Graphene-Based Heterogeneous Catalysis: Role of Graphene


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Graphene in Solid-State Catalysis
1.2. Why Graphene Is Attractive in Solid-State Catalysis
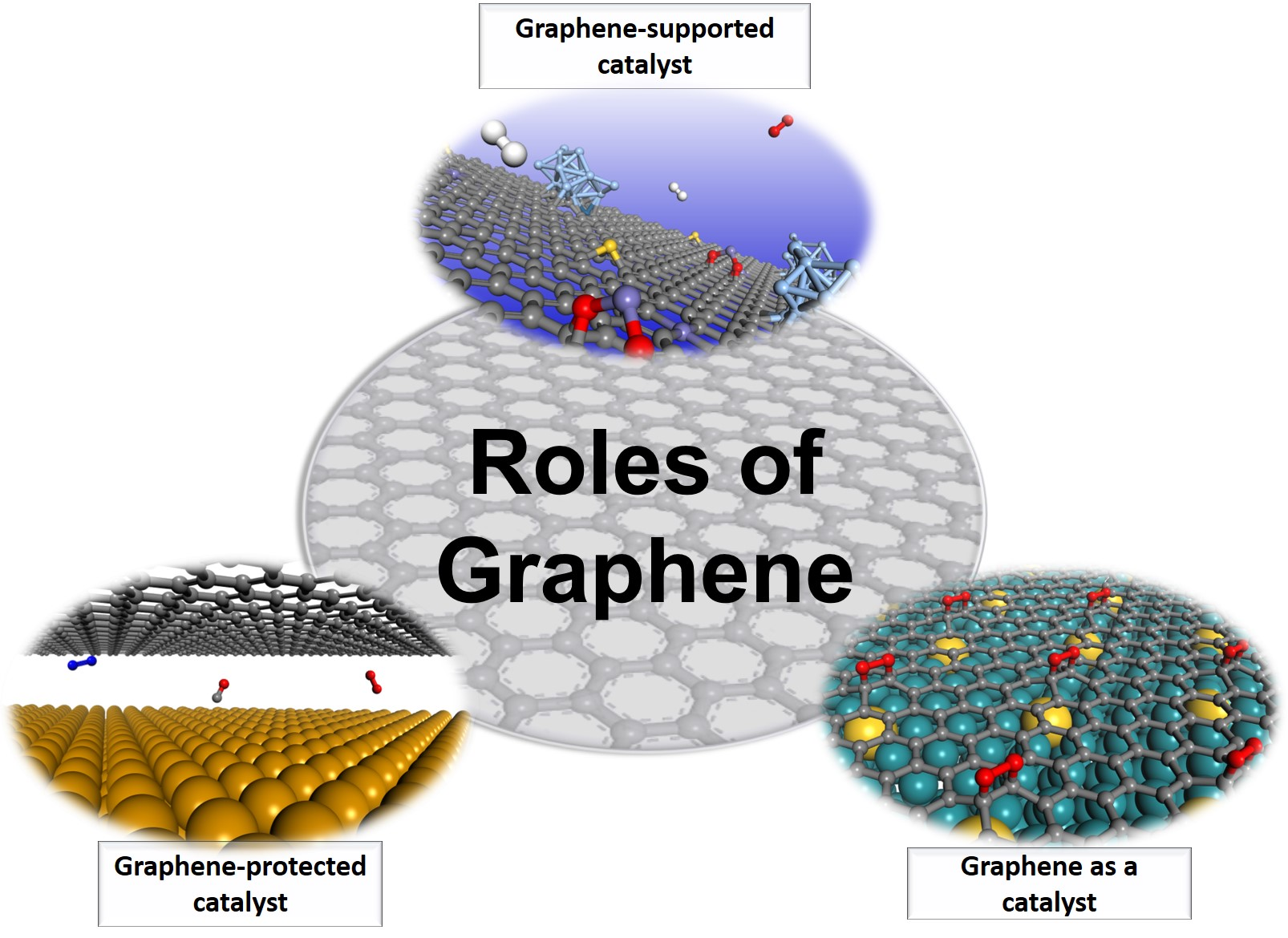
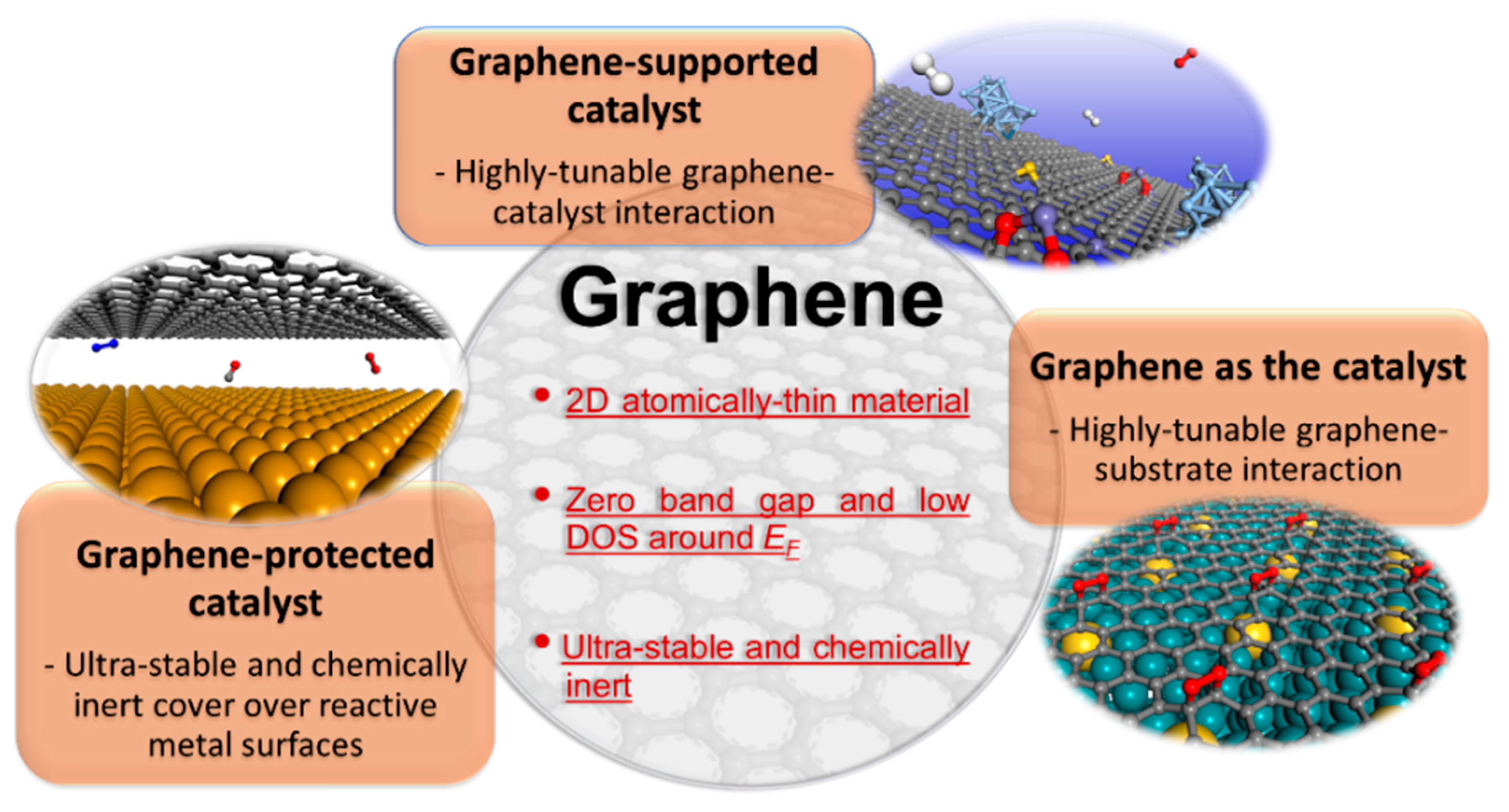
2. Different Roles of Graphene in Solid-State Catalysis
2.1. Graphene as a Support
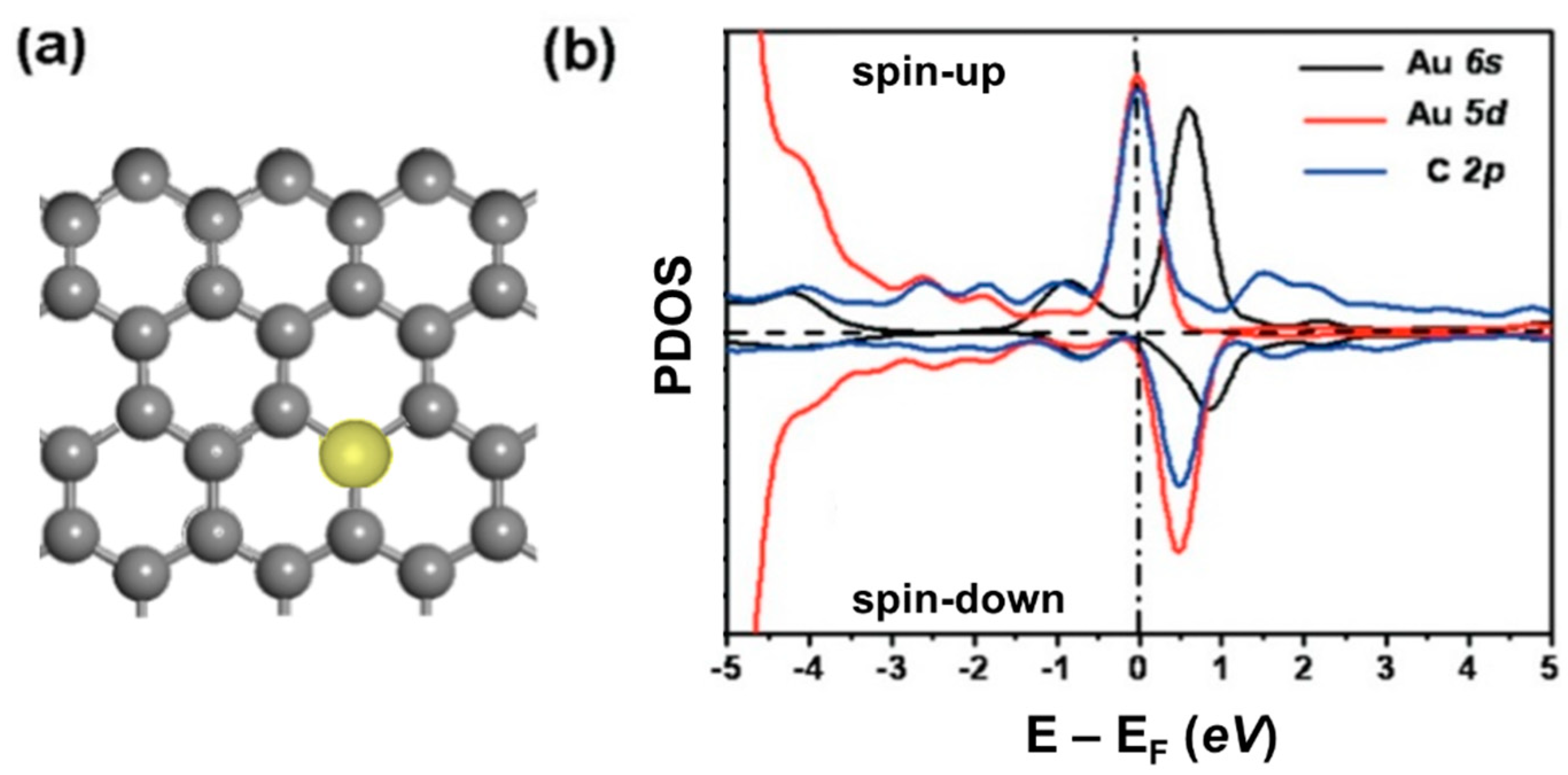
2.1.1. Vacancy Enhanced Graphene Reactivity
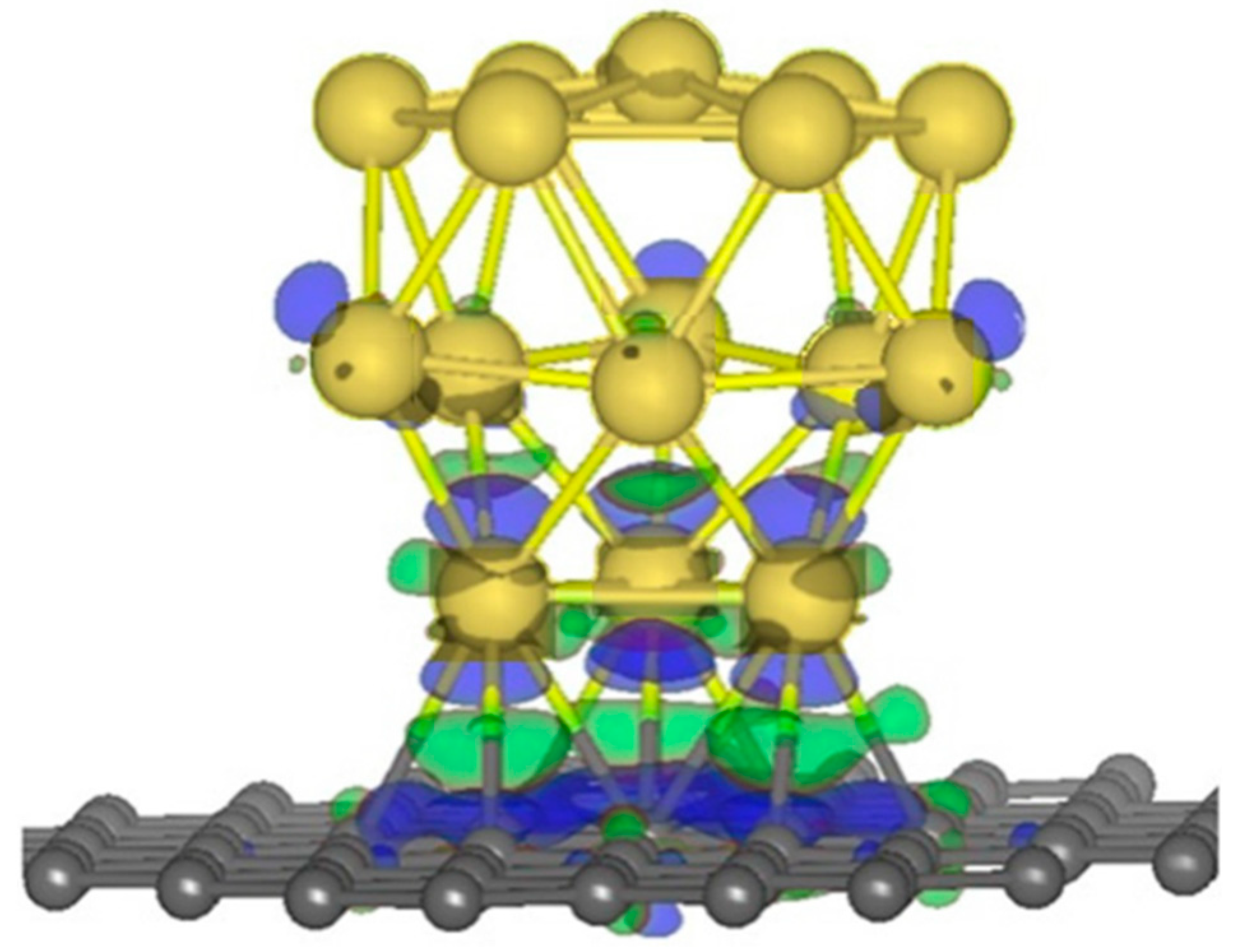
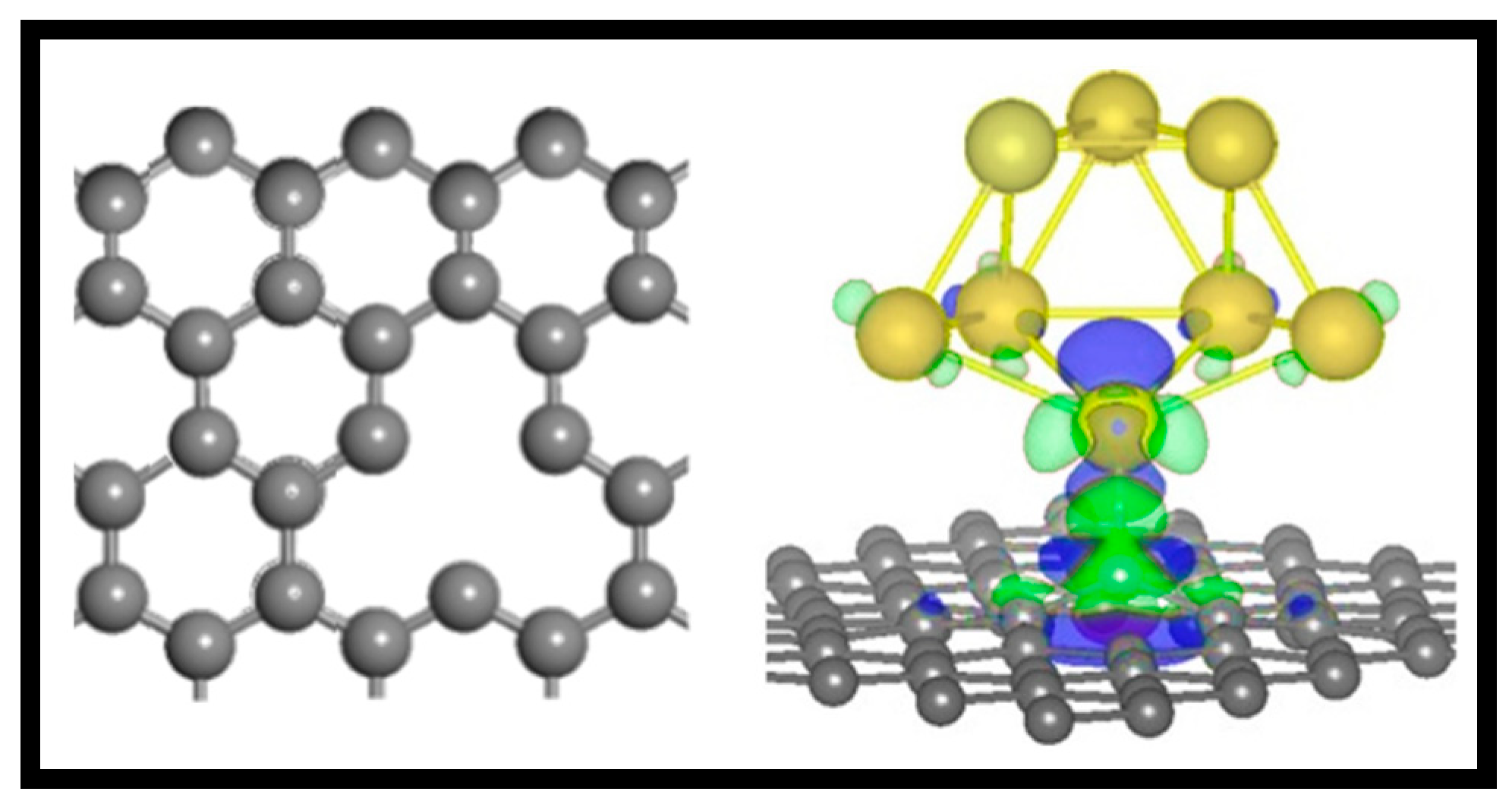
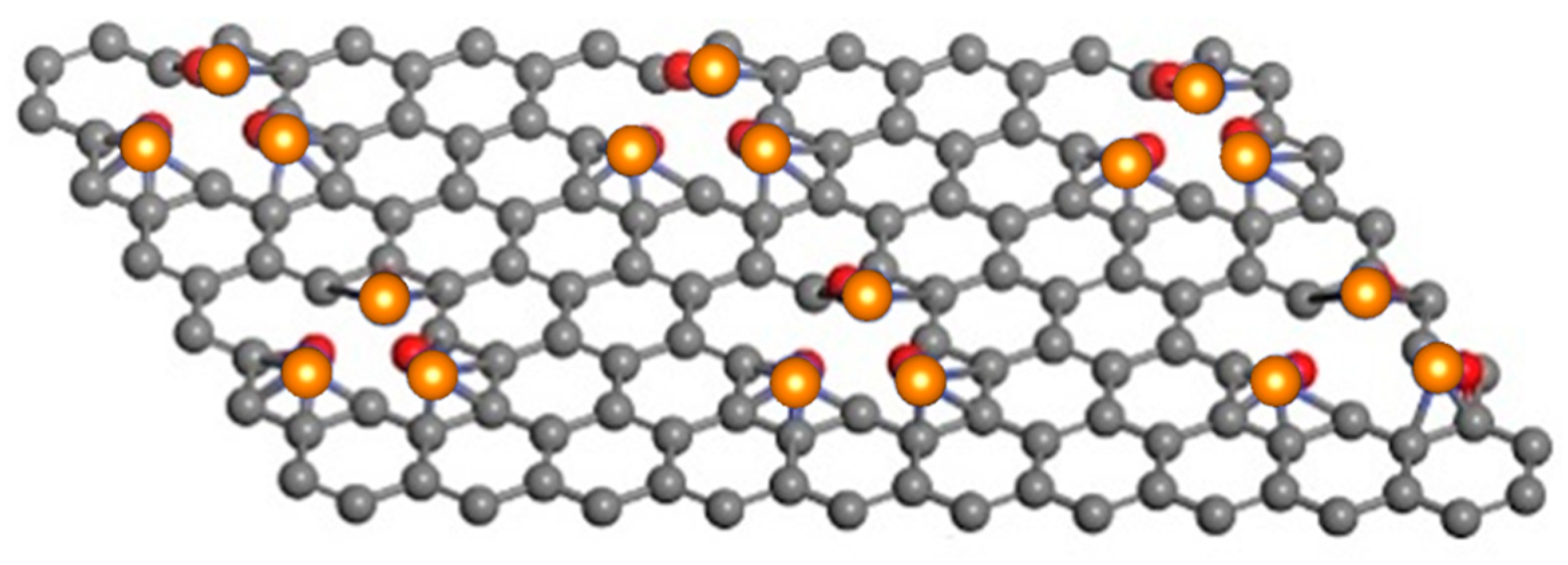
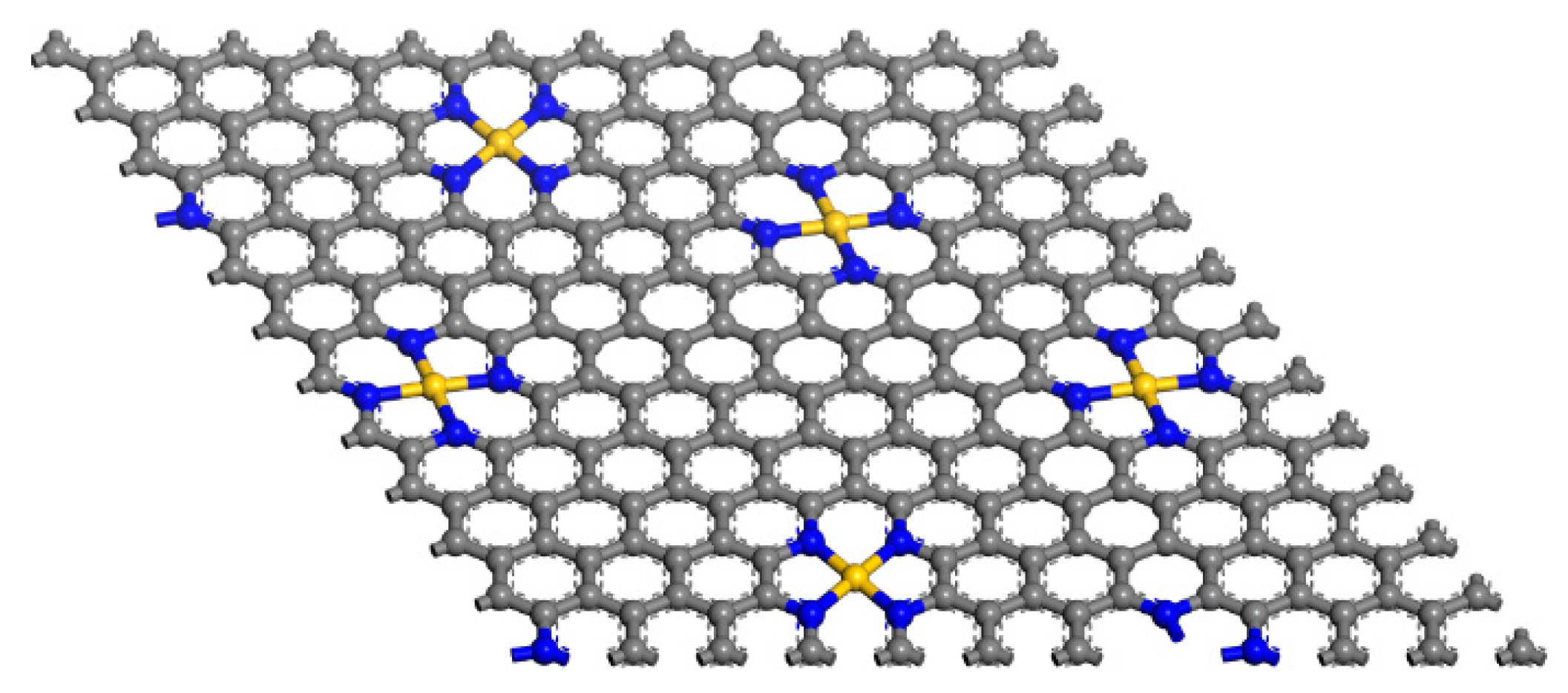
2.1.2. Anchor Atoms to Stabilize the Supported Atomic Scale Catalysts
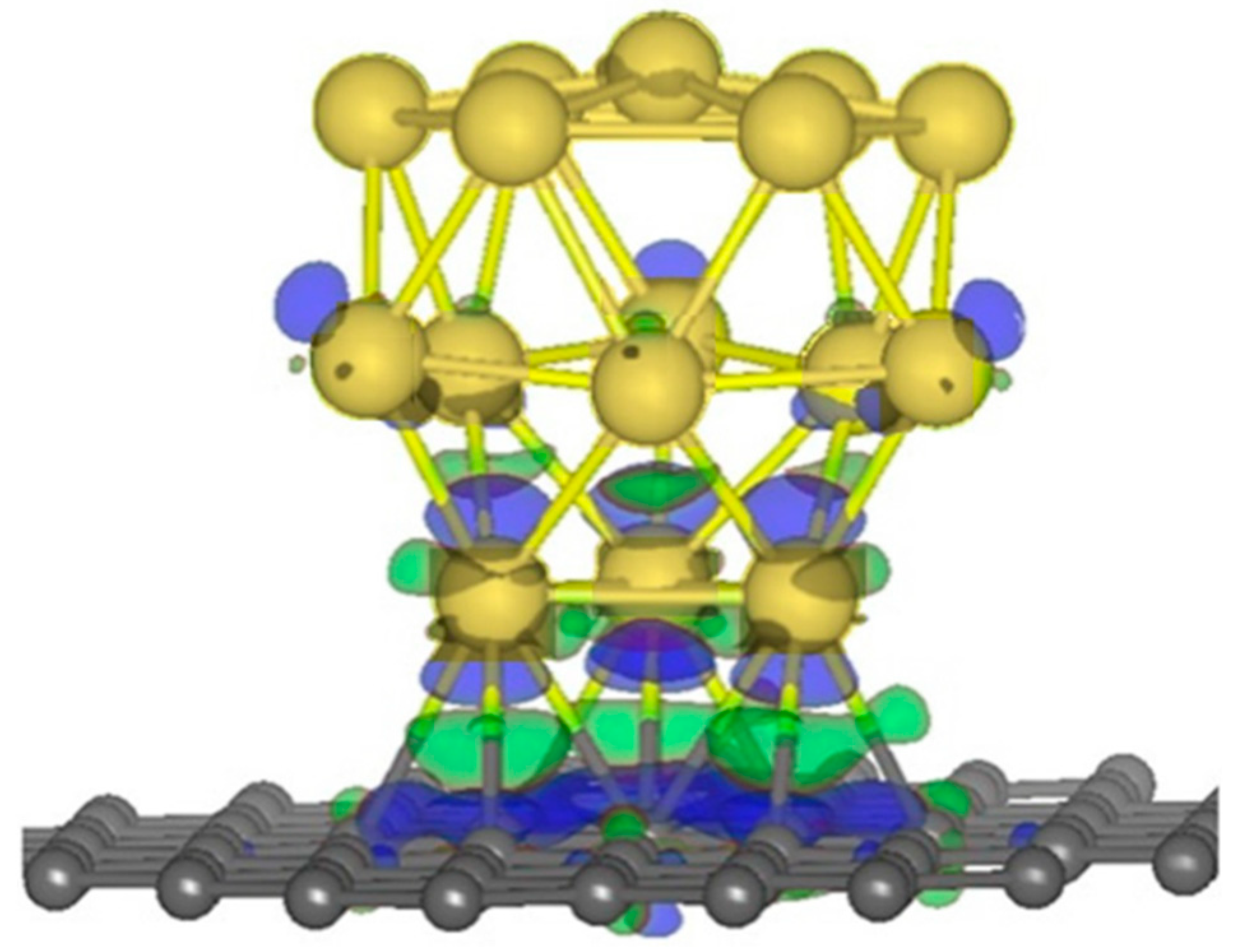
2.1.3. Strained-Enhanced Graphene Reactivity
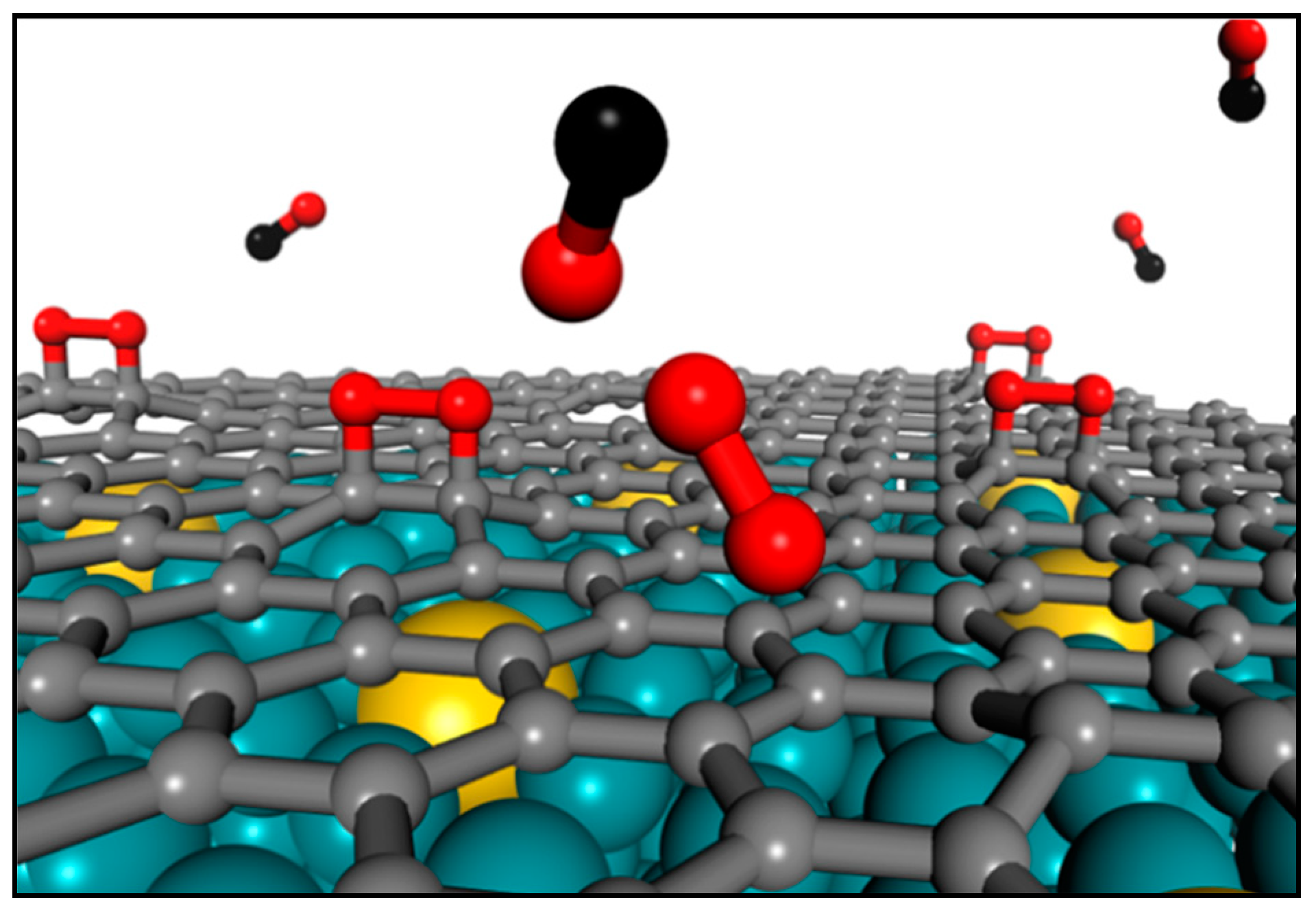
2.2. Graphene as a Cover
2.3. Graphene Itself as a Catalyst: Substrate Engineering of Graphene Reactivity
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Neto, A.H.C.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Das Sarma, S.; Adam, S.; Hwang, E.H.; Rossi, E. Electronic transport in two-dimensional graphene. Rev. Mod. Phys. 2011, 83, 407–470. [Google Scholar] [CrossRef] [Green Version]
- Morozov, S.; Novoselov, K.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.; Geim, A.K.; Morozov, S.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Machado, B.F.; Serp, P. Graphene-based materials for catalysis. Catal. Sci. Technol. 2012, 2, 54–75. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, G.; Zhang, F. Multiple roles of graphene in heterogeneous catalysis. Chem. Soc. Rev. 2015, 44, 3023–3035. [Google Scholar] [CrossRef]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Metal nanoparticles supported on two-dimensional graphenes as heterogeneous catalysts. Coord. Chem. Rev. 2016, 312, 99–148. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, J.; Meng, X.; Yu, L.; Deng, D.; Bao, X. Catalysis with Two-Dimensional Materials Confining Single Atoms: Concept, Design, and Applications. Chem. Rev. 2019, 119, 1806–1854. [Google Scholar] [CrossRef]
- Lu, Y.-H.; Zhou, M.; Zhang, C.; Feng, Y.-P. Metal-Embedded Graphene: A Possible Catalyst with High Activity. J. Phys. Chem. C 2009, 113, 20156–20160. [Google Scholar] [CrossRef]
