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Predina, J.; Eruslanov, E.; Judy, B.; Kapoor, V.; Cheng, G.; Wang, L.C.; Sun, J.; Moon, E.K.; Fridlender, Z.G.; Albelda, S.; et al. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc. Natl. Acad. Sci. USA 2013, 110, E415–E424. [Google Scholar] [CrossRef] [PubMed]
- Sablotzki, A.; Ebel, H.; Muhling, J.; Dehne, M.G.; Nopens, H.; Giesselmann, H.; Hempelmann, G. Dysregulation of immune response following neurosurgical operations. Acta Anaesthesiol. Scand. 2000, 44, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Otvos, B.; Alban, T.J.; Grabowski, M.M.; Bayik, D.; Mulkearns-Hubert, E.E.; Radivoyevitch, T.; Rabljenovic, A.; Johnson, S.; Androjna, C.; Mohammadi, A.M.; et al. Preclinical Modeling of Surgery and Steroid Therapy for Glioblastoma Reveals Changes in Immunophenotype that are Associated with Tumor Growth and Outcome. Clin. Cancer Res. 2021, 27, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Wu, Z.F.; Lee, M.S.; Lou, Y.S.; Wu, K.L.; Cheng, K.I.; Lai, H.C. Propofol-based total intravenous anesthesia is associated with better survival than desflurane anesthesia in glioblastoma surgery. PLoS ONE 2021, 16, e0255627. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zeng, M.; Ji, N.; Hao, S.; Zhou, Y.; Gao, Z.; Gu, H.; Zhang, L.; Ma, D.; Peng, Y.; et al. Impact of Anesthesia on Long-term Outcomes in Patients With Supratentorial High-grade Glioma Undergoing Tumor Resection: A Retrospective Cohort Study. J. Neurosurg. Anesthesiol. 2020, 32, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Inada, T.; Yamanouchi, Y.; Jomura, S.; Sakamoto, S.; Takahashi, M.; Kambara, T.; Shingu, K. Effect of propofol and isoflurane anaesthesia on the immune response to surgery. Anaesthesia 2004, 59, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Kushida, A.; Inada, T.; Shingu, K. Enhancement of antitumor immunity after propofol treatment in mice. Immunopharmacol. Immunotoxicol. 2007, 29, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Schmoch, T.; Jungk, C.; Bruckner, T.; Haag, S.; Zweckberger, K.; von Deimling, A.; Brenner, T.; Unterberg, A.; Weigand, M.A.; Uhle, F.; et al. The anesthetist’s choice of inhalational vs. intravenous anesthetics has no impact on survival of glioblastoma patients. Neurosurg. Rev. 2021, 44, 2707–2715. [Google Scholar] [CrossRef] [PubMed]
- Grau, S.J.; Lohr, M.; Taurisano, V.; Trautner, H.; Timmer, M.; Schwab, S.G.; Hampl, J.; Annecke, T. The choice of anaesthesia for glioblastoma surgery does not impact the time to recurrence. Sci. Rep. 2020, 10, 5556. [Google Scholar] [CrossRef] [PubMed]
- Efremidou, E.; Zachou, M.-E.; Kaprana, A.; Koukourakis, M.; Chloropoulou, P. The effects of anesthesia on cancer progression and antitumor immunity. A review. J. Surg. Surg. Res. 2024, 10, 014–021. [Google Scholar] [CrossRef]
- Deegan, C.A.; Murray, D.; Doran, P.; Moriarty, D.C.; Sessler, D.I.; Mascha, E.; Kavanagh, B.P.; Buggy, D.J. Anesthetic technique and the cytokine and matrix metalloproteinase response to primary breast cancer surgery. Reg. Anesth. Pain. Med. 2010, 35, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Tavare, A.N.; Perry, N.J.; Benzonana, L.L.; Takata, M.; Ma, D. Cancer recurrence after surgery: Direct and indirect effects of anesthetic agents. Int. J. Cancer 2012, 130, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Stollings, L.M.; Jia, L.J.; Tang, P.; Dou, H.; Lu, B.; Xu, Y. Immune Modulation by Volatile Anesthetics. Anesthesiology 2016, 125, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Benzonana, L.L.; Zhao, H.; Watts, H.R.; Perry, N.J.; Bevan, C.; Brown, R.; Ma, D. Prostate cancer cell malignancy via modulation of HIF-1alpha pathway with isoflurane and propofol alone and in combination. Br. J. Cancer 2014, 111, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhao, H.; Hennah, L.; Ning, J.; Liu, J.; Tu, H.; Ma, D. Impact of isoflurane on malignant capability of ovarian cancer in vitro. Br. J. Anaesth. 2015, 114, 831–839. [Google Scholar] [CrossRef]
