Reactivity and Mechanism of Photo- and Electrocatalytic Hydrogen Evolution by a Diimine Copper(I) Complex

 , ,
, ,  , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
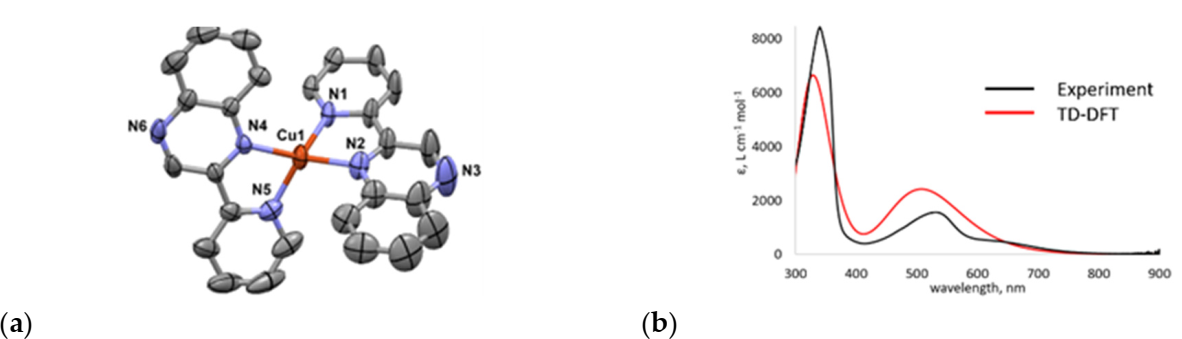
2.1. Synthesis
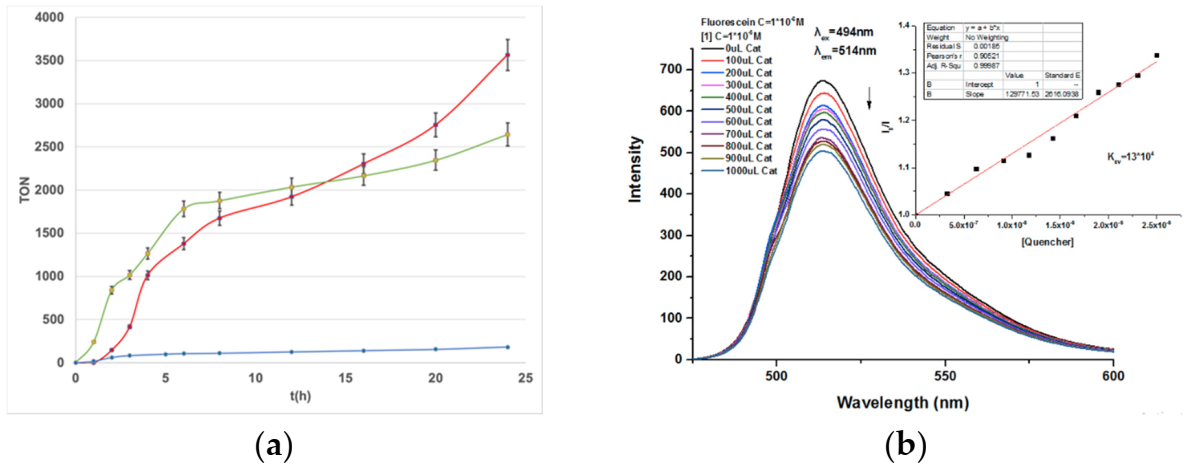
2.2. Photocatalytic H2 Production
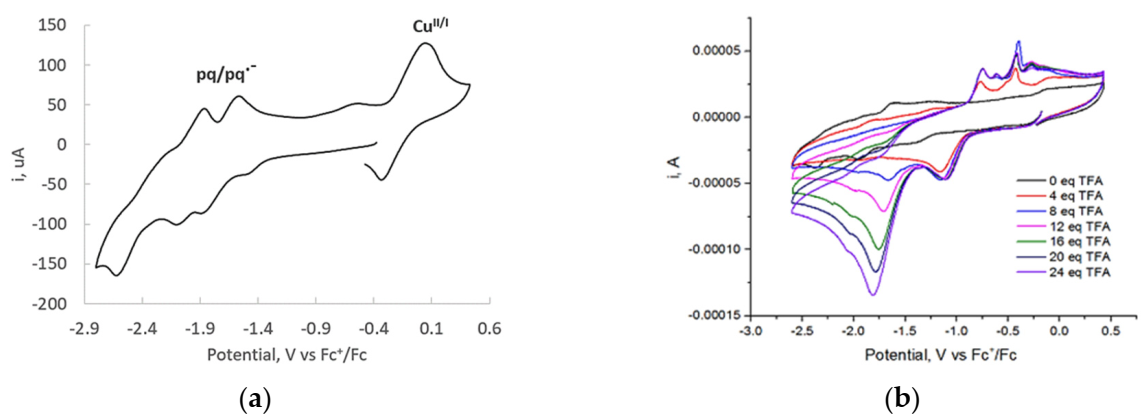
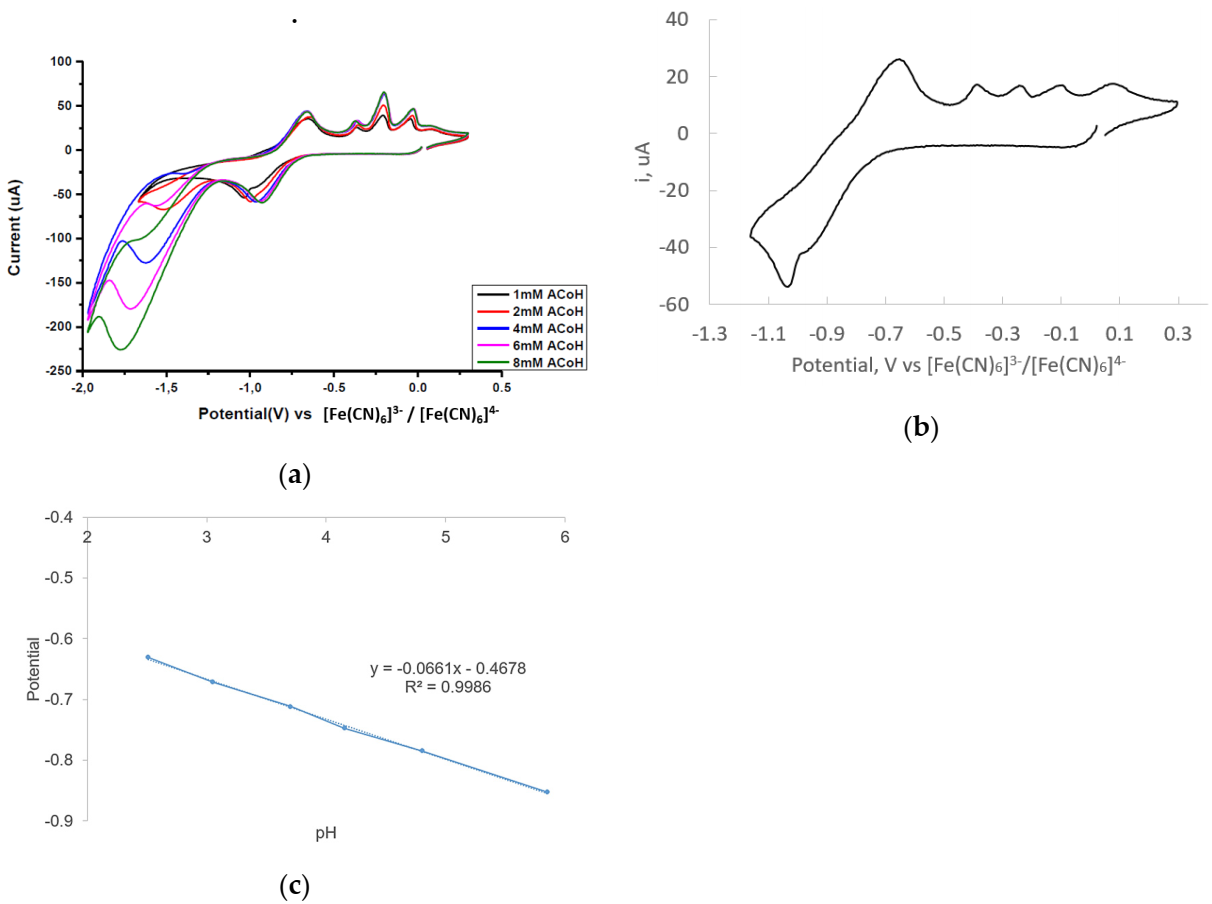
2.3. Electrocatalytic Proton Reduction
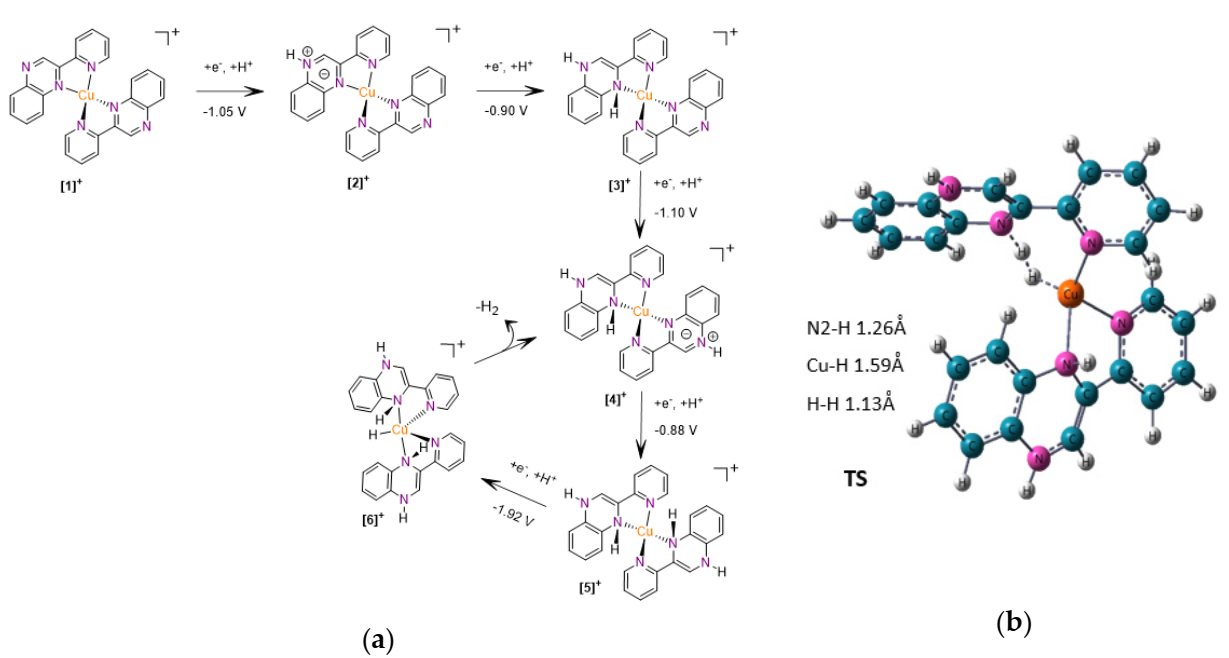
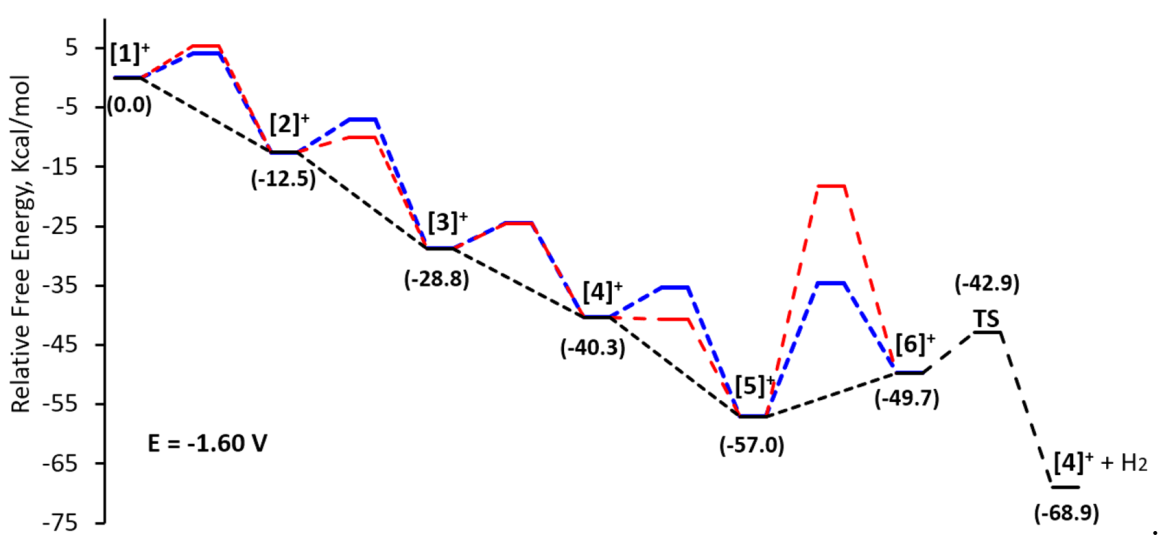
2.4. Computational Study of the Catalytic Mechanism
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Synthesis
4.3. Cyclic Voltammetry
4.4. Theoretical Studies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paria, S.; Reiser, O. Copper in Photocatalysis. ChemCatChem 2014, 6, 2477–2483. [Google Scholar] [CrossRef]
- Chen, Z.; Meyer, T.J. Copper(II) Catalysis of Water Oxidation. Angew. Chem. Int. Ed. 2013, 52, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Casitas, A.; Ribas, X. The role of organometallic copper(III) complexes in homogeneous catalysis. Chem. Sci. 2013, 4, 2301. [Google Scholar] [CrossRef]
- Liu, S.; Lei, Y.-J.; Xin, Z.-J.; Lu, Y.-B.; Wang, H.-Y. Water splitting based on homogeneous copper molecular catalysts. J. Photochem. Photobiol. A 2018, 355, 141–151. [Google Scholar] [CrossRef]
- Sandroni, M.; Pellegrin, Y.; Odobel, F. Heteroleptic bis-diimine copper(I) complexes for applications in solar energy conversion. Comptes Rendus Chim. 2016, 19, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Mejía, E.; Luo, S.-P.; Karnahl, M.; Friedrich, A.; Tschierlei, S.; Surkus, A.-E.; Junge, H.; Gladiali, S.; Lochbrunner, S.; Beller, M. A Noble-Metal-Free System for Photocatalytic Hydrogen Production from Water. Chem. Eur. J. 2013, 19, 15972–15978. [Google Scholar] [CrossRef]
