The HD-Domain Metalloprotein Superfamily: An Apparent Common Protein Scaffold with Diverse Chemistries


 ,
, 
 , ,
, , 
Abstract
1. Introduction
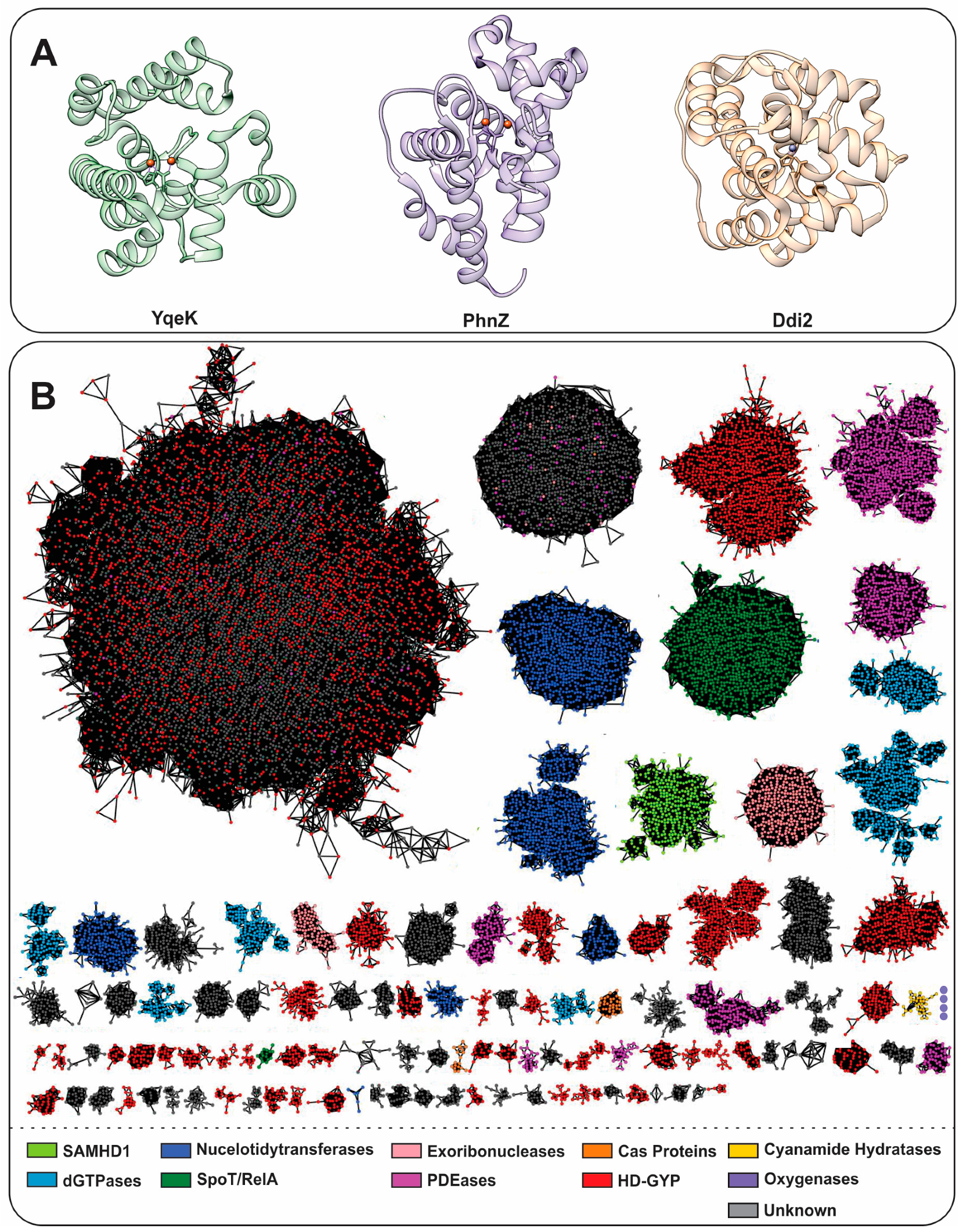
1.1. Characteristics and General Classification of HD-Domain Proteins
1.2. HD-Domain Hydrolases: The Phosphatase Subfamily
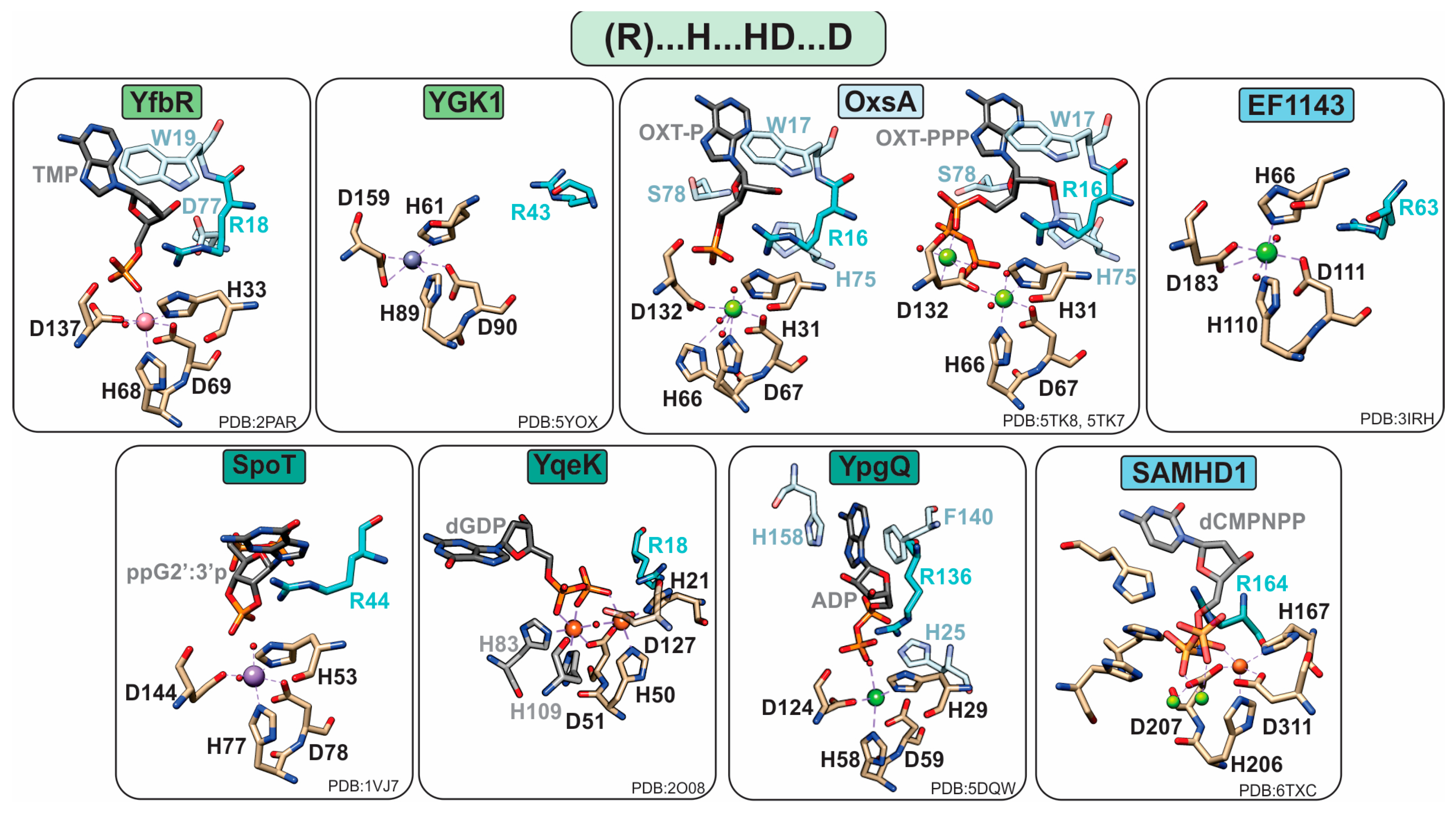
1.2.1. Monophosphatases
YfbR and YGK1
1.2.2. Diphosphatases
YqeK
YpgQ
SpoT/RelA
1.2.3. Triphosphatases
SAMHD1
EF1143
OxsA
1.3. HD-Domain Hydrolases: The PDE Subfamily
1.3.1. HD-Domain PDEs Acting on DNA
1.3.2. HD-Domain PDEs Acting on Cyclic Mononucleotides
1.3.3. HD-Domain PDEs Acting on c-di-AMP
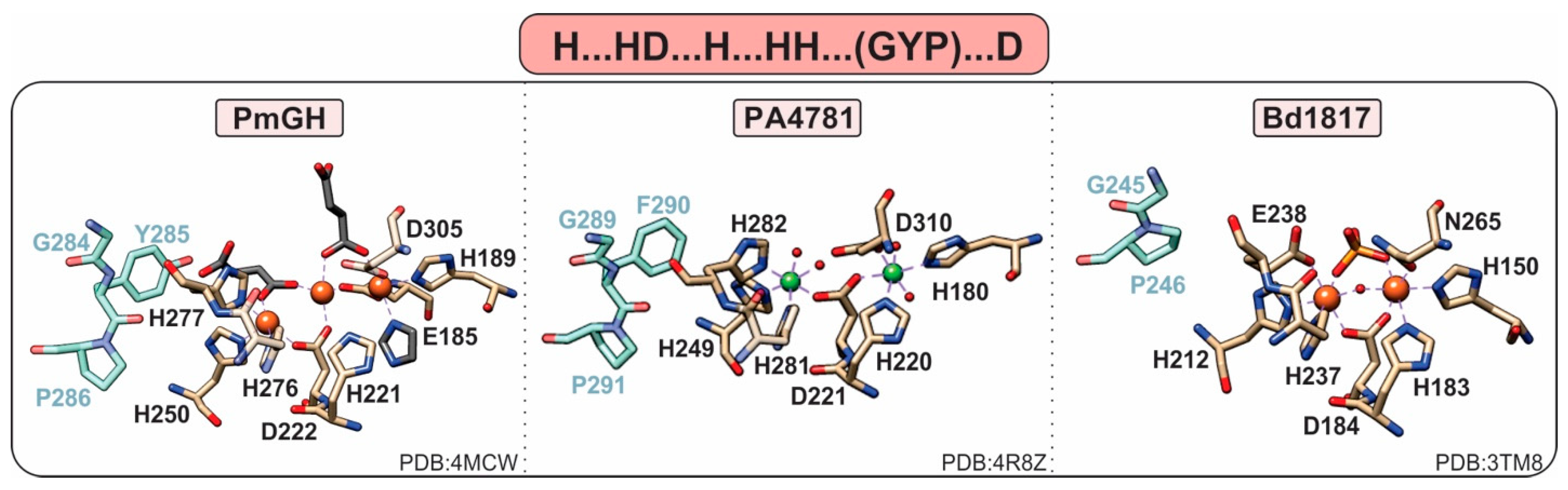
1.3.4. HD-Domain PDEs Acting on c-di-GMP and c-GAMP; the HD-GYP Subclass
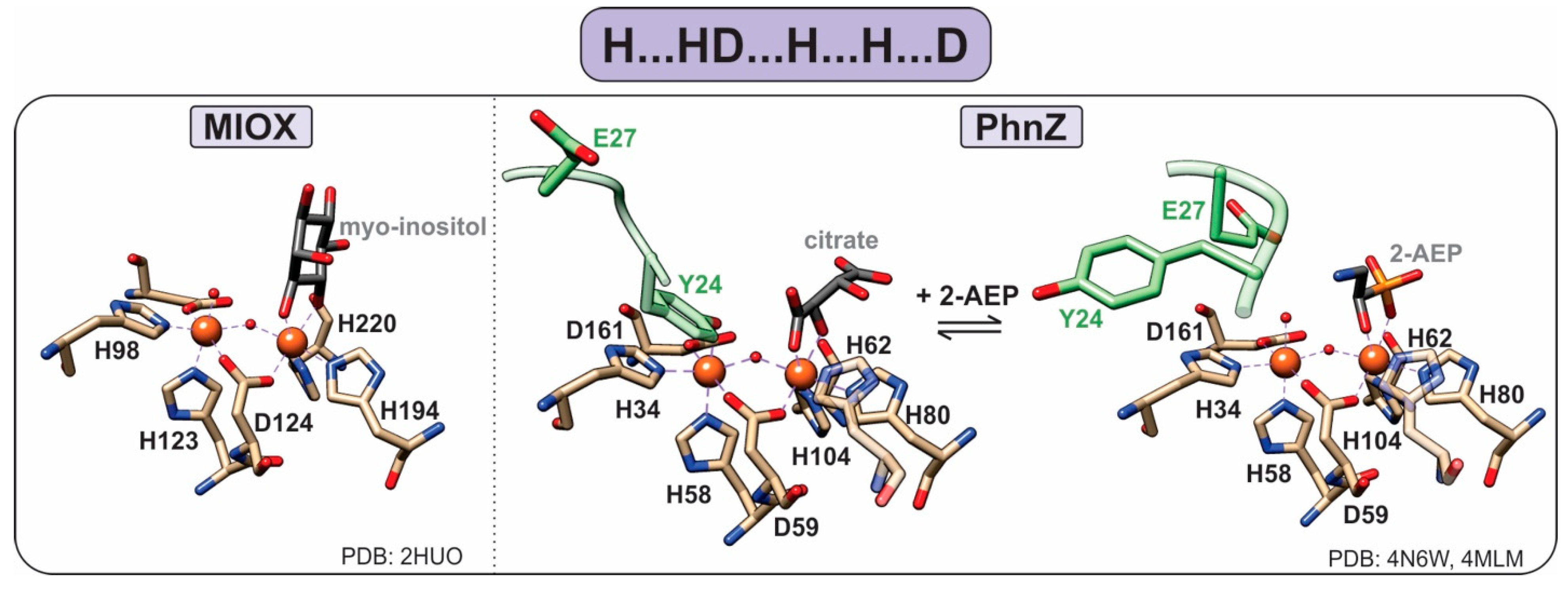
1.4. HD-Domain Oxygenases
1.5. Lyases
The HD-Domain Cyanamide Hydratase Ddi2
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Wu, L. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol. 2018, 27, 254–267. [Google Scholar] [CrossRef]
- Zimmerman, M.D.; Proudfoot, M.; Yakunin, A.; Minor, W. Structural Insight into the Mechanism of Substrate Specificity and Catalytic Activity of an HD-Domain Phosphohydrolase: The 5′-Deoxyribonucleotidase YfbR from Escherichia coli. J. Mol. Biol. 2008, 378, 215–226. [Google Scholar] [CrossRef]
- Ji, X.; Tang, C.; Zhao, Q.; Wang, W.; Xiong, Y. Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci. USA 2014, 111, E4305–E4314. [Google Scholar] [CrossRef]
- Yang, J.; Wang, F.; Yang, D.; Zhou, K.; Liu, M.; Gao, Z.; Liu, P.; Dong, Y.; Zhang, J.; Liu, Q. Structural and biochemical characterization of the yeast HD domain containing protein YGK1 reveals a metal-dependent nucleoside 5ʹ-monophosphatase. Biochem. Biophys. Res. Commun. 2018, 501, 674–681. [Google Scholar] [CrossRef]
- Huynh, T.; Luo, S.; Pensinger, D.; Sauer, J.-D.; Tong, L.; Woodward, J. An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc. Natl. Acad. Sci. USA 2015, 112, E747–E756. [Google Scholar] [CrossRef]
- Dow, J.M.; Fouhy, Y.; Lucey, J.F.; Ryan, R.P. The HD-GYP Domain, Cyclic Di-GMP Signaling, and Bacterial Virulence to Plants. Mol. Plant-Microbe Interact. 2006, 19, 1378–1384. [Google Scholar] [CrossRef]