- Muthukrishnan, S.D.; Kawaguchi, R.; Nair, P.; Prasad, R.; Qin, Y.; Johnson, M.; Wang, Q.; VanderVeer-Harris, N.; Pham, A.; Alvarado, A.G.; et al. P300 promotes tumor recurrence by regulating radiation-induced conversion of glioma stem cells to vascular-like cells. Nat. Commun. 2022, 13, 6202. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro-Oncology 2014, 16, 671–685. [Google Scholar] [CrossRef] [PubMed]
- McAbee, J.H.; Rath, B.H.; Valdez, K.; Young, D.L.; Wu, X.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. Radiation Drives the Evolution of Orthotopic Xenografts Initiated from Glioblastoma Stem-like Cells. Cancer Res. 2019, 79, 6032–6043. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Gupta, K.; Vuckovic, I.; Zhang, S.; Xiong, Y.; Carlson, B.L.; Jacobs, J.; Olson, I.; Petterson, X.M.; Macura, S.I.; Sarkaria, J.; et al. Radiation Induced Metabolic Alterations Associate With Tumor Aggressiveness and Poor Outcome in Glioblastoma. Front. Oncol. 2020, 10, 535. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Bailleul, J.; Ruan, Y.; Abdulrahman, L.; Scott, A.J.; Yazal, T.; Sung, D.; Park, K.; Hoang, H.; Nathaniel, J.; Chu, F.I.; et al. M2 isoform of pyruvate kinase rewires glucose metabolism during radiation therapy to promote an antioxidant response and glioblastoma radioresistance. Neuro-Oncology 2023, 25, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Daviaud, C.; Minns, H.E.; Lazarian, A.; Wacker, A.; Costa, A.P.; Attarwala, N.; Chen, Q.; Choi, S.W.; Rabadan, R.; et al. Radiation therapy promotes unsaturated fatty acids to maintain survival of glioblastoma. Cancer Lett. 2023, 570, 216329. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kim, H.S.; Shin, T.H.; Kang, I.; Lee, J.Y.; Kim, J.J.; Kang, H.K.; Seo, Y.; Lee, S.; Yu, K.R.; et al. PGE maintains self-renewal of human adult stem cells via EP2-mediated autocrine signaling and its production is regulated by cell-to-cell contact. Sci. Rep. 2016, 6, 26298. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.M.; Kim, J.Y.; Cho, H.J.; Lee, W.J.; Nguyen, D.; Kim, S.S.; Oh, Y.T.; Kim, H.J.; Hubert, C.G.; Jung, C.W.; et al. Tissue factor is a critical regulator of radiation therapy-induced glioblastoma remodeling. Cancer Cell 2023, 41, 1480–1497.e9. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Sananikone, E.; Kanji, S.; Tomimatsu, N.; Di Cristofaro, L.F.M.; Kollipara, R.K.; Saha, D.; Floyd, J.R.; Sung, P.; Hromas, R.; Burns, T.C.; et al. Elimination of Radiation-Induced Senescence in the Brain Tumor Microenvironment Attenuates Glioblastoma Recurrence. Cancer Res. 2021, 81, 5935–5947. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.C.; Suh, Y.; An, Y.; Lee, H.J.; Jeong, Y.J.; Uddin, N.; Cui, Y.H.; Roh, T.H.; Shim, J.K.; Chang, J.H.; et al. Proinvasive extracellular matrix remodeling in tumor microenvironment in response to radiation. Oncogene 2018, 37, 3317–3328. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Ko, I.O.; Park, H.; Jeong, Y.J.; Park, J.A.; Kim, K.S.; Park, M.J.; Lee, H.J. Radiation-Induced Changes in Tumor Vessels and Microenvironment Contribute to Therapeutic Resistance in Glioblastoma. Front. Oncol. 2019, 9, 1259. [Google Scholar] [CrossRef] [PubMed]
