Determining the Location of Co2+ in Zeolites by UV-Vis Diffuse Reflection Spectroscopy: A Critical View


Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalysts
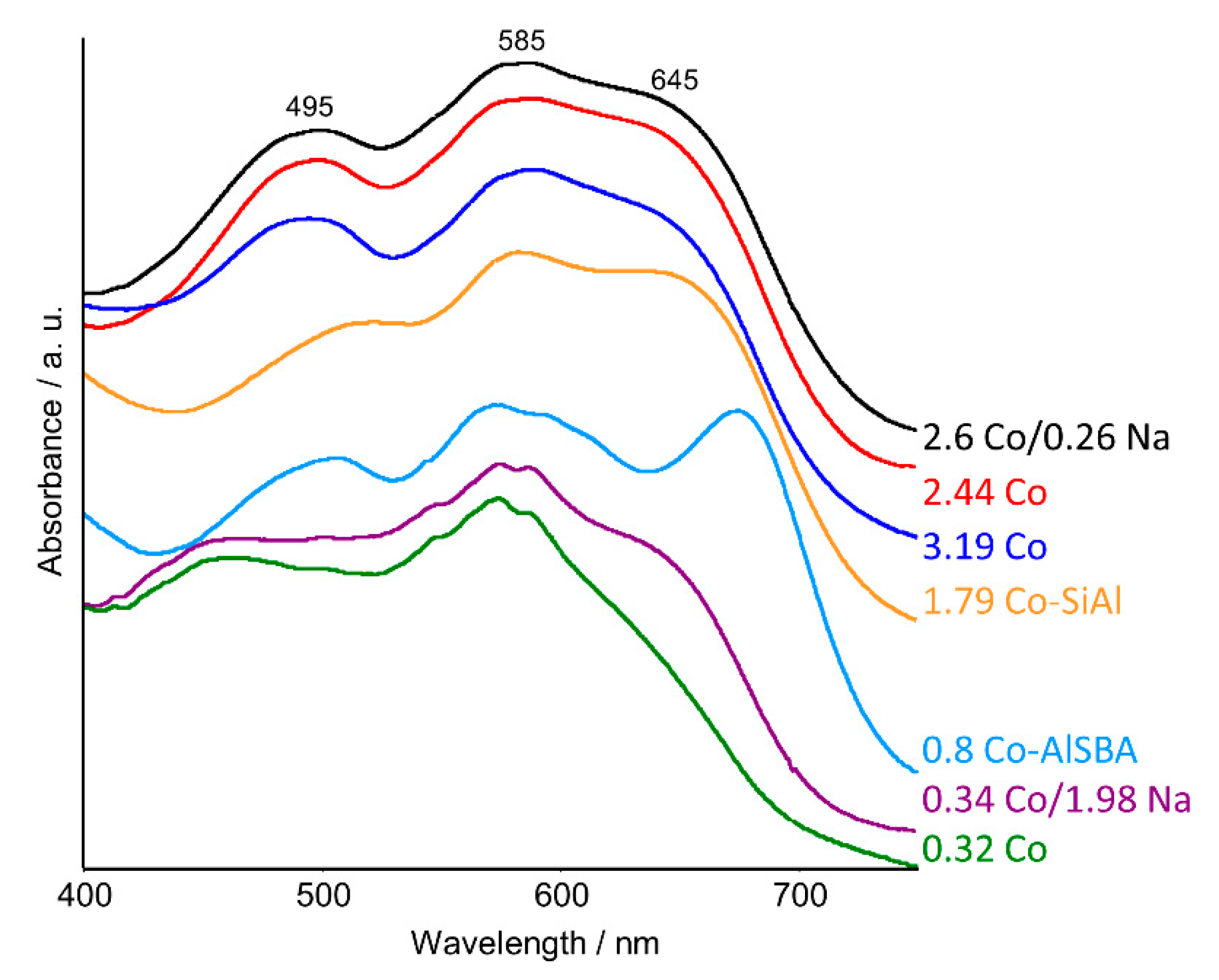
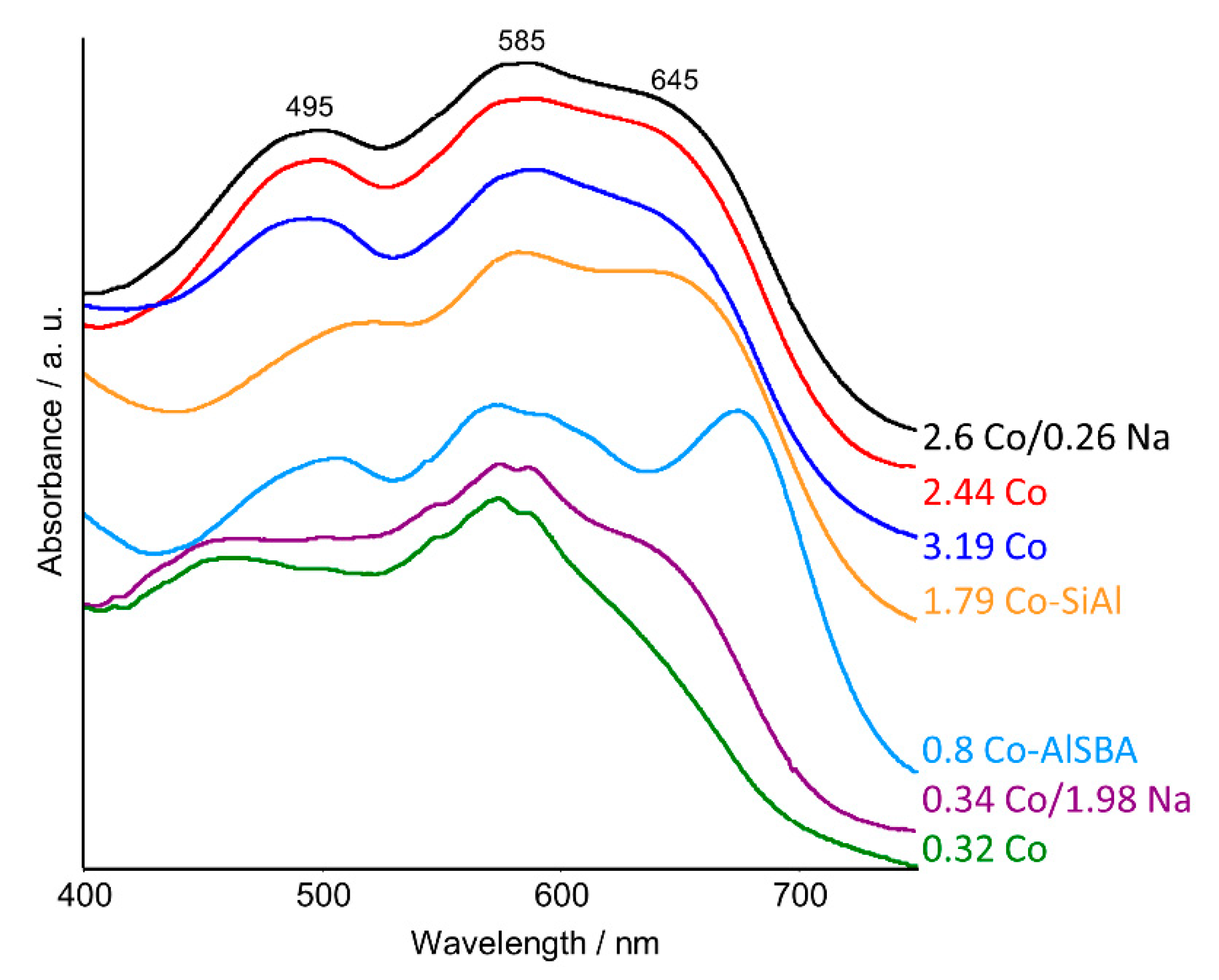
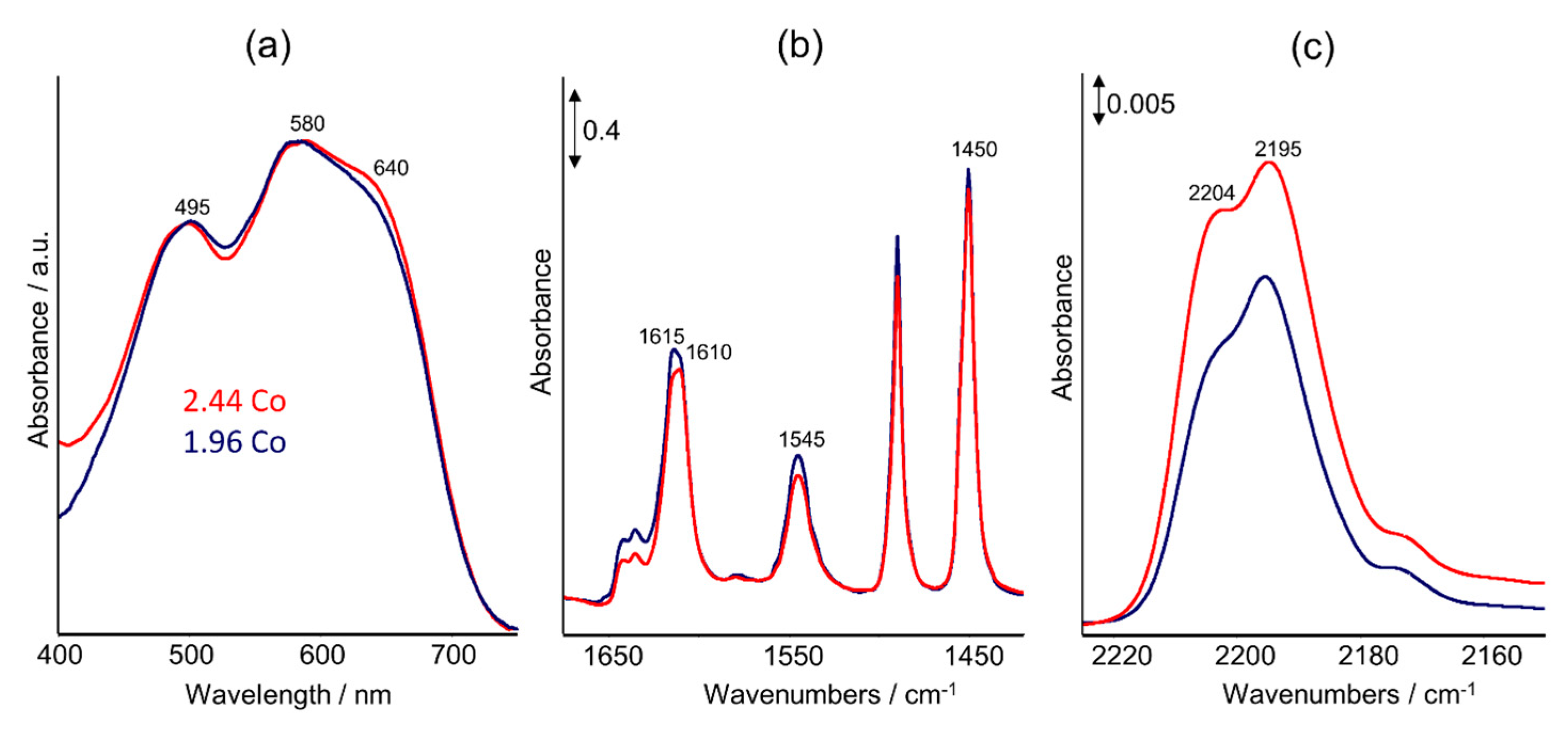
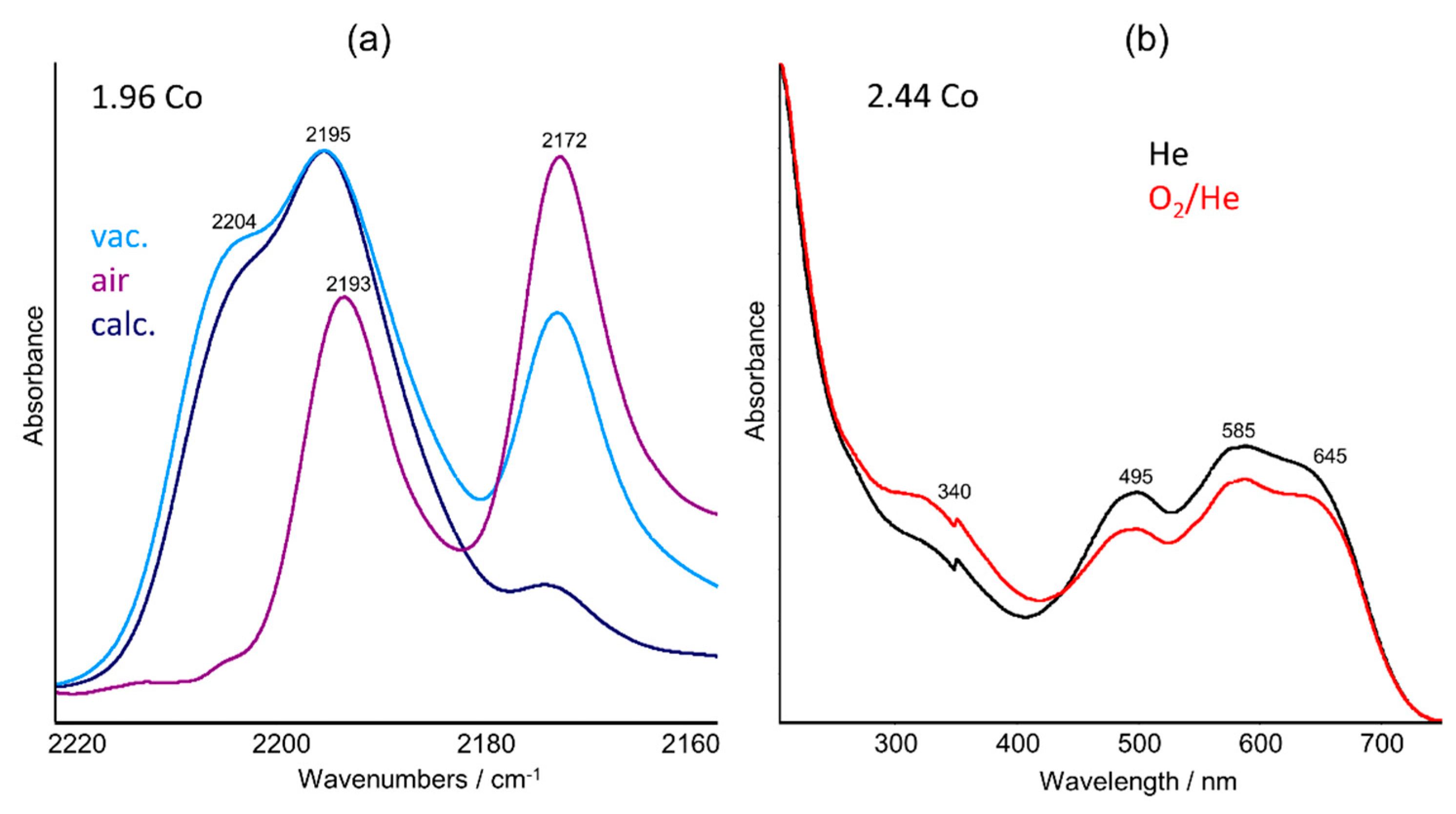
2.2. UV–Vis DRS Studies
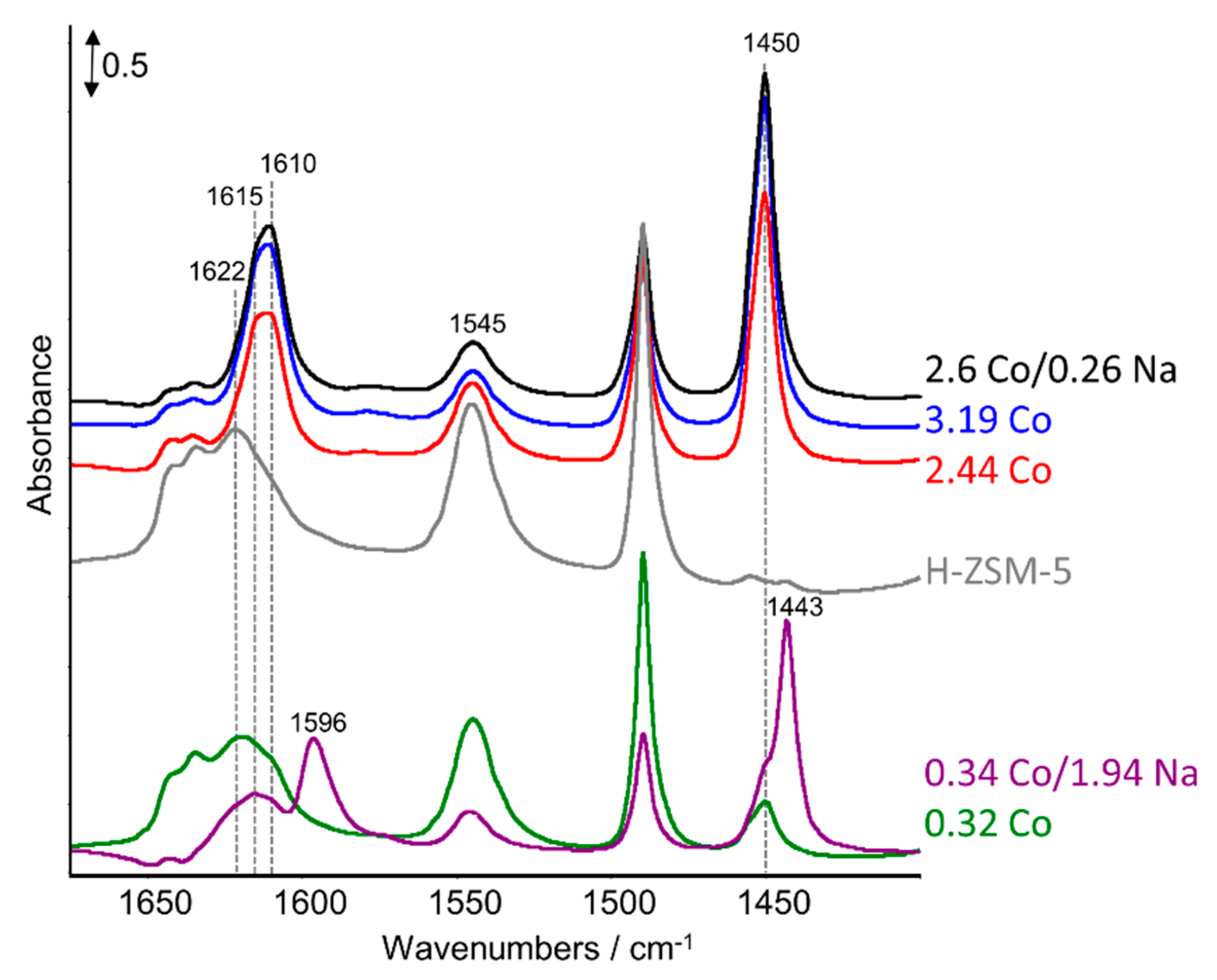
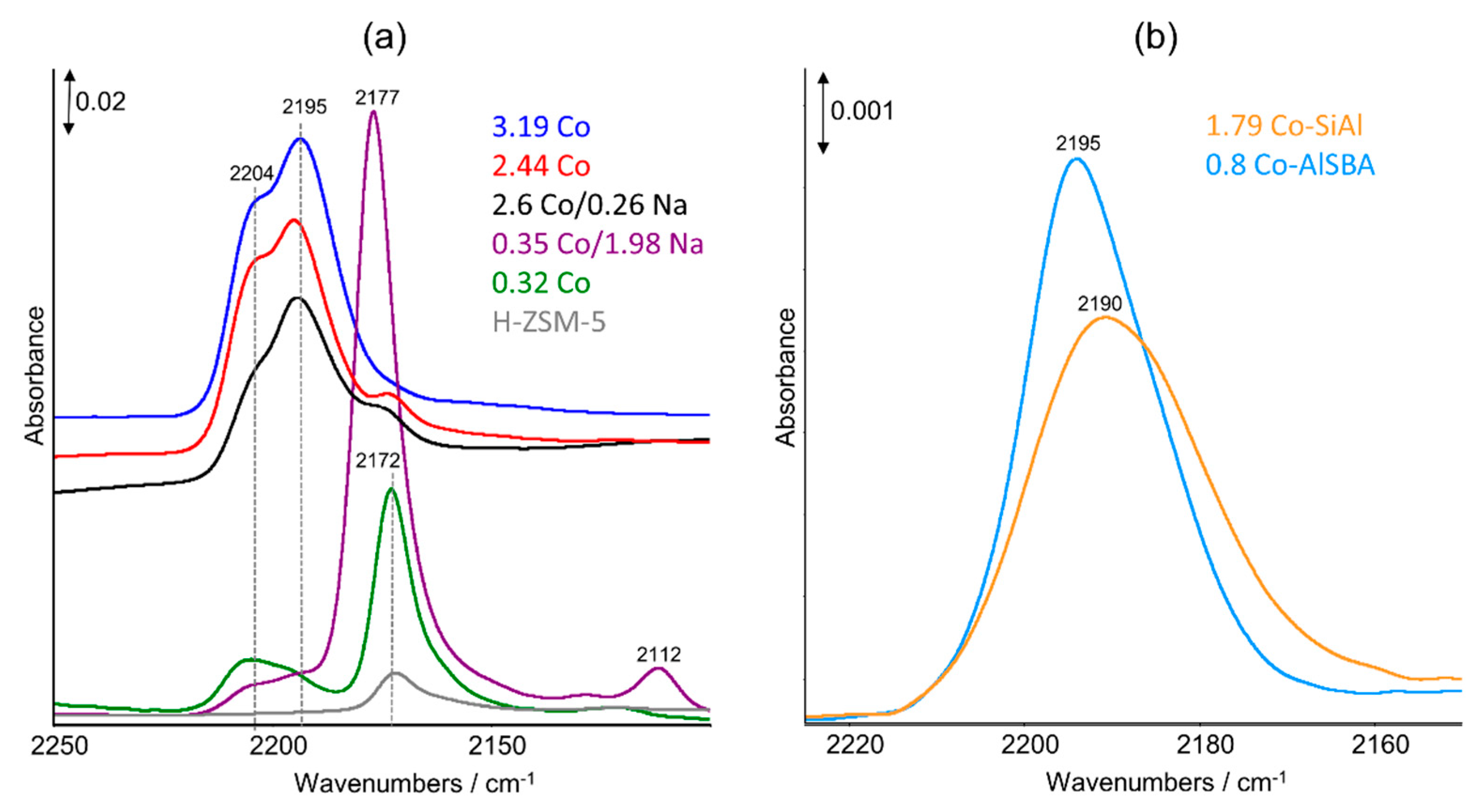
2.3. FTIR Studies: Pyridine and CO Adsorption
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armor, J.N. Catalytic reduction of nitrogen-oxides with methane in the presence of excess oxygen–A review. Catal. Today 1995, 26, 147–158. [Google Scholar] [CrossRef]
- Lonyi, F.; Solt, H.E.; Paszti, Z.; Valyon, J. Mechanism of NO-SCR by methane over Co,H–ZSM-5 and Co,H-mordenite catalysts. Appl. Catal. B Environ. 2014, 150, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, H.Y.; Sachtler, W.M.H. Catalytic reduction of NOx by hydrocarbons over Co/ZSM-5 catalysts prepared with different methods. Appl. Catal. B Environ. 2000, 26, L227–L239. [Google Scholar] [CrossRef]
- Wang, X.; Chen, H.-Y.; Sachtler, W.M.H. Mechanism of the selective reduction of NOx over Co/MFI: Comparison with Fe/MFI. J. Catal. 2001, 197, 281–291. [Google Scholar] [CrossRef]
- Chupin, C.; Vanveen, A.; Konduru, M.; Despres, J.; Mirodatos, C. Identity and location of active species for NO reduction by CH4 over Co-ZSM-5. J. Catal. 2006, 241, 103–114. [Google Scholar] [CrossRef]
- Sadovskaya, E.M.; Suknev, A.P.; Pinaeva, L.G.; Goncharov, V.B.; Bal’zhinimaev, B.S.; Chupin, C.; Pérez-Ramírez, J.; Mirodatos, C. Mechanism and kinetics of the selective NO reduction over Co-ZSM-5 studied by the SSITKA technique: 2. Reactivity of NOx-adsorbed species with methane. J. Catal. 2004, 225, 179–189. [Google Scholar] [CrossRef]