- Ryan, R.P.; McCarthy, Y.; Andrade, M.; Farah, C.S.; Armitage, J.P.; Dow, J.M. Cell-cell signal-dependent dynamic interactions between HD-GYP and GGDEF domain proteins mediate virulence in Xanthomonas campestris. Proc. Natl. Acad. Sci. USA 2010, 107, 5989–5994. [Google Scholar] [CrossRef]
- Atkinson, G.C.; Tenson, T.; Hauryliuk, V. The RelA/SpoT Homolog (RSH) Superfamily: Distribution and Functional Evolution of ppGpp Synthetases and Hydrolases across the Tree of Life. PLoS ONE 2011, 6, e23479. [Google Scholar] [CrossRef]
- Minazzato, G.; Gasparrini, M.; Amici, A.; Cianci, M.; Mazzola, F.; Orsomando, G.; Sorci, L.; Raffaelli, N. Functional Characterization of COG1713 (YqeK) as a Novel Diadenosine Tetraphosphate Hydrolase Family. J. Bacteriol. 2020, 202, e00053-20. [Google Scholar] [CrossRef]
- Brown, P.M.; Caradoc-Davies, T.T.; Dickson, J.M.J.; Cooper, G.J.S.; Loomes, K.M.; Baker, E.N. Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc. Natl. Acad. Sci. USA 2006, 103, 15032–15037. [Google Scholar] [CrossRef]
- Wörsdörfer, B.; Lingaraju, M.; Yennawar, N.H.; Boal, A.K.; Krebs, C.; Bollinger, J.M.; Pandelia, M.-E. Organophosphonate-degrading PhnZ reveals an emerging family of HD domain mixed-valent diiron oxygenases. Proc. Natl. Acad. Sci. USA 2013, 110, 18874–18879. [Google Scholar] [CrossRef] [PubMed]
- Rajakovich, L.J.; Pandelia, M.-E.; Mitchell, A.J.; Chang, W.-C.; Zhang, B.; Boal, A.K.; Krebs, C.; Bollinger, J.M. A New Microbial Pathway for Organophosphonate Degradation Catalyzed by Two Previously Misannotated Non-Heme-Iron Oxygenases. Biochemistry 2019, 58, 1627–1647. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, S.; Paiardini, A.; Stelitano, V.; Brunotti, P.; Cervoni, L.; Fernicola, S.; Protano, C.; Vitali, M.; Cutruzzolà, F.; Giardina, G. Structural Basis of Functional Diversification of the HD-GYP Domain Revealed by the Pseudomonas aeruginosa PA4781 Protein, Which Displays an Unselective Bimetallic Binding Site. J. Bacteriol. 2015, 197, 1525–1535. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Staalduinen, L.M.; McSorley, F.R.; Schiessl, K.; Séguin, J.; Wyatt, P.B.; Hammerschmidt, F.; Zechel, D.L.; Jia, Z. Crystal structure of PhnZ in complex with substrate reveals a di-iron oxygenase mechanism for catabolism of organophosphonates. Proc. Natl. Acad. Sci. USA 2014, 111, 5171–5176. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Park, S.C.; Song, W.S.; Kim, O.-H.; Oh, B.-C.; Yoon, S.-I. Structural and biochemical characterization of bacterial YpgQ protein reveals a metal-dependent nucleotide pyrophosphohydrolase. J. Struct. Biol. 2016, 195, 113–122. [Google Scholar] [CrossRef]