- Kavan, L.; Saygili, Y.; Freitag, M.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Electrochemical Properties of Cu(II/I)-Based Redox Mediators for Dye-Sensitized Solar Cells. Electrochim. Acta 2017, 227, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Magni, M.; Biagini, P.; Colombo, A.; Dragonetti, C.; Roberto, D.; Valore, A. Versatile copper complexes as a convenient springboard for both dyes and redox mediators in dye sensitized solar cells. Coord. Chem. Rev. 2016, 322, 69–93. [Google Scholar] [CrossRef]
- Muthuramalingam, S.; Khamrang, T.; Velusamy, M.; Mayilmurugan, R. Catalytic fixation of atmospheric carbon dioxide by copper(II) complexes of bidentate ligands. Dalton Trans. 2017, 46, 16065–16076. [Google Scholar] [CrossRef]
- Liu, W.; Huang, H.; Ouyang, T.; Jiang, L.; Zhong, D.; Zhang, W.; Lu, T. A Copper(II) Molecular Catalyst for Efficient and Selective Photochemical Reduction of CO2 to CO in a Water Containing System. Chem. Eur. J. 2018, 24, 4503–4508. [Google Scholar] [CrossRef]
- Barnett, S.M.; Goldberg, K.I.; Mayer, J.M. A soluble copper–bipyridine water-oxidation electrocatalyst. Nature Chem. 2012, 4, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, C.; Liu, S.; Wang, J.-L.; Lin, W. A Biomimetic Copper Water Oxidation Catalyst with Low Overpotential. J. Am. Chem. Soc. 2014, 136, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-T.; Chen, Z.; Kang, P.; Meyer, T.J. Electrocatalytic Water Oxidation with a Copper(II) Polypeptide Complex. J. Am. Chem. Soc. 2013, 135, 2048–2051. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Balamurugan, J.; Vinothkannan, M.; Kim, A.R.; Sengodan, S.; Yoo, D.J. Nitrogen-doped graphene encapsulated FeCoMoS nanoparticles as advanced trifunctional catalyst for water splitting devices and zinc–air batteries. Appl. Catal. B 2020, 279, 119381. [Google Scholar] [CrossRef]
