Synergistic Palladium-Phosphoric Acid Catalysis in (3 + 2) Cycloaddition Reactions between Vinylcyclopropanes and Imines

 , and
, and 
Abstract
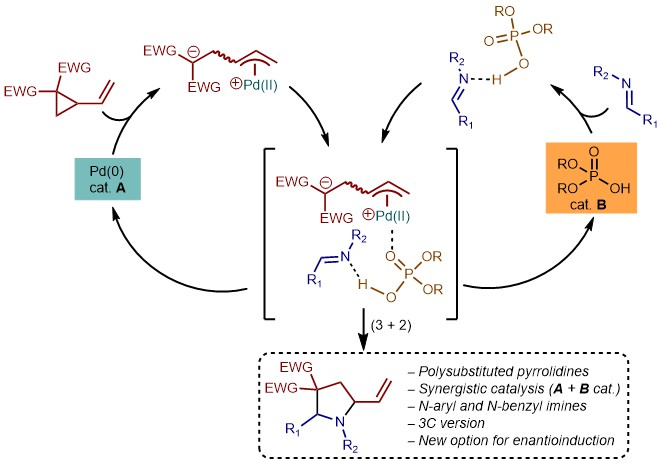
: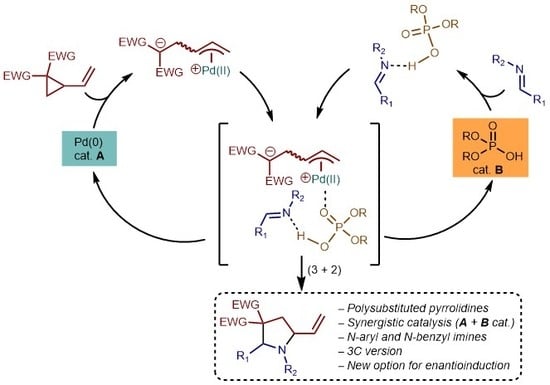
1. Introduction
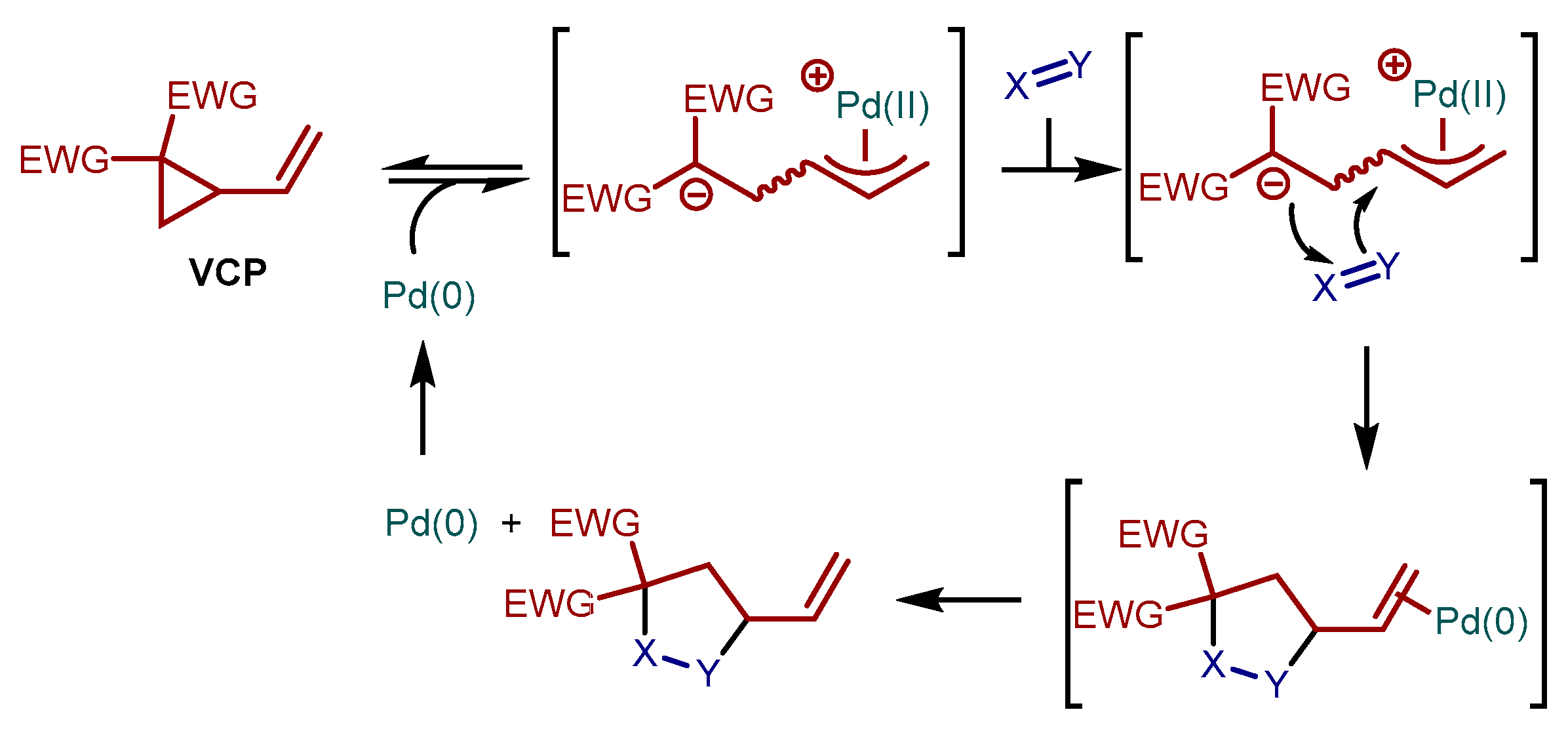
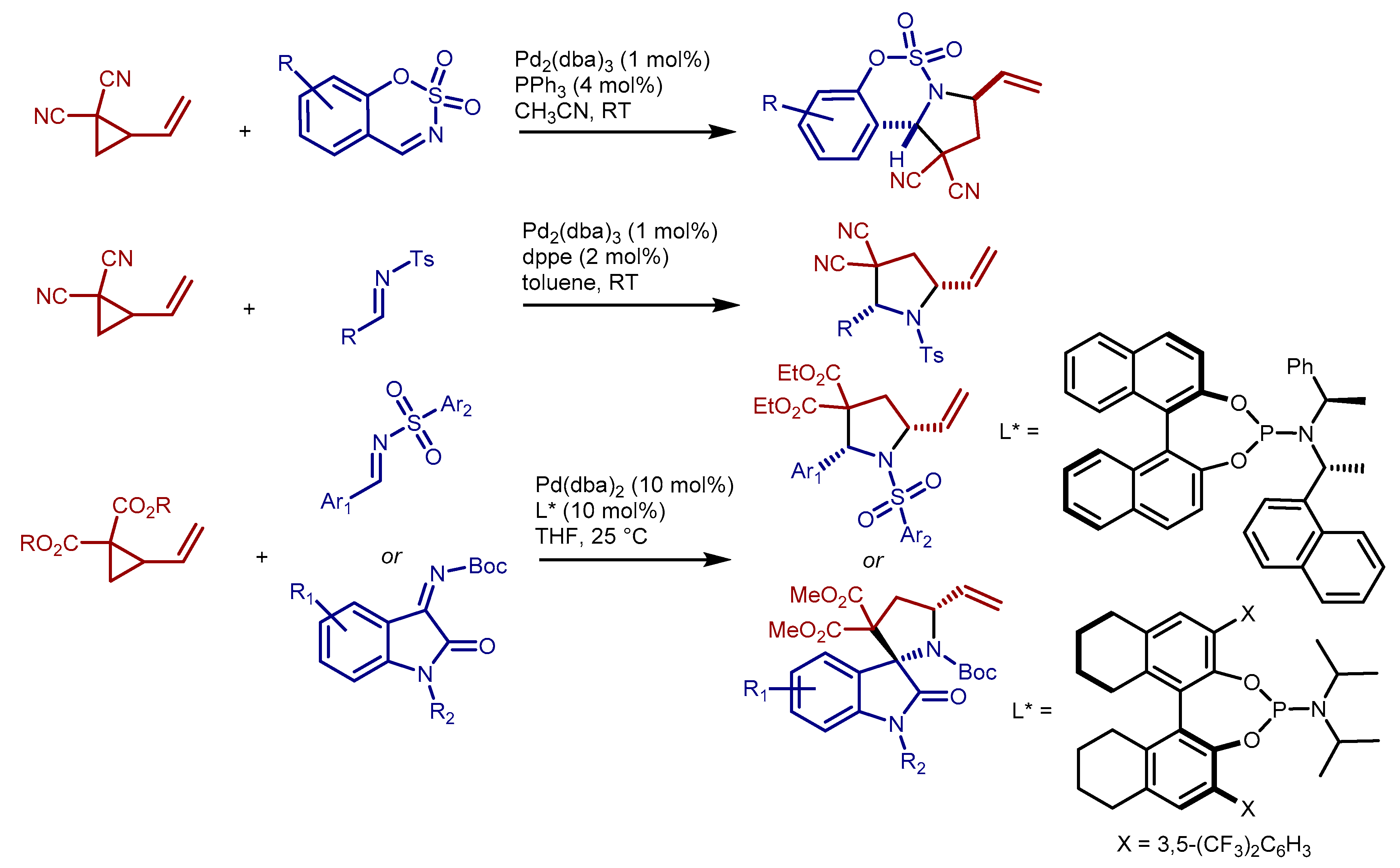
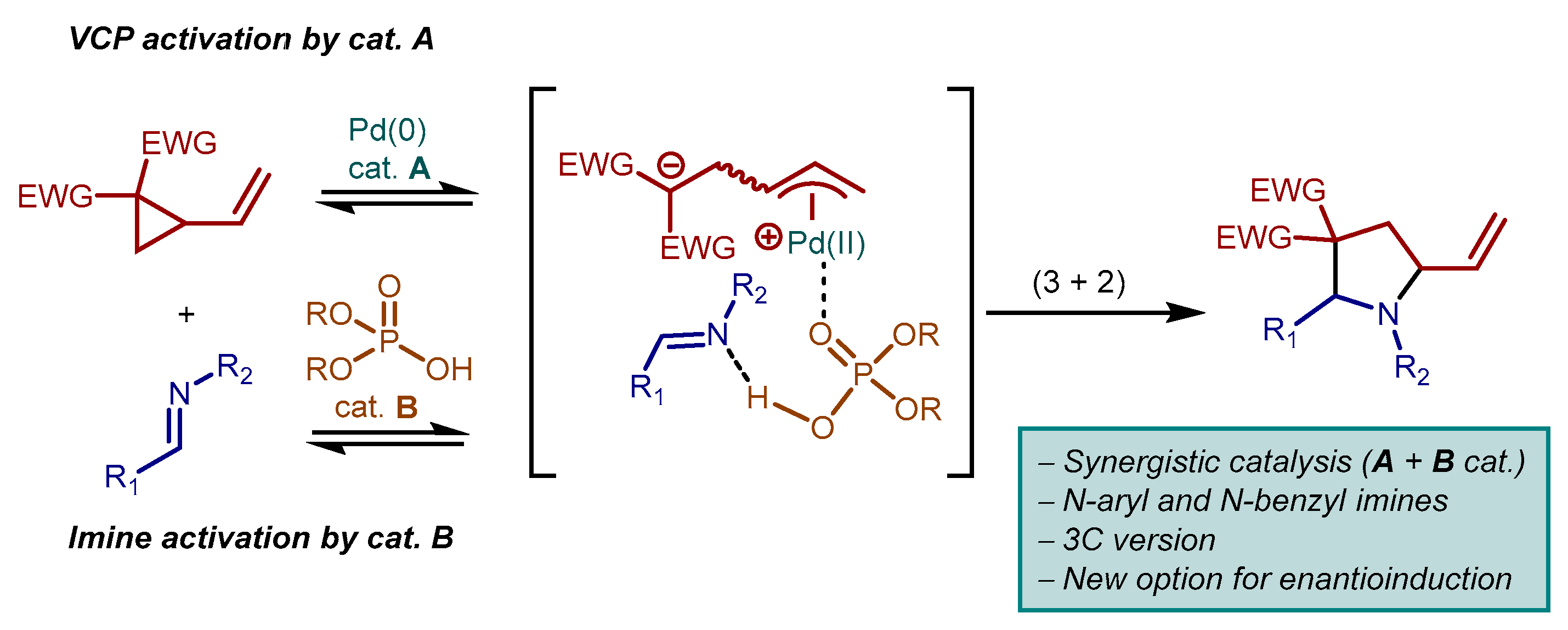
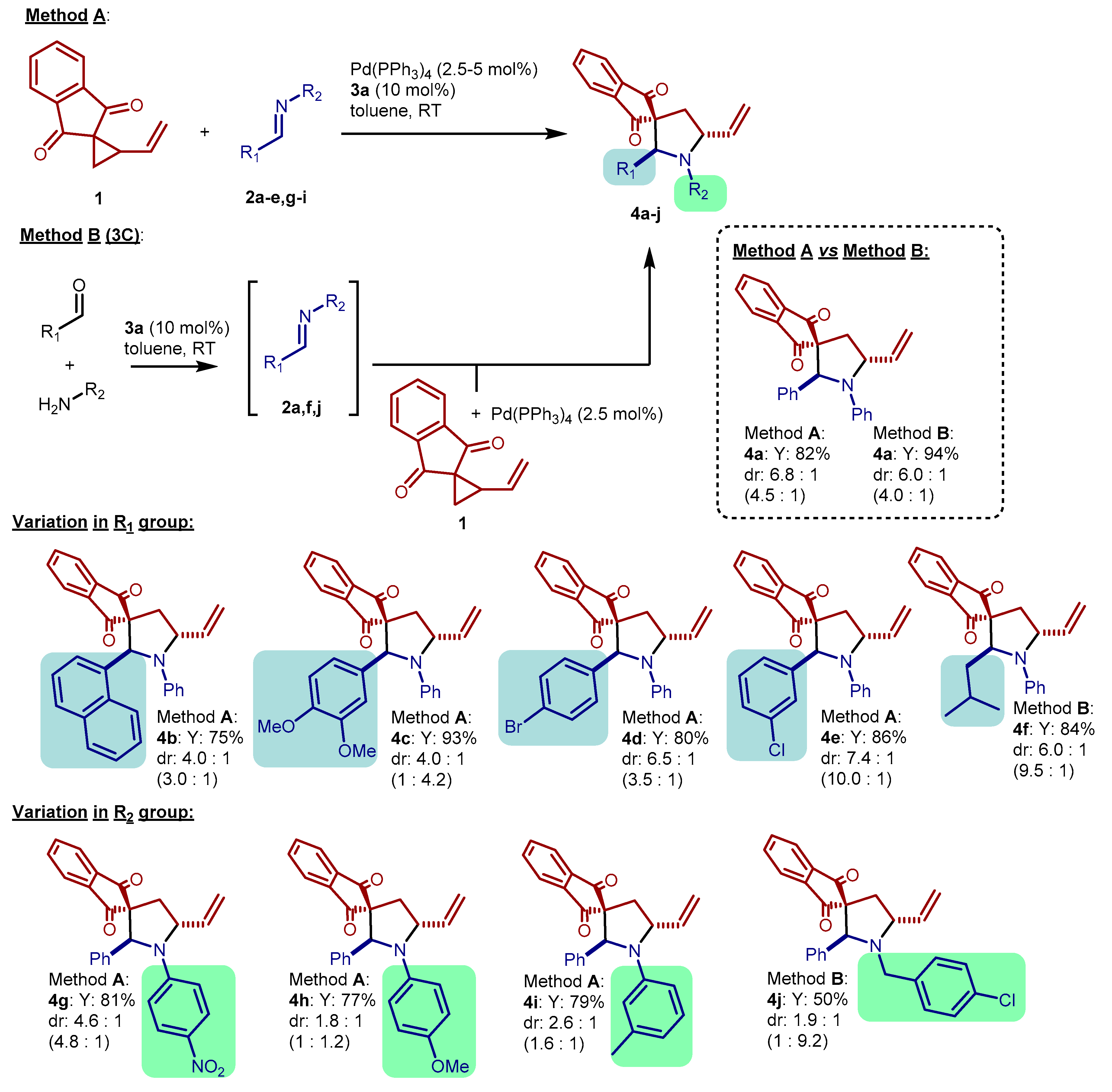
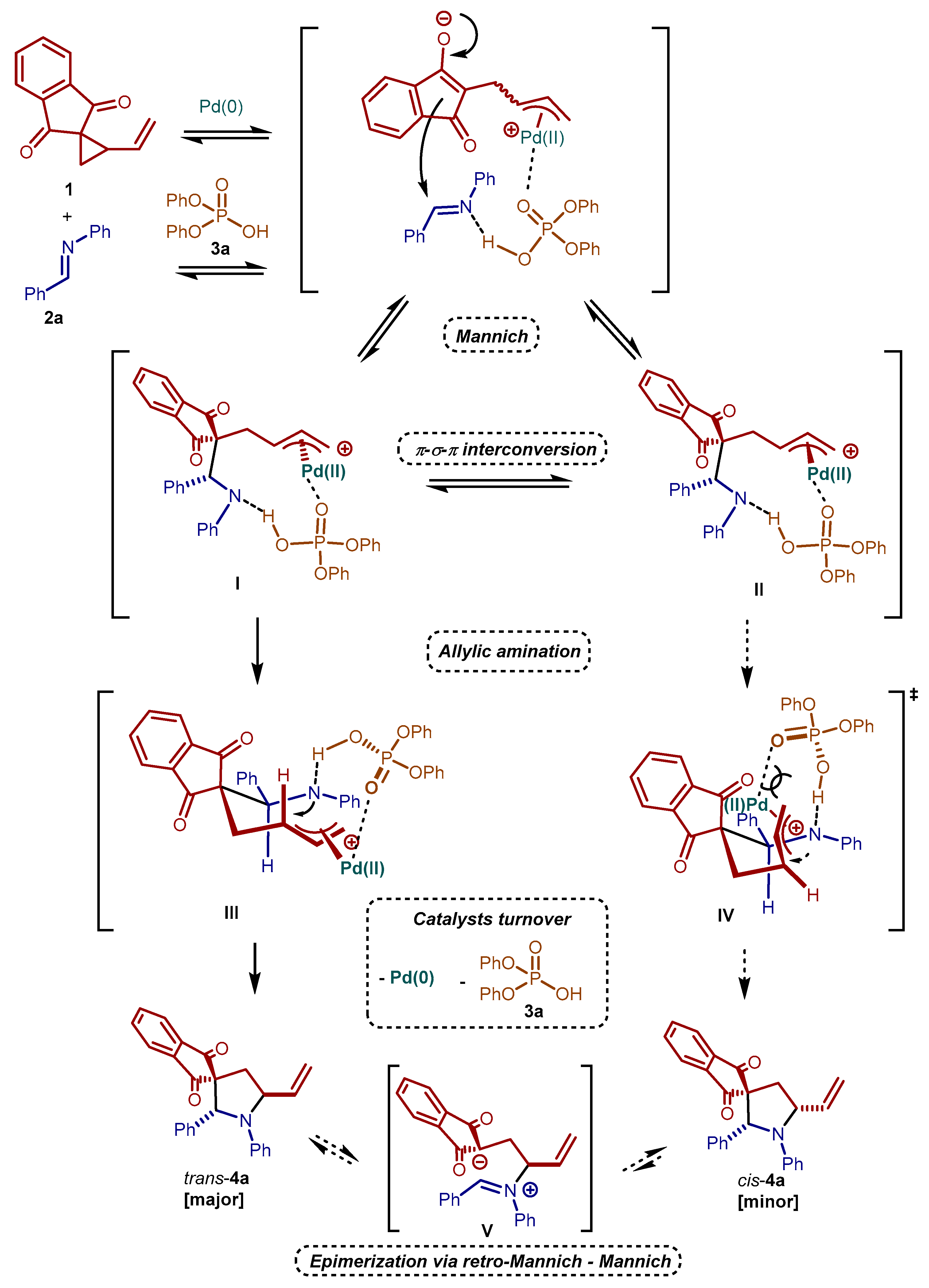
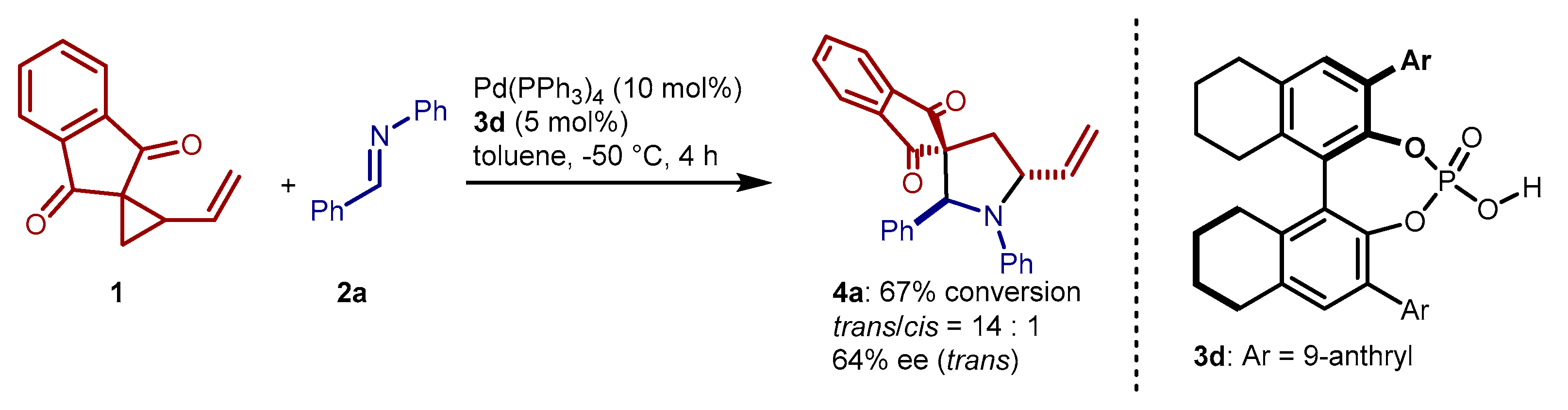
2. Results and Discussion
3. Experimental Section
3.1. Materials and Methods
3.2. General Procedures for the Synthesis of Pyrrolidines 4
3.3. Experimental Results and Characterization Data of Pyrrolidines 4