- Sun, T.; Fokema, M.D.; Ying, J.Y. Mechanistic study of NO reduction with methane over Co2+ modified ZSM-5 catalysts. Catal. Today 1997, 33, 251–261. [Google Scholar] [CrossRef]
- Campa, M.C.; DeRossi, S.; Ferraris, G.; Indovina, V. Catalytic activity of Co-ZSM-5 for the abatement of NOx with methane in the presence of oxygen. Appl. Catal. B Environ. 1996, 8, 315–331. [Google Scholar] [CrossRef]
- Campa, M.C.; Indovina, V. Cobalt-exchanged mordenites: Preparation, characterization and catalytic activity for the abatement of NO with CH4 in the presence of excess O-2. J. Porous Mater. 2007, 14, 251–261. [Google Scholar] [CrossRef]
- Campa, M.C.; Luisetto, I.; Pietrogiacomi, D.; Indovina, V. The catalytic activity of cobalt-exchanged mordenites for the abatement of NO with CH4 in the presence of excess O-2. Appl. Catal. B Environ. 2003, 46, 511–522. [Google Scholar] [CrossRef]
- Resini, C.; Montanari, T.; Nappi, L.; Bagnasco, G.; Turco, M.; Busca, G.; Bregani, F.; Notaro, M.; Rocchini, G. Selective catalytic reduction of NOx by methane over Co-H-MFI and Co-H-FER zeolite catalysts: Characterisation and catalytic activity. J. Catal. 2003, 214, 179–190. [Google Scholar] [CrossRef]
- Montanari, T.; Marie, O.; Daturi, M.; Busca, G. Searching for the active sites of Co-H-MFI catalyst for the selective catalytic reduction of NO by methane: A FT-IR in situ and operando study. Appl. Catal. B Environ. 2007, 71, 216–222. [Google Scholar] [CrossRef]
- Montanari, T.; Marie, O.; Daturi, M.; Busca, G. Cobalt on and in zeolites and silica–alumina: Spectroscopic characterization and reactivity. Catal. Today 2005, 110, 339–344. [Google Scholar] [CrossRef]