- Hogg, T.; Mechold, U.; Malke, H.; Cashel, M.; Hilgenfeld, R. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response. Cell 2004, 117, 57–68. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Morris, E.R.; Caswell, S.J.; Kunzelmann, S.; Arnold, L.H.; Purkiss, A.G.; Kelly, G.; Taylor, I.A. Crystal structures of SAMHD1 inhibitor complexes reveal the mechanism of water-mediated dNTP hydrolysis. Nat. Commun. 2020, 11, 3165. [Google Scholar] [CrossRef]
- Vorontsov, I.I.; Minasov, G.; Kiryukhina, O.; Brunzelle, J.S.; Shuvalova, L.; Anderson, W.F. Characterization of the deoxynucleotide triphosphate triphosphohydrolase (dNTPase) activity of the EF1143 protein from Enterococcus faecalis and crystal structure of the activator-substrate complex. J. Biol. Chem. 2011, 286, 33158–33166. [Google Scholar] [CrossRef]
- Vorontsov, I.I.; Wu, Y.; DeLucia, M.; Minasov, G.; Mehrens, J.; Shuvalova, L.; Anderson, W.F.; Ahn, J. Mechanisms of Allosteric Activation and Inhibition of the Deoxyribonucleoside Triphosphate Triphosphohydrolase from Enterococcus faecalis. J. Biol. Chem. 2014, 289, 2815–2824. [Google Scholar] [CrossRef]
- Bridwell-Rabb, J.; Kang, G.; Zhong, A.; Liu, H.-w.; Drennan, C.L. An HD domain phosphohydrolase active site tailored for oxetanocin-A biosynthesis. Proc. Natl. Acad. Sci. USA 2016, 113, 13750–13755. [Google Scholar] [CrossRef]
- Huo, Y.; Nam, K.H.; Ding, F.; Lee, H.; Wu, L.; Xiao, Y.; Farchione, M.D.; Zhou, S.; Rajashankar, K.; Kurinov, I.; et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014, 21, 771–777. [Google Scholar] [CrossRef]
- Gong, B.; Shin, M.; Sun, J.; Jung, C.H.; Bolt, E.L.; van der Oost, J.; Kim, J.S. Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc. Natl. Acad. Sci. USA 2014, 111, 16359–16364. [Google Scholar] [CrossRef]
- Beloglazova, N.; Petit, P.; Flick, R.; Brown, G.; Savchenko, A.; Yakunin, A.F. Structure and activity of the Cas3 HD nuclease MJ0384, an effector enzyme of the CRISPR interference. EMBO J. 2011, 30, 4616–4627. [Google Scholar] [CrossRef]