- Guo, N.; Yam, K.M.; Zhang, C. Substrate engineering of graphene reactivity: Towards high-performance graphene-based catalysts. npj 2D Mater. Appl. 2018, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Pop, E.; Varshney, V.; Roy, A.K. Thermal properties of graphene: Fundamentals and applications. MRS Bull. 2012, 37, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y. Density-functional calculations of icosahedral M13 (M=Pt and Au) clusters on graphene sheets and flakes. Chem. Phys. Lett. 2006, 420, 382–386. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B 2008, 77, 125416. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Li, F.; Zhu, Z.H.; Zhao, M.W.; Xu, X.G.; Su, X.Y. An ab initio study on gas sensing properties of graphene and Si-doped graphene. Eur. Phys. J. B 2011, 81, 475–479. [Google Scholar] [CrossRef]
- Antiochia, R.; Tortolini, C.; Tasca, F.; Gorton, L.; Bollella, P. Chapter 1—Graphene and 2D-Like Nanomaterials: Different Biofunctionalization Pathways for Electrochemical Biosensor Developmen. In Graphene Bioelectronics; Tiwari, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–35. [Google Scholar]
- Dai, J.; Yuan, J. Adsorption of molecular oxygen on doped graphene: Atomic, electronic, and magnetic properties. Phys. Rev. B 2010, 81, 165414. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lu, Y.-H.; Cai, Y.-Q.; Zhang, C.; Feng, Y.-P. Adsorption of gas molecules on transition metal embedded graphene: A search for high-performance graphene-based catalysts and gas sensors. Nanotechnology 2011, 22, 385502. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Q.; Cheng, Y.; Li, K.; Yao, Y.; Zhang, Q.; Dong, C.; Wang, P.; Schwingenschlögl, U.; Yang, W.; et al. Doping monolayer graphene with single atom substitutions. Nano Lett. 2012, 12, 141–144. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Routh, P.; Kim, D.-H.; Huang, W.; Chen, P. Heteroatom-doped graphene materials: Syntheses, properties and applications. Chem. Soc. Rev. 2014, 43, 7067–7098. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Xi, Y.; Liu, S.; Zhang, C. Greatly Enhancing Catalytic Activity of Graphene by Doping the Underlying Metal Substrate. Sci. Rep. 2015, 5, 12058. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Bao, X. Surface chemistry and catalysis confined under two-dimensional materials. Chem. Soc. Rev. 2017, 46, 1842–1874. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Meng, X.; Deng, D.; Bao, X. Confinement Catalysis with 2D Materials for Energy Conversion. Adv. Mater. 2019, 31, e1901996. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.G.; Tománek, D. Catalytic Growth of Single-Wall Carbon Nanotubes: An Ab Initio Study. Phys. Rev. Lett. 1997, 78, 2393–2396. [Google Scholar] [CrossRef] [Green Version]
- Gierz, I.; Riedl, C.; Starke, U.; Ast, C.R.; Kern, K. Atomic Hole Doping of Graphene. Nano Lett. 2008, 8, 4603–4607. [Google Scholar] [CrossRef] [Green Version]