- Bellmann, A.; Atia, H.; Bentrup, U.; Brückner, A. Mechanism of the selective reduction of NOx by methane over Co-ZSM-5. Appl. Catal. B Environ. 2018, 230, 184–193. [Google Scholar] [CrossRef]
- Dedecek, J.; Sobalik, Z.; Wichterlova, B. Siting and distribution of framework aluminium atoms in silicon-rich zeolites and impact on catalysis. Catal. Rev. 2012, 54, 135–223. [Google Scholar] [CrossRef]
- Dědeček, J.; Kaucký, D.; Wichterlová, B.; Gonsiorová, O. Co2+ ions as probes of Al distribution in the framework of zeolites. ZSM-5 study. Phys. Chem. Chem. Phys. 2002, 4, 5406–5413. [Google Scholar]
- Dědeček, J.; Wichterlová, B. Co2+Ion siting in pentasil-containing zeolites. I. Co2+Ion sites and their occupation in mordenite. A Vis−NIR diffuse reflectance spectroscopy study. J. Phys. Chem. B 1999, 103, 1462–1476. [Google Scholar]
- Kaucký, D.; Dědeček, J.; Wichterlová, B. Co2+ ion siting in pentasil-containing zeolites. Microporous Mesoporous Mater. 1999, 31, 75–87. [Google Scholar] [CrossRef]
- Dedecek, J.; Kaucky, D.; Wichterlova, B. Co2+ ion siting in pentasil-containing zeolites, part 3. Co2+ ion sites and their occupation in ZSM-5: A VIS diffuse reflectance spectroscopy study. Microporous Mesoporous Mater. 2000, 35–36, 483–494. [Google Scholar] [CrossRef]
- Dědeček, J.; Čapek, L.; Kaucký, D.; Sobalík, Z.; Wichterlová, B. Siting and distribution of the Co Ions in beta zeolite: A UV–Vis–NIR and FTIR study. J. Catal. 2002, 211, 198–207. [Google Scholar] [CrossRef]
- Kim, J.; Jentys, A.; Maier, S.M.; Lercher, J.A. Characterization of Fe-exchanged BEA zeolite under NH3 selective catalytic reduction conditions. J. Phys. Chem. C 2013, 117, 986–993. [Google Scholar] [CrossRef]