- Salmaggi, A.; Eoli, M.; Frigerio, S.; Silvani, A.; Gelati, M.; Corsini, E.; Broggi, G.; Boiardi, A. Intracavitary VEGF, bFGF, IL-8, IL-12 levels in primary and recurrent malignant glioma. J. Neurooncol. 2003, 62, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Kil, W.J.; Tofilon, P.J.; Camphausen, K. Post-radiation increase in VEGF enhances glioma cell motility in vitro. Radiat. Oncol. 2012, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Hart, E.; Ode, Z.; Derieppe, M.P.P.; Groenink, L.; Heymans, M.W.; Otten, R.; Lequin, M.H.; Janssens, G.O.R.; Hoving, E.W.; van Vuurden, D.G. Blood-brain barrier permeability following conventional photon radiotherapy—A systematic review and meta-analysis of clinical and preclinical studies. Clin. Transl. Radiat. Oncol. 2022, 35, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Tsien, C.I.; Lawrence, T.S.; Cao, Y. Blood-tumor barrier opening changes in brain metastases from pre to one-month post radiation therapy. Radiother. Oncol. 2017, 125, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Li, P.J.; Lai, S.Z.; Jin, T.; Ying, H.J.; Chen, Y.M.; Zhang, P.; Hang, Q.Q.; Deng, H.; Wang, L.; Feng, J.G.; et al. Radiotherapy opens the blood-brain barrier and synergizes with anlotinib in treating glioblastoma. Radiother. Oncol. 2023, 183, 109633. [Google Scholar] [CrossRef] [PubMed]
- Blethen, K.E.; Sprowls, S.A.; Arsiwala, T.A.; Wolford, C.P.; Panchal, D.M.; Fladeland, R.A.; Glass, M.J.; Dykstra, L.P.; Kielkowski, B.N.; Blackburn, J.R.; et al. Effects of whole-brain radiation therapy on the blood-brain barrier in immunocompetent and immunocompromised mouse models. Radiat. Oncol. 2023, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Lumniczky, K.; Szatmari, T.; Safrany, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Jung, J.S.; Kim, T.H.; Lim, S.J.; Oh, E.S.; Kim, J.Y.; Ji, K.A.; Joe, E.H.; Cho, K.H.; Han, I.O. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol. Dis. 2006, 21, 457–467. [Google Scholar] [CrossRef]
- McDonald, J.T.; Gao, X.; Steber, C.; Lee Breed, J.; Pollock, C.; Ma, L.; Hlatky, L. Host mediated inflammatory influence on glioblastoma multiforme recurrence following high-dose ionizing radiation. PLoS ONE 2017, 12, e0178155. [Google Scholar] [CrossRef]
- Tabatabaei, P.; Visse, E.; Bergstrom, P.; Brannstrom, T.; Siesjo, P.; Bergenheim, A.T. Radiotherapy induces an immediate inflammatory reaction in malignant glioma: A clinical microdialysis study. J. Neurooncol. 2017, 131, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Auffinger, B.; Guo, D.; Hasan, T.; Deheeger, M.; Tobias, A.L.; Kim, J.Y.; Atashi, F.; Zhang, L.; Lesniak, M.S.; et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol. Cancer Ther. 2016, 15, 3064–3076. [Google Scholar] [CrossRef] [PubMed]
- Feldheim, J.; Kessler, A.F.; Feldheim, J.J.; Schulz, E.; Wend, D.; Lazaridis, L.; Kleinschnitz, C.; Glas, M.; Ernestus, R.I.; Brandner, S.; et al. Effects of Long-Term Temozolomide Treatment on Glioblastoma and Astrocytoma WHO Grade 4 Stem-like Cells. Int. J. Mol. Sci. 2022, 23, 5238. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yao, F.; Lu, X.; Li, Q.; Su, Z.; Lee, J.H.; Wang, C.; Du, L. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am. J. Cancer Res. 2019, 9, 1161–1171. [Google Scholar] [PubMed]
- Tsidulko, A.Y.; Shevelev, O.B.; Khotskina, A.S.; Kolpakova, M.A.; Suhovskih, A.V.; Kazanskaya, G.M.; Volkov, A.M.; Aidagulova, S.V.; Zavyalov, E.L.; Grigorieva, E.V. Chemotherapy-Induced Degradation of Glycosylated Components of the Brain Extracellular Matrix Promotes Glioblastoma Relapse Development in an Animal Model. Front. Oncol. 2021, 11, 713139. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Foss, K.; Lichtor, T.; Phillips, H.; Roy, E. Resection of orthotopic murine brain glioma. Neuroimmunol. Neuroinflamm. 2021, 8, 64–69. [Google Scholar] [CrossRef]
- Tam, K.; Alhiyari, Y.; Huang, S.; Han, A.; Stafsudd, O.; Shori, R.; St John, M. Label-free, real-time detection of perineural invasion and cancer margins in a murine model of head and neck cancer surgery. Sci. Rep. 2022, 12, 12871. [Google Scholar] [CrossRef] [PubMed]