- Johll, H.; Kang, H.C.; Tok, E.S. Density functional theory study of Fe, Co, and Ni adatoms and dimers adsorbed on graphene. Phys. Rev. B 2009, 79, 245416. [Google Scholar] [CrossRef]
- Krasheninnikov, A.; Lehtinen, P.O.; Foster, A.S.; Pyykkö, P.; Nieminen, R.M. Embedding Transition-Metal Atoms in Graphene: Structure, Bonding, and Magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 2008, 77, 235430. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Solis, A.; Vasiliev, I. Ab initio study of K adsorption on graphene and carbon nanotubes: Role of long-range ionic forces. Phys. Rev. B 2007, 76, 235431. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.Z.; Yao, Y.X.; Lu, W.C.; Hupalo, M.; Tringides, M.C.; Ho, K.M. Bonding and charge transfer by metal adatom adsorption on graphene. Phys. Rev. B 2011, 83, 235411. [Google Scholar] [CrossRef]
- Sevinçli, H.; Topsakal, M.; Durgun, E.; Ciraci, S. Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys. Rev. B 2008, 77, 195434. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P.; Kawazoe, Y. Clustering of Ti on a C60 Surface and Its Effect on Hydrogen Storage. J. Am. Chem. Soc. 2005, 127, 14582–14583. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Martinez, I.; Felten, A.; Pireaux, J.J.; Bittencourt, C.; Ewels, C.P. Transition metal deposition on graphene and carbon nanotubes. J. Nanosci. Nanotechnol. 2009, 9, 6171–6175. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, Q.; Cheng, Y.; Yao, Y.; Wang, Q.; Li, K.; Schwingenschlögl, U.; Zhang, X.X.; Yang, W. Atomic Bonding between Metal and Graphene. J. Phys. Chem. C 2013, 117, 4632–4638. [Google Scholar] [CrossRef]
- Mao, Y.; Yuan, J.; Zhong, J. Density functional calculation of transition metal adatom adsorption on graphene. J. Phys. Condens. Matter 2008, 20, 115209. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Jhi, S.-H.; Park, N. Effective metal dispersion in pyridinelike nitrogen doped graphenes for hydrogen storage. Appl. Phys. Lett. 2008, 92, 013106. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Jhi, S.-H.; Lim, S.; Park, N. Effect of vacancy defects in graphene on metal anchoring and hydrogen adsorption. Appl. Phys. Lett. 2009, 94, 173102. [Google Scholar] [CrossRef] [Green Version]
- Nakada, K.; Ishii, A. Migration of adatom adsorption on graphene using DFT calculation. Solid State Commun. 2011, 151, 13–16. [Google Scholar] [CrossRef]
- Banhart, F.; Kotakoski, J.; Krasheninnikov, A.V. Structural Defects in Graphene. ACS Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Ugeda, M.M.; Brihuega, I.; Guinea, F.; Gómez-Rodríguez, J.M. Missing Atom as a Source of Carbon Magnetism. Phys. Rev. Lett. 2010, 104, 096804. [Google Scholar] [CrossRef]