- Kim, S.; Park, G.; Woo, M.H.; Kwak, G.; Kim, S.K. Control of hierarchical structure and framework-Al distribution of ZSM-5 via adjusting crystallization temperature and their effects on methanol conversion. ACS Catal. 2019, 9, 2880–2892. [Google Scholar] [CrossRef]
- Gil, B.; Janas, J.; Włoch, E.; Olejniczak, Z.; Datka, J.; Sulikowski, B. The influence of the initial acidity of HFER on the status of Co species and catalytic performance of CoFER and InCoFER in CH4-SCR-NO. Catal. Today 2008, 137, 174–178. [Google Scholar] [CrossRef]
- Gil, B.; Pietrzyk, P.; Datka, J.; Kozyra, P.; Sojka, Z. Speciation of cobalt in CoZSM-5 upon thermal treatment. In Studies in Surface Science and Catalysis; Čejka, J., Žilková, N., Nachtigall, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 158, pp. 893–900. [Google Scholar]
- Bagnasco, G.; Turco, M.; Resini, C.; Montanari, T.; Bevilacqua, M.; Busca, G. On the role of external Co sites in NO oxidation and reduction by methane over Co?H-MFI catalysts. J. Catal. 2004, 225, 536–540. [Google Scholar] [CrossRef]
- Montanari, T.; Bevilacqua, M.; Resini, C.; Busca, G. UV–Vis and FT-IR study of the nature and location of the active sites of partially exchanged Co−H zeolites. J. Phys. Chem. B 2004, 108, 2120–2127. [Google Scholar] [CrossRef]
- Beznis, N.V.; Weckhuysen, B.M.; Bitter, J.H. Partial oxidation of methane over Co-ZSM-5: Tuning the oxygenate selectivity by altering the preparation route. Catal. Lett. 2010, 136, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Llusar, M.; Forés, A.; Badenes, J.A.; Calbo, J.; Tena, M.A.; Monrós, G. Colour analysis of some cobalt-based blue pigments. J. Eur. Ceram. Soc. 2001, 21, 1121–1130. [Google Scholar] [CrossRef]