- Sathiskumar, C.; Ramakrishnan, S.; Vinothkannan, M.; Rhan Kim, A.; Karthikeyan, S.; Yoo, D.J. Nitrogen-Doped Porous Carbon Derived from Biomass Used as Trifunctional Electrocatalyst toward Oxygen Reduction, Oxygen Evolution and Hydrogen Evolution Reactions. Nanomaterials 2019, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elayappan, V.; Shanmugam, R.; Chinnusamy, S.; Yoo, D.J.; Mayakrishnan, G.; Kim, K.; Noh, H.S.; Kim, M.K.; Lee, H. Three-dimensional bimetal TMO supported carbon based electrocatalyst developed via dry synthesis for hydrogen and oxygen evolution. Appl. Surf. Sci. 2020, 505, 144642. [Google Scholar] [CrossRef]
- Kügler, M.; Scholz, J.; Kronz, A.; Siewert, I. Copper complexes as catalyst precursors in the electrochemical hydrogen evolution reaction. Dalton Trans. 2016, 45, 6974–6982. [Google Scholar] [CrossRef]
- Liu, X.; Cui, S.; Sun, Z.; Du, P. A Robust and Highly Active Copper-Based Electrocatalyst for Hydrogen Production at Low Overpotential in Neutral Water. Chem. Comm. 2015, 51, 12954–12957. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, M.; Chen, H.; Liang, Y.; Sun, J.; Sun, L. A Cu-Based Nanoparticulate Film as Super-Active and Robust Catalyst Surpasses Pt for Electrochemical H2 Production from Neutral and Weak Acidic Aqueous Solutions. Adv. Energy Mater. 2016, 6, 1502319. [Google Scholar] [CrossRef]
- Zhao, J.; Tran, P.D.; Chen, Y.; Loo, J.S.C.; Barber, J.; Xu, Z.J. Achieving High Electrocatalytic Efficiency on Copper: A Low-Cost Alternative to Platinum for Hydrogen Generation in Water. ACS Catal. 2015, 5, 4115–4120. [Google Scholar] [CrossRef]
- Du, J.; Wang, J.; Ji, L.; Xu, X.; Chen, Z. A Highly Active and Robust Copper-Based Electrocatalyst toward Hydrogen Evolution Reaction with Low Overpotential in Neutral Solution. ACS Appl. Mater. Interfaces 2016, 8, 30205–30211. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zheng, H.; Sun, Z.; Han, A.; Du, P. Earth-Abundant Copper-Based Bifunctional Electrocatalyst for Both Catalytic Hydrogen Production and Water Oxidation. ACS Catal. 2015, 5, 1530–1538. [Google Scholar] [CrossRef]