- Gan, Y.; Sun, L.; Banhart, F. One- and Two-Dimensional Diffusion of Metal Atoms in Graphene. Small 2008, 4, 587–591. [Google Scholar] [CrossRef]
- Rodríguez-Manzo, J.A.; Cretu, O.; Banhart, F. Trapping of Metal Atoms in Vacancies of Carbon Nanotubes and Graphene. ACS Nano 2010, 4, 3422–3428. [Google Scholar] [CrossRef] [PubMed]
- Malola, S.; Häkkinen, H.; Koskinen, P. Gold in graphene: In-plane adsorption and diffusion. Appl. Phys. Lett. 2009, 94, 43106. [Google Scholar] [CrossRef] [Green Version]
- Yoo, E.; Okata, T.; Akita, T.; Kohyama, M.; Nakamura, J.; Honma, I. Enhanced Electrocatalytic Activity of Pt Subnanoclusters on Graphene Nanosheet Surface. Nano Lett. 2009, 9, 2255–2259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, A.; Dai, Z.; Zhang, C.; Feng, Y.P. Greatly enhanced adsorption and catalytic activity of Au and Pt clusters on defective graphene. J. Chem. Phys. 2010, 132, 194704. [Google Scholar] [CrossRef] [PubMed]
- Fampiou, I.; Ramasubramaniam, A. Binding of Pt Nanoclusters to Point Defects in Graphene: Adsorption, Morphology, and Electronic Structure. J. Phys. Chem. C 2012, 116, 6543–6555. [Google Scholar] [CrossRef]
- Fampiou, I.; Ramasubramaniam, A. CO Adsorption on Defective Graphene-Supported Pt13 Nanoclusters. J. Phys. Chem. C 2013, 117, 19927–19933. [Google Scholar] [CrossRef]
- Fampiou, I.; Ramasubramaniam, A. Influence of Support Effects on CO Oxidation Kinetics on CO-Saturated Graphene-Supported Pt13 Nanoclusters. J. Phys. Chem. C 2015, 119, 8703–8710. [Google Scholar] [CrossRef]
- Lim, D.-H.; Negreira, A.S.; Wilcox, J. DFT Studies on the Interaction of Defective Graphene-Supported Fe and Al Nanoparticles. J. Phys. Chem. C 2011, 115, 8961–8970. [Google Scholar] [CrossRef]
- Song, W.; Wang, J.-L.; Wang, B.; Hu, W.-P.; Wang, Y. First-principles study on the structures and electronic properties of graphene-supported Nin (n = 1–6) clusters. Mol. Simul. 2018, 44, 1529–1538. [Google Scholar] [CrossRef]
- Wang, L.; Ma, S.; Jiao, Z.; Yuan, D. Capability of defective graphene-supported Co4 nanoparticle toward ammonia dehydrogenation. Appl. Surf. Sci. 2019, 465, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Yu, G.; Chen, W.; Chen, Z. CO Catalytic Oxidation on Iron-Embedded Graphene: Computational Quest for Low-Cost Nanocatalysts. J. Phys. Chem. C 2010, 114, 6250–6254. [Google Scholar] [CrossRef]