- Benda, C.; Ebert, J.; Scheltema, R.A.; Schiller, H.B.; Baumgärtner, M.; Bonneau, F.; Mann, M.; Conti, E. Structural Model of a CRISPR RNA-Silencing Complex Reveals the RNA-Target Cleavage Activity in Cmr4. Mol. Cell 2014, 56, 43–54. [Google Scholar] [CrossRef]
- Pandit, J.; Forman, M.D.; Fennell, K.F.; Dillman, K.S.; Menniti, F.S. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. USA 2009, 106, 18225–18230. [Google Scholar] [CrossRef]
- Scapin, G.; Patel, S.B.; Chung, C.; Varnerin, J.P.; Edmondson, S.D.; Mastracchio, A.; Parmee, E.R.; Singh, S.B.; Becker, J.W.; Van der Ploeg, L.H.T.; et al. Crystal Structure of Human Phosphodiesterase 3B: Atomic Basis for Substrate and Inhibitor Specificity. Biochemistry 2004, 43, 6091–6100. [Google Scholar] [CrossRef]
- Xu, R.X.; Hassell, A.M.; Vanderwall, D.; Lambert, M.H.; Holmes, W.D.; Luther, M.A.; Rocque, W.J.; Milburn, M.V.; Zhao, Y.; Ke, H.; et al. Atomic Structure of PDE4: Insights into Phosphodiesterase Mechanism and Specificity. Science 2000, 288, 1822–1825. [Google Scholar] [CrossRef]
- Zhang, K.Y.J.; Card, G.L.; Suzuki, Y.; Artis, D.R.; Fong, D.; Gillette, S.; Hsieh, D.; Neiman, J.; West, B.L.; Zhang, C.; et al. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol. Cell 2004, 15, 279–286. [Google Scholar] [CrossRef]
- Kawai, K.; Endo, Y.; Asano, T.; Amano, S.; Sawada, K.; Ueo, N.; Takahashi, N.; Sonoda, Y.; Nagai, M.; Kamei, N.; et al. Discovery of 2-(cyclopentylamino)thieno [3,2-d]pyrimidin-4(3H)-one derivatives as a new series of potent phosphodiesterase 7 inhibitors. J. Med. Chem. 2014, 57, 9844–9854. [Google Scholar] [CrossRef]
- Wang, H.; Yan, Z.; Yang, S.; Cai, J.; Robinson, H.; Ke, H. Kinetic and Structural Studies of Phosphodiesterase-8A and Implication on the Inhibitor Selectivity. Biochemistry 2008, 47, 12760–12768. [Google Scholar] [CrossRef]
- Liu, S.; Mansour, M.N.; Dillman, K.S.; Perez, J.R.; Danley, D.E.; Aeed, P.A.; Simons, S.P.; LeMotte, P.K.; Menniti, F.S. Structural basis for the catalytic mechanism of human phosphodiesterase 9. Proc. Natl. Acad. Sci. USA 2008, 105, 13309–13314. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Hou, J.; Zheng, M.; Robinson, H.; Ke, H. Structural insight into substrate specificity of phosphodiesterase 10. Proc. Natl. Acad. Sci. USA 2007, 104, 5782–5787. [Google Scholar] [CrossRef]