- Fan, M.; Gao, R.; Zou, Y.-C.; Wang, D.; Bai, N.; Li, G.-D.; Zou, X. An efficient nanostructured copper(I) sulfide-based hydrogen evolution electrocatalyst at neutral pH. Electrochim. Acta 2016, 215, 366–373. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.; Zhou, Q.; Wang, W.; Hou, Y.; Zhang, B.; Wang, X. Photocatalytic hydrogen evolution by Cu(II) complexes. Dalton Trans. 2016, 45, 5439–5443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Yang, Y.; Yao, T.; Sun, L. A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution. Angew. Chem. 2014, 126, 14023–14027. [Google Scholar] [CrossRef]
- Liao, R.-Z.; Wang, M.; Sun, L.; Siegbahn, P.E.M. Mechanism of Hydrogen Evolution in Cu(bztpen)—Catalysed Water Reduction: A DFT Study. Dalton Trans. 2015, 44, 9736–9739. [Google Scholar] [CrossRef]
- Ekanayake, D.M.; Kulesa, K.M.; Singh, J.; Kpogo, K.K.; Mazumder, S.; Schlegel, H.B.; Verani, C.N. A pentadentate nitrogen-rich copper electrocatalyst for water reduction with pH-dependent molecular mechanisms. Dalton Trans. 2017, 46, 16812–16820. [Google Scholar] [CrossRef]
- Lei, H.; Fang, H.; Han, Y.; Lai, W.; Fu, X.; Cao, R. Reactivity and Mechanism Studies of Hydrogen Evolution Catalyzed by Copper Corroles. ACS Catal. 2015, 5, 5145–5153. [Google Scholar] [CrossRef]
- Haddad, A.Z.; Cronin, S.P.; Mashuta, M.S.; Buchanan, R.M.; Grapperhaus, C.A. Metal-Assisted Ligand-Centered Electrocatalytic Hydrogen Evolution upon Reduction of a Bis (thiosemicarbazonato) Cu(II) Complex. Inorg. Chem. 2017, 56, 11254–11265. [Google Scholar] [CrossRef]
- Fang, T.; Lu, H.-X.; Zhao, J.-X.; Zhan, S.-Z.; Lv, Q.-Y. A new copper(I)–triazenido electro-catalyst for catalyzing hydrogen evolution from acetic acid and water. J. Mol. Catal. A Chem. 2015, 396, 304–309. [Google Scholar] [CrossRef]
- Beyene, B.B.; Das, K.; Kerayu, B.A.; Datta, A.; Hung, C.-H. Electrocatalytic H2 evolution of a Schiff-base assisted Cu(II) derivative as catalyst on homogeneous and heterogeneous phase. Catal. Commun. 2019, 119, 111–114. [Google Scholar] [CrossRef]