- Wannakao, S.; Nongnual, T.; Khongpracha, P.; Maihom, T.; Limtrakul, J. Reaction Mechanisms for CO Catalytic Oxidation by N2O on Fe-Embedded Graphene. J. Phys. Chem. C 2012, 116, 16992–16998. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Y.; Shen, Z.; Chen, W.; Li, C.; Dai, X. High catalytic activity for CO oxidation on single Fe atom stabilized in graphene vacancies. RSC Adv. 2016, 6, 93985–93996. [Google Scholar] [CrossRef]
- Song, E.H.; Wen, Z.; Jiang, Q. CO Catalytic Oxidation on Copper-Embedded Graphene. J. Phys. Chem. C 2011, 115, 3678–3683. [Google Scholar] [CrossRef]
- Tang, Y.; Yang, Z.; Dai, X. A theoretical simulation on the catalytic oxidation of CO on Pt/graphene. Phys. Chem. Chem. Phys. 2012, 14, 16566–16572. [Google Scholar] [CrossRef]
- Jiang, Q.G.; Ao, Z.M.; Li, S.; Wen, Z. Density functional theory calculations on the CO catalytic oxidation on Al-embedded graphene. RSC Adv. 2014, 4, 20290–20296. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D.; Nematollahi, P.; Abdollahpour, H. A comparative DFT study on the CO oxidation reaction over Al- and Ge-embedded graphene as efficient metal-free catalysts. Appl. Surf. Sci. 2016, 378, 418–425. [Google Scholar] [CrossRef]
- Tang, Y.; Dai, X.; Yang, Z.; Liu, Z.; Pan, L.; Ma, D.; Lu, Z. Tuning the catalytic property of non-noble metallic impurities in graphene. Carbon 2014, 71, 139–149. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Saeidi, N. Sn-embedded graphene: An active catalyst for CO oxidation to CO2? Physica E 2015, 74, 382–387. [Google Scholar] [CrossRef]
- Tang, Y.; Pan, L.; Chen, W.; Li, C.; Shen, Z.; Dai, X. Reaction mechanisms for CO catalytic oxidation on monodisperse Mo atom-embedded graphene. Appl. Phys. A 2015, 119, 475–485. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Nematollahi, P.; Nurazar, R. Pd-embedded graphene: An efficient and highly active catalyst for oxidation of CO. Superlattices Microstruct. 2016, 92, 60–67. [Google Scholar] [CrossRef]
- Xu, G.; Wang, R.; Yang, F.; Ma, D.; Yang, Z.; Lu, Z. CO oxidation on single Pd atom embedded defect-graphene via a new termolecular Eley-Rideal mechanism. Carbon 2017, 118, 35–42. [Google Scholar] [CrossRef]
- Dai, G.; Chen, L.; Zhao, X. Tungsten-Embedded Graphene: Theoretical Study on a Potential High-Activity Catalyst toward CO Oxidation. Materials 2018, 11, 1848. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, J.; Ao, Z.; Huang, H.; He, H.; Wu, Y. First Principles Study on the CO Oxidation on Mn-Embedded Divacancy Graphene. Front. Chem. 2018, 6, 187. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Chen, L.; Zhao, X. Catalytic oxidation mechanisms of carbon monoxide over single and double vacancy Cr-embedded graphene. J. Mater. Sci. 2019, 54, 1395–1408. [Google Scholar] [CrossRef]