- Bianco, J.; Bastiancich, C.; Joudiou, N.; Gallez, B.; des Rieux, A.; Danhier, F. Novel model of orthotopic U-87 MG glioblastoma resection in athymic nude mice. J. Neurosci. Methods 2017, 284, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Schiapparellia, P.; Zhang, P.; Lara-Velazqueza, M.; Guerrero-Cazaresa, H.; Linb, R.; Sub, H.; Chakrounb, R.W.; Tusaa, M.; Quiñones-Hinojosaa, A.; Cui, H. Self-assembling and self-formulating prodrug hydrogelator extends survival in a glioblastoma resection and recurrence model. J. Control. Release 2020, 319, 311–321. [Google Scholar] [CrossRef]
- Wang, F.; Huang, Q.; Su, H.; Sun, M.; Wang, Z.; Chen, Z.; Zheng, M.; Chakroun, R.W.; Monroe, M.K.; Chen, D.; et al. Self-assembling paclitaxel-mediated stimulation of tumor-associated macrophages for postoperative treatment of glioblastoma. Proc. Natl. Acad. Sci. USA 2023, 120, e2204621120. [Google Scholar] [CrossRef] [PubMed]
- Kauer, T.M.; Figueiredo, J.L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011, 15, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Sheets, K.T.; Bago, J.R.; Paulk, I.L.; Hingtgen, S.D. Image-Guided Resection of Glioblastoma and Intracranial Implantation of Therapeutic Stem Cell-seeded Scaffolds. J. Vis. Exp. 2018, 137, e57452. [Google Scholar] [CrossRef]
- Rogers, S.; Hii, H.; Huang, J.; Ancliffe, M.; Gottardo, N.G.; Dallas, P.; Lee, S.; Endersby, R. A novel technique of serial biopsy in mouse brain tumour models. PLoS ONE 2017, 12, e0175169. [Google Scholar] [CrossRef] [PubMed]
- Belykh, E.; Miller, E.J.; Hu, D.; Martirosyan, N.L.; Woolf, E.C.; Scheck, A.C.; Byvaltsev, V.A.; Nakaji, P.; Nelson, L.Y.; Seibel, E.J.; et al. Scanning Fiber Endoscope Improves Detection of 5-Aminolevulinic Acid-Induced Protoporphyrin IX Fluorescence at the Boundary of Infiltrative Glioma. World Neurosurg. 2018, 113, e51–e69. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, N.L.; Georges, J.; Eschbacher, J.M.; Cavalcanti, D.D.; Elhadi, A.M.; Abdelwahab, M.G.; Scheck, A.C.; Nakaji, P.; Spetzler, R.F.; Preul, M.C. Potential application of a handheld confocal endomicroscope imaging system using a variety of fluorophores in experimental gliomas and normal brain. Neurosurg. Focus. 2014, 36, E16. [Google Scholar] [CrossRef]
- Hingtgen, S.; Figueiredo, J.L.; Farrar, C.; Duebgen, M.; Martinez-Quintanilla, J.; Bhere, D.; Shah, K. Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J. Neurooncol 2013, 111, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Stocker, S.; Wagner, S.; Stepp, H.; Fritsch, C.; Goetz, C.; Goetz, A.E.; Kiefmann, R.; Reulen, H.J. Intraoperative Detection of Malignant Gliomas by 5-Aminolevulinic Acid-induced Porphyrin Fluorescence. Neurosurgery 1998, 42, 518–526. [Google Scholar] [PubMed]
- Ma, G.; Ikeda, H.; Inokuchi, T.; Sano, K. Effect of photodynamic therapy using 5-aminolevulinic acid on 4-nitroquinoline-1-oxide-induced premalignant and malignant lesions of mouse tongue. Oral. Oncol. 1999, 35, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Bogaards, A.; Varma, A.; Collens, S.P.; Lin, A.; Giles, A.; Yang, V.X.; Bilbao, J.M.; Lilge, L.D.; Muller, P.J.; Wilson, B.C. Increased brain tumor resection using fluorescence image guidance in a preclinical model. Lasers Surg. Med. 2004, 35, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.J.; Dios, R.R.; Hattab, E.M.; Burrell, K.; Rakopoulos, P.; Sabha, N.; Hawkins, C.; Zadeh, G.; Rutka, J.T.; Cohen-Gadol, A.A. Study of the biodistribution of fluorescein in glioma-infiltrated mouse brain and histopathological correlation of intraoperative findings in high-grade gliomas resected under fluorescein fluorescence guidance. J. Neurosurg. 2015, 122, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.A.; Spence, A.M.; Carski, T.; Berger, M.S. Indocyanine green (ICG) staining and demarcation of tumor margins in a rat glioma model. Surg. Neurol. 1993, 40, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Zeh, R.; Sheikh, S.; Xia, L.; Pierce, J.; Newton, A.; Predina, J.; Cho, S.; Nasrallah, M.; Singhai, S.; Dorsey, J.; et al. The second window ICG technique demonstrates a broad plateau period for near infrared fluorescence tumor contrast in glioblastoma. PLoS ONE 2017, 12, e0182034. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.R.; Martirosyan, N.; Lemole, G.M.; Trouard, T.P.; Romanowski, M. Intraoperative brain tumor resection with indocyanine green using augmented microscopy. J. Biomed. Opt. 2018, 23, 090501. [Google Scholar] [CrossRef] [PubMed]