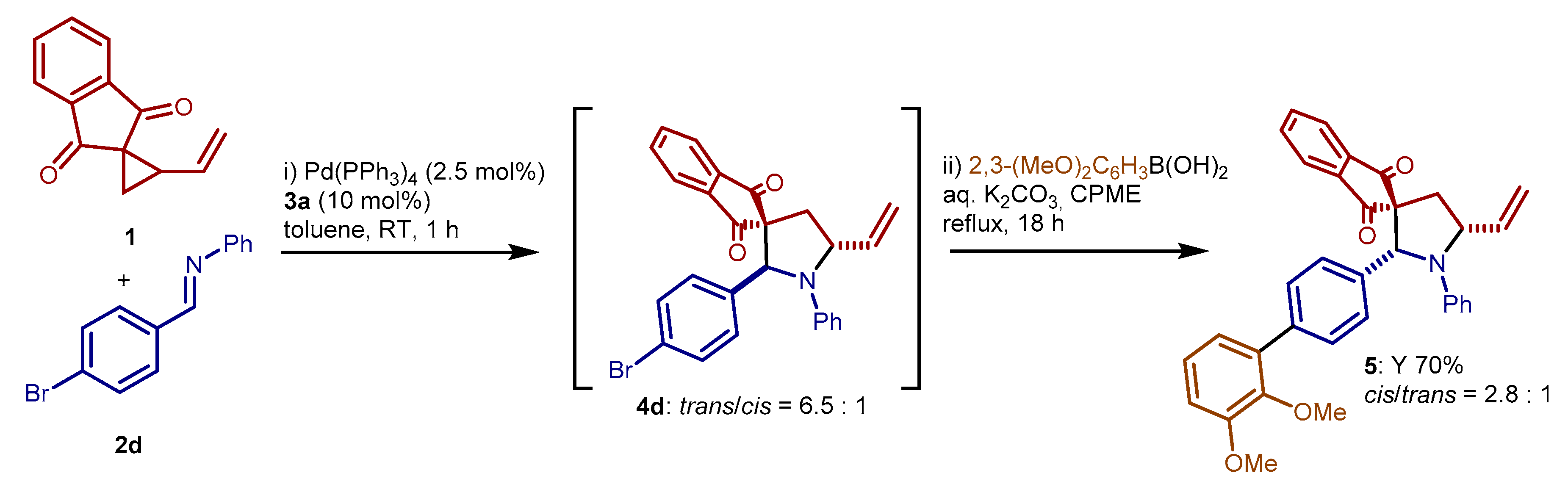
3.4. One Pot Cycloaddition—Suzuki–Miyaura Reaction: Preparation of Compound 5
3.5. Enantioselective Cycloaddition with Chiral Phosphoric Acid 3d
1′,2′-Diphenyl-5′-vinylspiro[indene-2,3′-pyrrolidine]-1,3-dione (4a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiao, L.; Yu, Z.-X. Vinylcyclopropane derivatives in transition-metal-catalyzed cycloadditions for the synthesis of carbocyclic compounds. J. Org. Chem. 2013, 78, 6842–6848. [Google Scholar] [CrossRef]
- Venkataraman, G.; Chandrasekaran, S. Recent advances in the synthesis and reactivity of vinylcyclopropanes. Synthesis 2016, 4347–4380. [Google Scholar]