- Zhao, J.-X.; Chen, Y.; Fu, H.-G. Si-embedded graphene: An efficient and metal-free catalyst for CO oxidation by N2O or O2. Theor. Chem. Acc. 2012, 131, 1242. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Z.; Dai, X.; Yang, Z.; Chen, W.; Ma, D.; Lu, Z. Theoretical study on the Si-doped graphene as an efficient metal-free catalyst for CO oxidation. Appl. Surf. Sci. 2014, 308, 402–407. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammad-Valipour, R.; Mousavi-Khoshdel, S.M.; Nematollahi, P. A Comparative Study of CO Oxidation on Nitrogen- and Phosphorus-Doped Graphene. ChemPhysChem 2015, 16, 3719–3727. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P. Probing reaction pathways for oxidation of CO by O2 molecule over P-doped divacancy graphene: A DFT study. Appl. Surf. Sci. 2018, 440, 580–585. [Google Scholar] [CrossRef]
- Deng, D.; Novoselov, K.S.; Fu, Q.; Zheng, N.; Tian, Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016, 11, 218–230. [Google Scholar] [CrossRef]
- Deng, D.; Pan, X.; Yu, L.; Cui, Y.; Jiang, Y.; Qi, J.; Li, W.-X.; Fu, Q.; Ma, X.; Xue, Q.; et al. Toward N-Doped Graphene via Solvothermal Synthesis. Chem. Mater. 2011, 23, 1188–1193. [Google Scholar] [CrossRef]
- Gao, Y.; Hu, G.; Zhong, J.; Shi, Z.; Zhu, Y.; Su, D.S.; Wang, J.; Bao, X.; Ma, D. Nitrogen-Doped sp2-Hybridized Carbon as a Superior Catalyst for Selective Oxidation. Angew. Chem. Int. Ed. Engl. 2013, 52, 2109–2113. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, Y.; Chen, W.; Tang, P.; Li, W.; Shi, Z.; Su, D.; Wang, J.; Ma, D. Catalytic Epoxidation Reaction over N-Containing sp2 Carbon Catalysts. ACS Catal. 2014, 4, 1261–1266. [Google Scholar] [CrossRef]
- Bangert, U.; Bleloch, A.; Gass, M.H.; Seepujak, A.; Berg, J.V.D. Doping of few-layered graphene and carbon nanotubes using ion implantation. Phys. Rev. B 2010, 81, 245423. [Google Scholar] [CrossRef]
- Robertson, A.W.; Allen, C.S.; Wu, Y.A.; He, K.; Olivier, J.; Neethling, J.; Kirkland, A.I.; Warner, J.H. Spatial control of defect creation in graphene at the nanoscale. Nat. Commun. 2012, 3, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.W.; Montanari, B.; He, K.; Kim, J.; Allen, C.S.; Wu, Y.A.; Olivier, J.; Neethling, J.; Harrison, N.; Kirkland, A.I.; et al. Dynamics of Single Fe Atoms in Graphene Vacancies. Nano Lett. 2013, 13, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Cheng, H.; Yi, H.; Lin, Y.; Yao, T.; Wang, C.; Li, J.; Wei, S.; Lu, J. Single-Atom Pd(1)/Graphene Catalyst Achieved by Atomic Layer Deposition: Remarkable Performance in Selective Hydrogenation of 1,3-Butadiene. J. Am. Chem. Soc. 2015, 137, 10484–10487. [Google Scholar] [CrossRef]