- Senders, J.T.; Muskens, I.S.; Schnoor, R.; Karhade, A.V.; Cote, D.J.; Smith, T.R.; Broekman, M.L. Agents for fluorescence-guided glioma surgery: A systematic review of preclinical and clinical results. Acta Neurochir. 2017, 159, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, J.J.C.; Stalpers, L.J.A.; Claes, A.; Hovinga, K.E.; Musters, G.D.; Peter Vandertop, W.; Richel, D.J.; Leenders, W.P.J.; van Furth, W.R. Tumor control by whole brain irradiation of anti-VEGF-treated mice bearing intracerebral glioma. Eur. J. Cancer 2009, 45, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, B.S.; Tanny, S.; Johnston, C.J.; Majewska, A.K.; O’Banion, M.K.; Marples, B. In Vivo Imaging of the Microglial Landscape After Whole Brain Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Institoris, A.; Murphy-Royal, C.; Tarantini, S.; Yabluchanskiy, A.; Haidey, J.N.; Csiszar, A.; Ungvari, Z.; Gordon, G.R. Whole brain irradiation in mice causes long-term impairment in astrocytic calcium signaling but preserves astrocyte-astrocyte coupling. Geroscience 2021, 43, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, A.; Stevenson, K.; Tulk, A.; Chalmers, A. Evaluation of four different small animal radiation plans on tumour and normal tissue dosimetry in a glioblastoma mouse model. Br. J. Radiol. 2018, 92, 20180469. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Xu, X.; Garzon-Muvdi, T.; Yuanxuan Xia, M.; Eileen Kim, M.; Zineb Belcaid, M.; Andrew Luksik, M.; Russell Maxwell, M.; Choi, J.; Wang, H.; et al. In vivo Bioluminescence Tomography Center of Mass-Guided Conformal Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2020, 106, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Stackhouse, C.T.; Anderson, J.C.; Yue, Z.; Nguyen, T.; Eustace, N.J.; Langford, C.P.; Wang, J.; Rowland, J.R.t.; Xing, C.; Mikhail, F.M.; et al. An in vivo model of glioblastoma radiation resistance identifies long noncoding RNAs and targetable kinases. JCI Insight 2022, 7, e148717. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Rabkin, S.D.; Martuza, R.L. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J. Immunother. Cancer 2020, 8, e000345. [Google Scholar] [CrossRef]
- Park, J.; Kim, C.G.; Shim, J.K.; Kim, J.H.; Lee, H.; Lee, J.E.; Kim, M.H.; Haam, K.; Jung, I.; Park, S.H.; et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. Oncoimmunology 2019, 8, e1525243. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Qi, N.; Li, J.; Zhang, G. Temozolomide combined with PD-1 Antibody therapy for mouse orthotopic glioma model. Biochem. Biophys. Res. Commun. 2018, 501, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert. Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.L.; Ricks, C.B.; Bohm, A.K.; Lun, X.; Maxwell, L.; Safdar, S.; Bukhari, S.; Gerber, A.; Sayeed, W.; Bering, E.A.; et al. ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PLoS ONE 2018, 13, e0202860. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cai, S.; Bailey, B.J.; Reza Saadatzadeh, M.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Zachary Gunter, T.; Long, E.C.; Minto, R.E.; et al. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg. 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gong, K.; Puliyappadamba, V.T.; Panchani, N.; Pan, E.; Mukherjee, B.; Damanwalla, Z.; Bharia, S.; Hatanpaa, K.J.; Gerber, D.E.; et al. Efficacy of EGFR plus TNF inhibition in a preclinical model of temozolomide-resistant glioblastoma. Neuro-Oncology 2019, 21, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Rabé, M.; Dumont, S.; Alvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Séry, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Plowman, J.; Waud, W.R.; Koutsoukos, A.D.; Rubinstein, L.V.; Moore, T.D.; Grever, M.R. Preclinical antitumor activity of temozolomide in mice: Efficacy against human brain tumor xenografts and synergism with 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1994, 54, 3793–3799. [Google Scholar] [PubMed]