- Meazza, M.; Guo, H.; Rios, R. Synthetic applications of vinyl cyclopropane opening. Org. Biomol. Chem. 2017, 15, 2479–2490. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.D.W.; Lakeland, C.P.; Harrity, J.P.A. Utilizing palladium-stabilized zwitterions for the construction of N-heterocycles. Chem. Eur. J. 2017, 23, 13830–13857. [Google Scholar] [CrossRef]
- Fumagalli, G.; Stanton, S.; Bower, J.F. Recent methodologies that exploit C–C single-bond cleavage of strained ring systems by transition metal complexes. Chem. Rev. 2017, 117, 9404–9432. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Ohashi, Y.; Tsuji, J. Palladium-catalyzed [3+2] cycloaddition reaction of vinylcyclopropanes with α,β-unsaturated esters or ketones. Tetrahedron Lett. 1985, 26, 3825–3828. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, C.; Shi, W.; Xiao, Y.; Guo, H. Pd-catalyzed diastereoselective [3+2] cycloaddition of vinylcyclopropanes with sulfamate-derived cyclic imines. Org. Biomol. Chem. 2018, 16, 4881–4887. [Google Scholar] [CrossRef]
- Ling, J.; Laugeois, M.; Ratovelomanana-Vidal, V.; Vitale, M.R. Palladium(0)-catalyzed diastereroselective (3+2) cycloadditions of vinylcyclopropanes with sulfonyl-activated imines. Synlett 2018, 29, 2288–2292. [Google Scholar]