- Yan, H.; Lin, Y.; Wu, H.; Zhang, W.; Sun, Z.; Cheng, H.; Liu, W.; Wang, C.; Li, J.; Huang, X.; et al. Bottom-up precise synthesis of stable platinum dimers on graphene. Nat. Commun. 2017, 8, 1070. [Google Scholar] [CrossRef]
- Yan, H.; Zhao, X.; Guo, N.; Lyu, Z.; Du, Y.; Xi, S.; Guo, R.; Chen, C.; Chen, Z.; Liu, W.; et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 2018, 9, 3197. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Siahrostami, S.; Akey, A.J.; Li, Y.; Lu, Z.; Lattimer, J.; Hu, Y.; Stokes, C.; Gangishetty, M.; Chen, G.; et al. Transition-Metal Single Atoms in a Graphene Shell as Active Centers for Highly Efficient Artificial Photosynthesis. Chem 2017, 3, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jia, Y.; Gao, G.; Yan, X.; Chen, N.; Chen, J.; Soo, M.T.; Wood, B.; Yang, D.; Du, A.; et al. Graphene Defects Trap Atomic Ni Species for Hydrogen and Oxygen Evolution Reactions. Chem 2018, 4, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhao, J.; Chen, Z. Fe-Anchored Graphene Oxide: A Low-Cost and Easily Accessible Catalyst for Low-Temperature CO Oxidation. J. Phys. Chem. C 2012, 116, 2507–2514. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, M.; Zhang, A.; Zhang, C. Graphene Oxide: An Ideal Support for Gold Nanocatalysts. J. Phys. Chem. C 2012, 116, 22336–22340. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; López, N.; Pérez-Ramírez, J. A Stable Single-Site Palladium Catalyst for Hydrogenations. Angew. Chem. Int. Ed. 2015, 54, 11265–11269. [Google Scholar] [CrossRef]
- Thomas, J.M. Tens of thousands of atoms replaced by one. Nature 2015, 525, 325–326. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, Y.; Shen, Y.; Liu, S.; Zhang, Y. Molecular engineering of polymeric carbon nitride: Advancing applications from photocatalysis to biosensing and more. Chem. Soc. Rev. 2018, 47, 2298–2321. [Google Scholar] [CrossRef]
- Rivera-Cárcamo, C.; Serp, P. Single Atom Catalysts on Carbon-Based Materials. ChemCatChem 2018, 10, 5058–5091. [Google Scholar] [CrossRef]
- Deng, D.; Chen, X.; Yu, L.; Wu, X.; Liu, Q.; Liu, Y.; Yang, H.; Tian, H.; Hu, Y.; Du, P.; et al. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 2015, 1, e1500462. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Huang, X.; Xi, S.; Miao, S.; Ding, J.; Cai, W.; Liu, S.; Yang, X.; Yang, H.; Gao, J.; et al. Single Cobalt Atoms Anchored on Porous N-Doped Graphene with Dual Reaction Sites for Efficient Fenton-like Catalysis. J. Am. Chem. Soc. 2018, 140, 12469–12475. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Xi, S.; Du, Y.; Hai, X.; Wang, J.; Xu, H.; Wu, G.; Zhang, J.; Lu, J.; et al. A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. Engl. 2019, 58, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, W.; Shen, Z.; Chang, S.; Zhao, M.; Dai, X. Nitrogen coordinated silicon-doped graphene as a potential alternative metal-free catalyst for CO oxidation. Carbon 2017, 111, 448–458. [Google Scholar] [CrossRef]