- Lovering, A.L.; Capeness, M.J.; Lambert, C.; Hobley, L.; Sockett, R.E. The Structure of an Unconventional HD-GYP Protein from Bdellovibrio Reveals the Roles of Conserved Residues in this Class of Cyclic-di-GMP Phosphodiesterases. mBio 2011, 2, e00163-11. [Google Scholar] [CrossRef]
- Bellini, D.; Caly, D.L.; McCarthy, Y.; Bumann, M.; An, S.-Q.; Dow, J.M.; Ryan, R.P.; Walsh, M.A. Crystal structure of an HD-GYP domain cyclic-di-GMP phosphodiesterase reveals an enzyme with a novel trinuclear catalytic iron centre. Mol. Microbiol. 2014, 91, 26–38. [Google Scholar] [CrossRef]
- Li, J.; Jia, Y.; Lin, A.; Hanna, M.; Chelico, L.; Xiao, W.; Moore, S.A. Structure of Ddi2, a highly inducible detoxifying metalloenzyme from Saccharomyces cerevisiae. J. Biol. Chem. 2019, 294, 10674–10685. [Google Scholar] [CrossRef]
- Barnes, C.O.; Wu, Y.; Song, J.; Lin, G.; Baxter, E.L.; Brewster, A.S.; Nagarajan, V.; Holmes, A.; Soltis, S.M.; Sauter, N.K.; et al. The crystal structure of dGTPase reveals the molecular basis of dGTP selectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 9333–9339. [Google Scholar] [CrossRef]
- Oussenko, I.A.; Sanchez, R.; Bechhofer, D.H. Bacillus subtilis YhaM, a member of a new family of 3′-to-5′ exonucleases in gram-positive bacteria. J. Bacteriol. 2002, 184, 6250–6259. [Google Scholar] [CrossRef]
- Sun, S.; Pandelia, M.-E. HD-[HD-GYP] Phosphodiesterases: Activities and Evolutionary Diversification within the HD-GYP Family. Biochemistry 2020, 59, 2340–2350. [Google Scholar] [CrossRef]
- Biswas, K.H.; Sopory, S.; Visweswariah, S.S. The GAF Domain of the cGMP-Binding, cGMP-Specific Phosphodiesterase (PDE5) Is a Sensor and a Sink for cGMP. Biochemistry 2008, 47, 3534–3543. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef]
- Ji, X.; Zou, J.; Peng, H.; Stolle, A.-S.; Xie, R.; Zhang, H.; Peng, B.; Mekalanos, J.J.; Zheng, J. Alarmone Ap4A is elevated by aminoglycoside antibiotics and enhances their bactericidal activity. Proc. Natl. Acad. Sci. USA 2019, 116, 9578–9585. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, F.C. (Ed.) The Stringent Response. In Escherichia coli and Salmonella: Cellular and Molecular Biology; ASM Press: Washington, DC, USA, 1996; pp. 1458–1496. [Google Scholar]
- Xiao, H.; Kalman, M.; Ikehara, K.; Zemel, S.; Glaser, G.; Cashel, M. Residual guanosine 3′,5′-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J. Biol. Chem. 1991, 266, 5980–5990. [Google Scholar]