- Hirst, T.C.; Vesterinen, H.M.; Sena, E.S.; Egan, K.J.; Macleod, M.R.; Whittle, I.R. Systematic review and meta-analysis of temozolomide in animal models of glioma: Was clinical efficacy predicted? Br. J. Cancer 2013, 108, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Goldwirt, L.; Zahr, N.; Farinotti, R.; Fernandez, C. Development of a new UPLC-MSMS method for the determination of temozolomide in mice: Application to plasma pharmacokinetics and brain distribution study. Biomed. Chromatogr. 2013, 27, 889–893. [Google Scholar] [CrossRef] [PubMed]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.M.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved brain penetration and antitumor efficacy of temozolomide by inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Yao, J.; Zhang, Y.; Li, M.; Yu, B.; Wang, K. β-elemene combined with temozolomide in treatment of brain glioma. Biochem. Biophys. Rep. 2021, 28, 101144. [Google Scholar] [CrossRef] [PubMed]
- Reydderman, L.; Statkevich, P.; Thonoor, C.M.; Patrick, J.; Batra, V.K.; Wirth, M. Disposition and pharmacokinetics of temozolomide in rat. Xenobiotica 2004, 34, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; McCully, C.; Godwin, K.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J. Neurooncol. 2003, 61, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic. Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, L.L.; Pagel, M.A.; Netto, J.P.; Neuwelt, E.A. Intra-arterial administration improves temozolomide delivery and efficacy in a model of intracerebral metastasis, but has unexpected brain toxicity. J. Neuro-Oncol. 2016, 126, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Guo, P.; Wang, X.; Nuthalapati, S.; Gallo, J.M. Preclinical pharmacokinetic and pharmacodynamic evaluation of metronomic and conventional temozolomide dosing regimens. J. Pharmacol. Exp. Ther. 2007, 321, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Segura-Collar, B.; Jiménez-Sánchez, J.; Gargini, R.; Dragoj, M.; Sepúlveda, J.M.; Pešić, M.; Sánchez-Gómez, P.; Pérez-García, V.M. On optimal temozolomide scheduling for slowly growing gliomas. bioRxiv 2022. [Google Scholar] [CrossRef]
- Delgado-Goni, T.; Julia-Sape, M.; Candiota, A.P.; Pumarola, M.; Arus, C. Molecular imaging coupled to pattern recognition distinguishes response to temozolomide in preclinical glioblastoma. NMR Biomed. 2014, 27, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Akbar, U.; Jones, T.; Winestone, J.; Michael, M.; Shukla, A.; Sun, Y.; Duntsch, C. Delivery of temozolomide to the tumor bed via biodegradable gel matrices in a novel model of intracranial glioma with resection. J. Neurooncol. 2009, 94, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Graham-Gurysh, E.; Moore, K.M.; Satterlee, A.B.; Sheets, K.T.; Lin, F.-C.; Bachelder, E.M.; Miller, C.R.; Hingtgen, S.D.; Ainslie, K.M. Sustained Delivery of Doxorubicin via Acetalated Dextran Scaffold Prevents Glioblastoma Recurrence after Surgical Resection. Mol. Pharm. 2018, 15, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Stuckey, D.W.; Pignatta, S.; Reinshagen, C.; Khalsa, J.K.; Roozendaal, N.; Martinez-Quintanilla, J.; Tamura, K.; Keles, E.; Shah, K. Tumor Resection Recruits Effector T Cells and Boosts Therapeutic Efficacy of Encapsulated Stem Cells Expressing IFNβ in Glioblastomas. Clin. Cancer Res. 2017, 23, 7047–7058. [Google Scholar] [CrossRef] [PubMed]