- Lee, K.; Ruddy, D.A.; Dukovic, G.; Neale, N.R. Synthesis, optical, and photocatalytic properties of cobalt mixed-metal spinel oxides Co(Al1−xGax)2O4. J. Mater. Chem. A 2015, 3, 8115–8122. [Google Scholar] [CrossRef]
- Rangappa, D.; Ohara, S.; Naka, T.; Kondo, A.; Ishii, M.; Adschiri, T. Synthesis and organic modification of CoAl2O4 nanocrystals under supercritical water conditions. J. Mater. Chem. 2007, 17, 4426–4429. [Google Scholar] [CrossRef]
- El Kabouss, K.; Kacimi, M.; Ziyad, M.; Ammar, S.; Ensuque, A.; Piquemal, J.-Y.; Bozon-Verduraz, F. Cobalt speciation in cobalt oxide-apatite materials: Structure–properties relationship in catalytic oxidative dehydrogenation of ethane and butan-2-ol conversion. J. Mater. Chem. 2006, 16, 2453–2463. [Google Scholar] [CrossRef]
- Liotta, L.F.; Pantaleo, G.; Macaluso, A.; Di Carlo, G.; Deganello, G. CoOx catalysts supported on alumina and alumina-baria: Influence of the support on the cobalt species and their activity in NO reduction by C3H6 in lean conditions. Appl. Catal. A Gen. 2003, 245, 167–177. [Google Scholar] [CrossRef]
- Radovanovic, P.V.; Norberg, N.S.; McNally, K.E.; Gamelin, D.R. Colloidal transition-metal-doped ZnO quantum dots. J. Am. Chem. Soc. 2002, 124, 15192–15193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, F.A.; Goodgame, D.M.L.; Goodgame, M. The electronic structures of tetrahedral cobalt(II) complexes. J. Am. Chem. Soc. 1961, 83, 4690–4699. [Google Scholar] [CrossRef]
- Klier, K.; Kellerman, R.; Hutta, P.J. Spectra of synthetic zeolites containing transition metal ions. V.* Π complexes of olefins and acetylene with Co(II)A molecular sieve. J. Chem. Phys. 1974, 61, 4224–4234. [Google Scholar] [CrossRef]