- Mauney, C.H.; Hollis, T. SAMHD1: Recurring roles in cell cycle, viral restriction, cancer, and innate immunity. Autoimmunity 2018, 51, 96–110. [Google Scholar] [CrossRef]
- Ji, X.; Wu, Y.; Yan, J.; Mehrens, J.; Yang, H.; DeLucia, M.; Hao, C.; Gronenborn, A.M.; Skowronski, J.; Ahn, J.; et al. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol. 2013, 20, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Hollenbaugh, J.A.; Shelton, J.; Tao, S.; Amiralaei, S.; Liu, P.; Lu, X.; Goetze, R.W.; Zhou, L.; Nettles, J.H.; Schinazi, R.F.; et al. Substrates and Inhibitors of SAMHD1. PLoS ONE 2017, 12, e0169052. [Google Scholar] [CrossRef]
- Hansen, E.C.; Seamon, K.J.; Cravens, S.L.; Stivers, J.T. GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc. Natl. Acad. Sci. USA 2014, 111, E1843–E1851. [Google Scholar] [CrossRef]
- Morisaka, H.; Yoshimi, K.; Okuzaki, Y.; Gee, P.; Kunihiro, Y.; Sonpho, E.; Xu, H.; Sasakawa, N.; Naito, Y.; Nakada, S.; et al. CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 2019, 10, 5302. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Boswell-Smith, V.; Spina, D.; Page, C.P. Phosphodiesterase inhibitors. Br. J. Pharmacol. 2006, 147, S252–S257. [Google Scholar] [CrossRef]
- Eskandari, N.; Mirmosayyeb, O.; Bordbari, G.; Bastan, R.; Yousefi, Z.; Andalib, A. A short review on structure and role of cyclic-3′,5′-adenosine monophosphate-specific phosphodiesterase 4 as a treatment tool. J. Res. Pharm. Pract. 2015, 4, 175–181. [Google Scholar] [CrossRef]
- Heikaus, C.C.; Pandit, J.; Klevit, R.E. Cyclic nucleotide binding GAF domains from phosphodiesterases: Structural and mechanistic insights. Structure 2009, 17, 1551–1557. [Google Scholar] [CrossRef]
- Tian, Y.; Cui, W.; Huang, M.; Robinson, H.; Wan, Y.; Wang, Y.; Ke, H. Dual specificity and novel structural folding of yeast phosphodiesterase-1 for hydrolysis of second messengers cyclic adenosine and guanosine 3′,5′-monophosphate. Biochemistry 2014, 53, 4938–4945. [Google Scholar] [CrossRef]
- Luo, Y.; Helmann, J.D. Analysis of the role of Bacillus subtilis σ(M) in β-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 2012, 83, 623–639. [Google Scholar] [CrossRef]