- Cao, J.-P.; Fang, T.; Fu, L.-Z.; Zhou, L.-L.; Zhan, S.-Z. First mononuclear copper(II) electro-catalyst for catalyzing hydrogen evolution from acetic acid and water. Int. J. Hydrog. Energy 2014, 39, 13972–13978. [Google Scholar] [CrossRef]
- Fu, L.-Z.; Fang, T.; Zhou, L.-L.; Zhan, S.-Z. A mononuclear copper electrocatalyst for both water reduction and oxidation. RSC Adv. 2014, 4, 53674–53680. [Google Scholar] [CrossRef]
- Zhou, L.-L.; Fang, T.; Cao, J.-P.; Zhu, Z.-H.; Su, X.-T.; Zhan, S.-Z. A dinuclear copper(II) electrocatalyst both water reduction and oxidation. J. Power Sources 2015, 273, 298–304. [Google Scholar] [CrossRef]
- Xin, Z.-J.; Liu, S.; Li, C.-B.; Lei, Y.-J.; Xue, D.-X.; Gao, X.-W.; Wang, H.-Y. Hydrogen production in a neutral aqueous solution with a water-soluble copper complex. Int. J. Hydrog. Energy 2017, 42, 4202–4207. [Google Scholar] [CrossRef]
- Nestke, S.; Kügler, M.; Scholz, J.; Wilken, M.; Jooss, C.; Siewert, I. A Copper Complex as Catalyst in Proton Reduction: A Copper Complex as Catalyst in Proton Reduction. Eur. J. Inorg. Chem. 2017, 2017, 3376–3382. [Google Scholar] [CrossRef]
- Kubas, G.J.; Monzyk, B.; Crumblis, A.L. Tetrakis (Acetonitrile) Copper (1+) Hexafluorophosphate (1-). In Inorganic Syntheses; Angelici, R.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 68–70. ISBN 9780470132593. [Google Scholar]
- Veroni, I.; Rontoyianni, A.; Mitsopoulou, C.A. Synthesis and characterization of a novel complex: Mo(CO)4[2-(2′pyridyl)quinoxaline]. An insight based on experimental and theoretical data. Dalton Trans. 2003, 255–260. [Google Scholar] [CrossRef]
- Garoufis, A.; Perlepes, S.P.; Vreugdenhil, A.; Butler, I.S.; Hadjiliadis, N. Synthesis and characterization of 1:2 metal complexes of the biheteroaromatic ligand 2-(2′-pyridyl)quinoxaline (L) and the X-ray structure of [CuL2](PF6). Inorganica Chim. Acta. 1995, 240, 673–677. [Google Scholar] [CrossRef]