- Bhere, D.; Choi, S.H.; van de Donk, P.; Hope, D.; Gortzak, K.; Kunnummal, A.; Khalsa, J.; Revai Lechtich, E.; Reinshagen, C.; Leon, V.; et al. Target receptor identification and subsequent treatment of resected brain tumors with encapsulated and engineered allogeneic stem cells. Nat. Commun. 2022, 13, 2810. [Google Scholar] [CrossRef] [PubMed]
- Datta, M.; Chatterjee, S.; Perez, E.M.; Gritsch, S.; Roberge, S.; Duquette, M.; Chen, I.X.; Naxerova, K.; Kumar, A.S.; Ghosh, M.; et al. Losartan controls immune checkpoint blocker-induced edema and improves survival in glioblastoma mouse models. Proc. Natl. Acad. Sci. USA 2023, 120, e2219199120. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Chunta, J.L.; Park, S.S.; Huang, J.; Martinez, A.A.; Grills, I.S.; Krueger, S.A.; Wilson, G.D.; Marples, B. Pulsed versus conventional radiation therapy in combination with temozolomide in a murine orthotopic model of glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 978–985. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Hudson, A.L.; Prasanna Kumar, R.; Wilmott, J.S.; Attrill, G.H.; Long, G.V.; Scolyer, R.A.; Clarke, S.J.; Wheeler, H.R.; Diakos, C.I.; et al. Temporal and spatial modulation of the tumor and systemic immune response in the murine Gl261 glioma model. PLoS ONE 2020, 15, e0226444. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.L.; Grogan, P.T.; Mladek, A.C.; Schroeder, M.A.; Kitange, G.J.; Decker, P.A.; Giannini, C.; Wu, W.; Ballman, K.A.; James, D.; et al. Radiosensitizing effects of TMZ observed in vivo only in a subset of MGMT methylated GBM xenografts. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, K.; Jacob, J.R.; Litzenberg, K.T.; Ray-Chaudhury, A.; Chakravarti, A. Cells isolated from residual intracranial tumors after treatment express iPSC genes and possess neural lineage differentiation plasticity. EBioMedicine 2018, 36, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.D.; Traver, G.; Roland, J.T.; Brockman, A.A.; Dean, D.; Johnson, L.; Boyd, K.; Ihrie, R.A.; Freeman, M.L. Bcl2-Expressing Quiescent Type B Neural Stem Cells in the Ventricular-Subventricular Zone Are Resistant to Concurrent Temozolomide/X-Irradiation. Stem Cells 2019, 37, 1629–1639. [Google Scholar] [CrossRef]
- Dey, D.; Parihar, V.K.; Szabo, G.G.; Klein, P.M.; Tran, J.; Moayyad, J.; Ahmed, F.; Nguyen, Q.A.; Murry, A.; Merriott, D.; et al. Neurological Impairments in Mice Subjected to Irradiation and Chemotherapy. Radiat. Res. 2020, 193, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Chaponis, D.; Barnes, J.W.; Dellagatta, J.L.; Kesari, S.; Fast, E.; Sauvageot, C.; Panagrahy, D.; Greene, E.R.; Ramakrishna, N.; Wen, P.Y.; et al. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. J. Neurooncol. 2011, 104, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Lemasson, B.; Wang, H.; Galban, S.; Li, Y.; Zhu, Y.; Heist, K.A.; Tsein, C.; Chenevert, T.L.; Rehemtulla, A.; Galban, C.J.; et al. Evaluation of Concurrent Radiation, Temozolomide and ABT-888 Treatment Followed by Maintenance Therapy with Temozolomide and ABT-888 in a Genetically Engineered Glioblastoma Mouse Model. Neoplasia 2016, 18, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Mancini, A.; Colapietro, A.; Gravina, G.L.; Vitale, F.; Marampon, F.; Monache, S.D.; Pompili, S.; Cristiano, L.; Vetuschi, A.; et al. The first-in-class alkylating deacetylase inhibitor molecule tinostamustine shows antitumor effects and is synergistic with radiotherapy in preclinical models of glioblastoma. J. Hematol. Oncol. 