- Li, X.-F.; Li, Q.-K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.; Zhang, X.-H.; Wang, Z.-Y.; Qiu, Q.; Luo, Y. Conversion of Dinitrogen to Ammonia by FeN3-Embedded Graphene. J. Am. Chem. Soc. 2016, 138, 8706–8709. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, A.; Dai, Z.; Feng, Y.P.; Zhang, C. Strain-Enhanced Stabilization and Catalytic Activity of Metal Nanoclusters on Graphene. J. Phys. Chem. C 2010, 114, 16541–16546. [Google Scholar] [CrossRef]
- Yin, H.; Tang, H.; Wang, D.; Gao, Y.; Tang, Z. Facile Synthesis of Surfactant-Free Au Cluster/Graphene Hybrids for High-Performance Oxygen Reduction Reaction. ACS Nano 2012, 6, 8288–8297. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Fu, Q.; Zhang, Y.-Y.; Weng, X.; Li, H.; Chen, M.; Jin, L.; Dong, A.; Mu, R.; Jiang, P.; et al. Graphene cover-promoted metal-catalyzed reactions. Proc. Natl. Acad. Sci. USA 2014, 111, 17023–17028. [Google Scholar] [CrossRef] [Green Version]
- Sutter, P.; Sadowski, J.T.; Sutter, E.A. Chemistry under Cover: Tuning Metal–Graphene Interaction by Reactive Intercalation. J. Am. Chem. Soc. 2010, 132, 8175–8179. [Google Scholar] [CrossRef]
- Sutter, P.W.; Flege, J.-I.; Sutter, E.A. Epitaxial graphene on ruthenium. Nat. Mater. 2008, 7, 406–411. [Google Scholar] [CrossRef]
- Stradi, D.; Barja, S.; Diaz, C.; Garnica, M.; Borca, B.; Hinarejos, J.J.; Sánchez-Portal, D.; Alcami, M.; Arnau, A.; De Parga, A.L.V.; et al. Lattice-matched versus lattice-mismatched models to describe epitaxial monolayer graphene on Ru(0001). Phys. Rev. B 2013, 88, 245401. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yam, K.M.; Guo, N.; Jiang, Z.; Li, S.; Zhang, C. Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts 2020, 10, 53. https://doi.org/10.3390/catal10010053
Yam KM, Guo N, Jiang Z, Li S, Zhang C. Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts. 2020; 10(1):53. https://doi.org/10.3390/catal10010053
Chicago/Turabian StyleYam, Kah Meng, Na Guo, Zhuoling Jiang, Shulong Li, and Chun Zhang. 2020. "Graphene-Based Heterogeneous Catalysis: Role of Graphene" Catalysts 10, no. 1: 53. https://doi.org/10.3390/catal10010053
APA StyleYam, K. M., Guo, N., Jiang, Z., Li, S., & Zhang, C. (2020). Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts, 10(1), 53. https://doi.org/10.3390/catal10010053