- Fahmi, T.; Port, G.C.; Cho, K.H. c-di-AMP: An Essential Molecule in the Signaling Pathways that Regulate the Viability and Virulence of Gram-Positive Bacteria. Genes 2017, 8, 197. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Natale, D.A.; Aravind, L.; Koonin, E.V. A specialized version of the HD hydrolase domain implicated in signal transduction. J. Mol. Microbiol. Biotechnol. 1999, 1, 303–305. [Google Scholar]
- Gao, J.; Tao, J.; Liang, W.; Zhao, M.; Du, X.; Cui, S.; Duan, H.; Kan, B.; Su, X.; Jiang, Z. Identification and characterization of phosphodiesterases that specifically degrade 3′3′-cyclic GMP-AMP. Cell Res. 2015, 25, 539–550. [Google Scholar] [CrossRef]
- Wright, T.A.; Jiang, L.; Park, J.J.; Anderson, W.A.; Chen, G.; Hallberg, Z.F.; Nan, B.; Hammond, M.C. Second messengers and divergent HD-GYP phosphodiesterases regulate 3′,3′-cGAMP signaling. Mol. Microbiol. 2019, 113, 222–236. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclasses | Protein | Nuclearity | Active Metal | Chemistry | Substrate | PDB ID | Origin | References |
|---|---|---|---|---|---|---|---|---|
| oxygenases | MIOX | dinuclear | Fe | Oxygenase | myo-inositol | 2HUO | Mus musculus | [11] |
| PhnZ | dinuclear | Fe | Oxygenase | OH-AEP | 4MLM | bacterium HF130_AEPn_1 | [12,15] | |
| TmpB | dinuclear | Fe | Oxygenase | OH-TMAEP | 6NPA | Leisingera caerulea | [13] | |
| phosphatases | YfbR | mononuclear | Co | Monophosphatase | dAMP | 2PAQ | Escherichia coli K-12 | [3] |
| YGK1 | mononuclear | Mn | Monophosphatase | dNMP | 5YOX | Saccharomyces cerevisiae | [5] | |
| YqeK | dinuclear | Fe | Diphosphatase | Ap4A | 2O08 | Bacillus halodurans | [10] | |
| YpgQ | mononuclear | Mn | Diphosphatase | dNTP | 5DQV | Bacillus subtilis | [16] | |
| SpoT | mononuclear | Mn | Diphosphatase | (p)ppGpp | 1VJ7 | Streptococcus dysgalactiae | [17] | |
| SAMHD1 | mononuclear | Mg | Triphosphatase | dNTP | 3U1N | Homo sapiens | [18,19] | |
| EF1143 | mononuclear | Mg | Triphosphatase | dNTP | 4LRL | Enterococcus faecalis V583 | [20,21] | |
| OxsA | Mono/dinuclear * | Co | Mono/Di/Triphosphatase | Oxetanocin-A | 5TK8 | Bacillus megaterium | [22] | |
| phosphodiesterases | Cas3 | dinuclear | Co | PDE | ssDNA | 4QQW | Thermobifida fusca YX | [23] |
| Cas3 | dinuclear | Ni | PDE | ssDNA | 4Q2C | Thermobaculum terrenum | [24] | |