- Klier, K. Transition-metal ions in zeolites: The perfect surface sites. Langmuir 1988, 4, 13–25. [Google Scholar] [CrossRef]
- Verberckmoes, A.A.; Uytterhoeven, M.G.; Schoonheydt, R.A. Framework and extra-framework Co2+ in CoAPO-5 by diffuse reflectance spectroscopy. Zeolites 1997, 19, 180–189. [Google Scholar] [CrossRef]
- Praliaud, H.; Coudurier, G. Optical spectroscopy of hydrated, dehydrated and ammoniated cobalt(II) exchanged zeolites X and Y. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1979, 75, 2601–2616. [Google Scholar] [CrossRef]
- Verberckmoes, A.A.; Weckhuysen, B.M.; Schoonheydt, R.A. Spectroscopy and coordination chemistry of cobalt in molecular sieves. Microporous Mesoporous Mater. 1998, 22, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Buzzoni, R.; Bordiga, S.; Ricchiardi, G.; Lamberti, C.; Zecchina, A.; Bellussi, G. Interaction of pyridine with acidic (H–ZSM5, H-β, H-MORD Zeolites) and superacidic (H-Nafion Membrane) systems: An IR investigation. Langmuir 1996, 12, 930–940. [Google Scholar] [CrossRef]
- Busca, G. The surface acidity of solid oxides and its characterization by IR spectroscopic methods. An attempt at systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Gora-Marek, K.; Gil, B.; Sliwa, M.; Datka, J. An IR spectroscopy study of Co sites in zeolites CoZSM-5. Appl. Catal. A Gen. 2007, 330, 33–42. [Google Scholar] [CrossRef]
- Góra-Marek, K.; Gil, B.; Datka, J. Quantitative IR studies of the concentration of Co2+ and Co3+ sites in zeolites CoZSM-5 and CoFER. Appl. Catal. A Gen. 2009, 353, 117–122. [Google Scholar] [CrossRef]
- Gora-Marek, K. The reduction and oxidation of Co species in CoZSM-5 zeolites studied by IR spectroscopy. Top. Catal. 2009, 52, 1023–1029. [Google Scholar] [CrossRef]