- Martínez, N.P.; Isaacs, M.; Oliver, A.G.; Ferraudi, G.; Lappin, A.; Guerrero, J. Effects of the non-covalent interactions on the electronic and electrochemical properties of Cu(I) biquinoline complexes. Dalton Trans. 2018, 47, 13171–13179. [Google Scholar] [CrossRef]
- Fourmond, V.; Canaguier, S.; Golly, B.; Field, M.J.; Fontecave, M.; Artero, V. A nickel–manganese catalyst as a biomimic of the active site of NiFe hydrogenases: A combined electrocatalytical and DFT mechanistic study. Energy Environ. Sci. 2011, 4, 2417. [Google Scholar] [CrossRef]
- Fourmond, V.; Jacques, P.-A.; Fontecave, M.; Artero, V. H 2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation. Inorg. Chem. 2010, 49, 10338–10347. [Google Scholar] [CrossRef] [PubMed]
- Zarkadoulas, A.; Field, M.J.; Papatriantafyllopoulou, C.; Fize, J.; Artero, V.; Mitsopoulou, C.A. Experimental and Theoretical Insight into Electrocatalytic Hydrogen Evolution with Nickel Bis(aryldithiolene) Complexes as Catalysts. Inorg. Chem. 2016, 55, 432–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, B.H.; Hammes-Schiffer, S. Proton-Coupled Electron Transfer in Molecular Electrocatalysis: Theoretical Methods and Design Principles. Inorg. Chem. 2014, 53, 6427–6443. [Google Scholar] [CrossRef] [PubMed]
- Oxford Diffraction. CrysAlis CCD and CrysAlis RED, Version 1.171.32.15; Oxford Diffraction Ltd.: Abingdon, UK, 2008. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Early finding of preferred orientation: A new method. J. Appl. Crystallogr. 1994, 27, 1045–1050. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97, Program for Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. A Found Crystallogr. 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Wu, S.; Dou, J.; Zhang, J.; Zhang, S. A simple and economical one-pot method to synthesize high-quality water soluble CdTe QDs. J. Mater. Chem. 2012, 22, 14573. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization type basis set for second row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsjui, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Namazian, M.; Lin, C.Y.; Coote, M.L. Benchmark Calculations of Absolute Reduction Potential of Ferricinium/Ferrocene Couple in Nonaqueous Solutions. J. Chem. Theory Comput. 2010, 6, 2721–2725. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Photosensitizer | e− Donor | Solvent | Evolved H2 |
|---|---|---|---|---|
| [1]+, μM | Fluorescein, mΜ | Triethanolamine, M | DMF:H2O | TON24 h |
| 100 | 1 | 0.5 | 1:2 | 28.04 |
| 10 | 1 | 0.5 | 1:2 | 154.96 |
| 1 | 1 | 0.5 | 1:2 | 855.45 |
| 0.1 | 1 | 0.5 | 1:2 | 1130.72 |
| 1 | 2 | 0.5 | 1:2 | 381.85 |
| 1 | 1.5 | 0.5 | 1:2 | 724.77 |
| 1 | 0.8 | 0.5 | 1:2 | 204.83 |
| 1 | 0.5 | 0.5 | 1:2 | 773.89 |
| 1 | 0.3 | 0.5 | 1:2 | 791.50 |
| 1 | 1 | 0.1 | 1:2 | 836.91 |
| 1 | 1 | 1 | 1:2 | 781.31 |
| 0.85 | 0.3 | 0.5 | 1:4 | 3564.31 |
| 8.5 | 0.3 | 0.5 | 1:4 | 547.98 |
| 0.85 | 0.3 | 0.5 | 1:13 | 2643.75 |
| 0.85 1 | 0.3 | 0.5 | 1:13 | 1837.82 |
| 0.85 | 0.3 | 0.5 | H2O | 182.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drosou, M.; Kamatsos, F.; Ioannidis, G.; Zarkadoulas, A.; Mitsopoulou, C.A.; Papatriantafyllopoulou, C.; Tzeli, D. Reactivity and Mechanism of Photo- and Electrocatalytic Hydrogen Evolution by a Diimine Copper(I) Complex. Catalysts 2020, 10, 1302. https://doi.org/10.3390/catal10111302
Drosou M, Kamatsos F, Ioannidis G, Zarkadoulas A, Mitsopoulou CA, Papatriantafyllopoulou C, Tzeli D. Reactivity and Mechanism of Photo- and Electrocatalytic Hydrogen Evolution by a Diimine Copper(I) Complex. Catalysts. 2020; 10(11):1302. https://doi.org/10.3390/catal10111302
Chicago/Turabian StyleDrosou, Maria, Fotios Kamatsos, George Ioannidis, Athanasios Zarkadoulas, Christiana A. Mitsopoulou, Constantina Papatriantafyllopoulou, and Demeter Tzeli. 2020. "Reactivity and Mechanism of Photo- and Electrocatalytic Hydrogen Evolution by a Diimine Copper(I) Complex" Catalysts 10, no. 11: 1302. https://doi.org/10.3390/catal10111302