| Cas3’’ | dinuclear | Ca | PDE | ssDNA | 3S4L | Methanocaldococcus jannaschii | [25] | |
| Cas10 | dinuclear | Ni, Mn | PDE | ssDNA | 4W8Y | Pyrococcus furiosus | [26] | |
| PDE1-3 | dinuclear # | Mg, Mn | PDE | cAMP, cGMP | 1TAZ, 3ITU, 1SO2 | Homo sapiens | [27,28] | |
| PDE4 | dinuclear # | Mg, Mn | PDE | cAMP | 1F0J | Homo sapiens | [29] | |
| PDE5 | dinuclear # | Mg | PDE | cGMP | 1TBF | Homo sapiens | [30] | |
| PDE7-8 | dinuclear # | Mn | PDE | cAMP | 4PM0, 3ECM | Homo sapiens | [31,32] | |
| PDE9 | dinuclear # | Mn, Mg | PDE | cGMP | 3DY8 | Homo sapiens | [33] | |
| PDE10 | dinuclear # | Mg | PDE | cAMP, cGMP | 2OUN | Homo sapiens | [34] | |
| PgpH | dinuclear | Mn | PDE | c-di-AMP | 4S1B | Listeria monocytogenes | [6] | |
| Bd1817 | dinuclear | Fe * | PDE | c-di-GMP | 3TM8 | Bdellovibrio bacteriovorus | [35] | |
| PmGH | trinuclear | Fe, Mn | PDE | c-di-GMP | 4MCW | Persephonella marina EX-H1 | [36] | |
| PA4781 | trinuclear & | Mg | PDE | c-di-GMP | 4R8Z | Pseudomonas aeruginosa PAO1 | [14] | |
| lyases | Ddi2 | mononuclear | Zn | Hydratase | Cyanamide | 6DKA | Saccharomyces cerevisiae | [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langton, M.; Sun, S.; Ueda, C.; Markey, M.; Chen, J.; Paddy, I.; Jiang, P.; Chin, N.; Milne, A.; Pandelia, M.-E. The HD-Domain Metalloprotein Superfamily: An Apparent Common Protein Scaffold with Diverse Chemistries. Catalysts 2020, 10, 1191. https://doi.org/10.3390/catal10101191
Langton M, Sun S, Ueda C, Markey M, Chen J, Paddy I, Jiang P, Chin N, Milne A, Pandelia M-E. The HD-Domain Metalloprotein Superfamily: An Apparent Common Protein Scaffold with Diverse Chemistries. Catalysts. 2020; 10(10):1191. https://doi.org/10.3390/catal10101191
Chicago/Turabian StyleLangton, Michelle, Sining Sun, Chie Ueda, Max Markey, Jiahua Chen, Isaac Paddy, Paul Jiang, Natalie Chin, Amy Milne, and Maria-Eirini Pandelia. 2020. "The HD-Domain Metalloprotein Superfamily: An Apparent Common Protein Scaffold with Diverse Chemistries" Catalysts 10, no. 10: 1191. https://doi.org/10.3390/catal10101191
APA StyleLangton, M., Sun, S., Ueda, C., Markey, M., Chen, J., Paddy, I., Jiang, P., Chin, N., Milne, A., & Pandelia, M.-E. (2020). The HD-Domain Metalloprotein Superfamily: An Apparent Common Protein Scaffold with Diverse Chemistries. Catalysts, 10(10), 1191. https://doi.org/10.3390/catal10101191