2018, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Burgenske, D.M.; Talele, S.; Pokorny, J.L.; Mladek, A.C.; Bakken, K.K.; Carlson, B.L.; Schroeder, M.A.; He, L.; Hu, Z.; Gampa, G.; et al. Preclinical modeling in glioblastoma patient-derived xenograft (GBM PDX) xenografts to guide clinical development of lisavanbulin-a novel tumor checkpoint controller targeting microtubules. Neuro-Oncology 2022, 24, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Reste, P.J.L.; Pineau, R.; Voutetakis, K.; Samal, J.; Jegou, G.; Lhomond, S.; Gorman, A.M.; Samali, A.; Patterson, J.B.; Zeng, Q.; et al. Local intracerebral inhibition of IRE1 by MKC8866 sensitizes glioblastoma to irradiation/chemotherapy in vivo. Cancer Lett. 2020, 494, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Kloepper, J.; Ren, J.; Tay, R.E.; Kazer, S.W.; Kiner, E.; Krishnan, S.; Posada, J.M.; Ghosh, M.; Mamessier, E.; et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun. 2021, 12, 2582. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Imaging Agent | Target | Tumor:Normal Tissue Ratio |
|---|---|---|
| IRDye 800CW-RGD | Integrin receptor | 16–18:1 |
| Anti-TRP-1-Alexa fluor 488 or 750 | TRP | unspecified |
| Anti-EGFR antibodies | EGFR | 200–1000:1 |
| Angiopep-2-Cy5.5 | LRP | 1.6:1 |
| Source | Animal | Sex | Route | Dose | t1/2 | Tmax | Cmax | AUC ** |
|---|---|---|---|---|---|---|---|---|
| [mg/kg] | [h] | [h] | [µg/mL] | [h × µg/mL] | ||||
| Plasma | ||||||||
| Goldwirt et al., 2013 [147] | Swiss mice | F | IP | 66 | 0.88 | 0.25 | 27.89 | 31 |
| de Gooijer et al., 2018 [148] | FVB mice | M | IV | 50 | 0.69 | ~55 | 65.4 | |
| Zhang et al., 2021 [149] | ICR mice | M | PO | 30 | 4.04 | 0.25 | 20.96 | 39.9 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | M | IV | 16.6 * | 1.2 | 0.08 | 34.7 | 55.6 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | F | IV | 16.6 * | 1.1 | 0.08 | 35.3 | 50.6 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | M | PO | 16.6 * | 1.17 | 0.75 | 21.5 | 55.2 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | F | PO | 16.6 * | 1.22 | 0.25 | 31.4 | 56.4 |
| Reyderman et al., 2004 [150] | Long–Evans rats | M | PO | 16.6 * | 1.4 | 0.25 | 27.5 | 78.8 |
| Reyderman et al., 2004 [150] | Long–Evans rats | F | PO | 16.6 * | 1.2 | 0.25 | 40.9 | 91.6 |
| Patel et al., 2003 [151] | Rhesus monkey | M | IV | 7.5 | 1.5 | 1.5 | 20.19 | 76.1 |
| Brain | ||||||||
| Goldwirt et al., 2013 [147] | Swiss mice | F | IP | 66 | 0.91 | 0.75 | 6.63 | 8.5 |
| de Gooijer et al., 2018 [148] | FVB mice | M | IV | 50 | ~23 | 36.8 | ||
| Zhang et al., 2021 [149] | ICR mice | M | PO | 30 | 4.2 | 0.5 | 20.62 | 53.3 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | M | IV | 16.6 * | 1.2 | 0.25 | 11.4 | 21.8 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | F | IV | 16.6 * | 1.1 | 0.08 | 11.1 | 18.5 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | M | PO | 16.6 * | 1.3 | 1 | 7.9 | 19.8 |
| Reyderman et al., 2004 [150] | Sprague–Dawley rats | F | PO | 16.6 * | 1.1 | 0.5 | 8.3 | 17.5 |
| CSF | ||||||||
| Patel et al., 2003 [151] | Rhesus monkey | M | IV | 7.5 | 1.5 | 2.5 | 5.05 | 24.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodgers, L.T.; Villano, J.L.; Hartz, A.M.S.; Bauer, B. Glioblastoma Standard of Care: Effects on Tumor Evolution and Reverse Translation in Preclinical Models. Cancers 2024, 16, 2638. https://doi.org/10.3390/cancers16152638
Rodgers LT, Villano JL, Hartz AMS, Bauer B. Glioblastoma Standard of Care: Effects on Tumor Evolution and Reverse Translation in Preclinical Models. Cancers. 2024; 16(15):2638. https://doi.org/10.3390/cancers16152638
Chicago/Turabian StyleRodgers, Louis T., John L. Villano, Anika M. S. Hartz, and Björn Bauer. 2024. "Glioblastoma Standard of Care: Effects on Tumor Evolution and Reverse Translation in Preclinical Models" Cancers 16, no. 15: 2638. https://doi.org/10.3390/cancers16152638
APA StyleRodgers, L. T., Villano, J. L., Hartz, A. M. S., & Bauer, B. (2024). Glioblastoma Standard of Care: Effects on Tumor Evolution and Reverse Translation in Preclinical Models. Cancers, 16(15), 2638. https://doi.org/10.3390/cancers16152638