- Kustov, L.M.; Kazanskii, V.B.; Beran, S.; Kubelkova, L.; Jiru, P. Adsorption of carbon monoxide on ZSM-5 zeolites: Infrared spectroscopic study and quantum-chemical calculations. J. Phys. Chem. 1987, 91, 5247–5251. [Google Scholar] [CrossRef]
- Zecchina, A.; Bordiga, S.; Lamberti, C.; Spoto, G.; Carnelli, L.; Otero Arean, C. Low-temperature fourier transform infrared study of the interaction of CO with cations in alkali-metal exchanged ZSM-5 zeolites. J. Phys. Chem. 1994, 98, 9577–9582. [Google Scholar] [CrossRef]
- Kraushaar-Czarnetzki, B.; Hoogervorst, W.G.M.; Andréa, R.R.; Emeis, C.A.; Stork, W.H.J. Characterisation of CoIIand CoIIIin CoAPO molecular sieves. J. Chem. Soc. Faraday Trans. 1991, 87, 891–895. [Google Scholar] [CrossRef]
- Drozdová, L.; Prins, R.; Dědeček, J.; Sobalík, Z.; Wichterlová, B. Bonding of Co ions in ZSM-5, ferrierite, and mordenite: An X-ray Absorption, UV–Vis, and IR study. J. Phys. Chem. B 2002, 106, 2240–2248. [Google Scholar] [CrossRef]
- Wu, S.; Han, Y.; Zou, Y.-C.; Song, J.-W.; Zhao, L.; Di, Y.; Liu, S.-Z.; Xiao, F.-S. Synthesis of heteroatom substituted SBA-15 by the “pH-Adjusting” method. Chem. Mater. 2004, 16, 486–492. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Support | Preparation Method | Content (wt%) | Co/Al |
|---|---|---|---|---|
| H–ZSM-5 | NH4–ZSM-5 | IE: water 100 mL/1 g zeolite, 60 °C, 24 h | Si: 34.90 Al: 3.25 | 0.00 |
| Na–ZSM-5 | NH4–ZSM-5 | IE: 0.1 M Na––nitrate 100 mL/1 g zeolite, 25 °C, 24 h | Na: 2.37 | 0.00 |
| 0.32 Co | NH4–ZSM-5 | IE: 0.0005 M Co–nitrate 100 mL/1 g zeolite, 60 °C, 24 h | Co: 0.32 | 0.04 |
| 3.19 Co | NH4–ZSM-5 | IE: 0.05 M Co–nitrate 100 mL/1 g zeolite, 25 °C, 24 h | Co: 3.19 | 0.46 |
| 2.44 Co | NH4–ZSM-5 | IE: 0.005 M Co–nitrate 100 mL/1 g zeolite, 60 °C, 24 h | Co: 2.44 | 0.30 |
| 1.96 Co 1 | NH4–ZSM-5 | IE: 0.05 M Co–acetate 16 mL/1 g zeolite, 70 °C, 12 h | Co: 1.96 | 0.28 |
| 0.34 Co/1.94 Na | Na–ZSM-5 | IE: 0.0005 M Co–nitrate 100 mL/1 g zeolite, 60 °C, 24 h | Co: 0.34 Na: 1.94 | 0.05 |
| 0.35 Co/1.98 Na | Na–ZSM-5 | IE: 0.0005 M Co–acetate 100 mL/1 g zeolite, 60 °C, 24 h | Co: 0.35 Na: 1.98 | 0.05 |
| 2.6 Co/0.26 Na | Na–ZSM-5 | IE: 0.05 M Co–nitrate 100 mL/1 g zeolite, 60 °C, 24 h | Co: 2.60 Na: 0.26 | 0.40 |
| 1.79 Co–SiAl | Siralox | IWI: 423 mg Co–acetate in 12.5 mL water, 5 g Siralox | Co: 1.79 | 0.06 |
| 0.8 Co–AlSBA | Al–SBA-15 | IE: 0.02 M, Co–acetate 100 mL/1 g AlSBA, 60 °C, 24 h | Co: 0.80 Al: 2.92 | 0.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellmann, A.; Rautenberg, C.; Bentrup, U.; Brückner, A. Determining the Location of Co2+ in Zeolites by UV-Vis Diffuse Reflection Spectroscopy: A Critical View. Catalysts 2020, 10, 123. https://doi.org/10.3390/catal10010123
Bellmann A, Rautenberg C, Bentrup U, Brückner A. Determining the Location of Co2+ in Zeolites by UV-Vis Diffuse Reflection Spectroscopy: A Critical View. Catalysts. 2020; 10(1):123. https://doi.org/10.3390/catal10010123
Chicago/Turabian StyleBellmann, Andrea, Christine Rautenberg, Ursula Bentrup, and Angelika Brückner. 2020. "Determining the Location of Co2+ in Zeolites by UV-Vis Diffuse Reflection Spectroscopy: A Critical View" Catalysts 10, no. 1: 123. https://doi.org/10.3390/catal10010123
APA StyleBellmann, A., Rautenberg, C., Bentrup, U., & Brückner, A. (2020). Determining the Location of Co2+ in Zeolites by UV-Vis Diffuse Reflection Spectroscopy: A Critical View. Catalysts, 10(1), 123. https://doi.org/10.3390/catal10010123