- Spielmann, K.; Tosi, E.; Lebrun, A.; Niel, G.; van der Lee, A.; de Figueiredo, R.M.; Campagne, J.-M. Vinylaziridines and cyclopropanes in Pd-catalyzed (3+2)-cycloaddition reactions with cyclic N-sulfonyl imines. Tetrahedron 2018, 74, 6497–6511. [Google Scholar] [CrossRef]
- Huang, X.-B.; Li, X.-J.; Li, T.-T.; Chen, B.; Chu, W.-D.; He, L.; Liu, Q.-Z. Palladium-catalyzed highly enantioselective cycloaddition of vinyl cyclopropanes with imines. Org. Lett. 2019, 21, 1713–1716. [Google Scholar] [CrossRef]
- Dieskau, A.P.; Holzwarth, M.S.; Plietker, B. Fe-Catalyzed allylic C−C-bond activation: Vinylcyclopropanes as versatile a1,a3,d5-synthons in traceless allylic substitutions and [3 + 2]-cycloadditions. J. Am. Chem. Soc. 2012, 134, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Pursley, D.; Plietker, B. Insertion of imines into vinylcyclopropanes catalyzed by nucleophilic iron complexes: A formal [3+2]-cycloaddition strategy for the synthesis of substituted pyrrolidine derivatives. Synlett 2014, 25, 2316–2318. [Google Scholar] [CrossRef]
- Tombe, R.; Kurahashi, T.; Matsubara, S. Nickel-catalyzed cycloaddition of vinylcyclopropanes to imines. Org. Lett. 2013, 15, 1791–1793. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Fandrick, D.R. Dynamic kinetic asymmetric cycloadditions of isocyanates to vinylaziridines. J. Am. Chem. Soc. 2003, 125, 11836–11837. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.E.; MacMillan, D.W.C. Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem. Sci. 2012, 3, 633–658. [Google Scholar] [CrossRef]
- Chen, D.-F.; Han, Z.-Y.; Zhou, X.-L.; Gong, L.-Z. Asymmetric organocatalysis combined with metal catalysis: Concept, proof of concept, and beyond. Acc. Chem. Res. 2014, 47, 2365–2377. [Google Scholar] [CrossRef]
- Mukherjee, S.; List, B. Chiral counteranions in asymmetric transition-metal catalysis: Highly enantioselective Pd/Brønsted acid-catalyzed direct α-allylation of aldehydes. J. Am. Chem. Soc. 2007, 129, 11336–11337. [Google Scholar] [CrossRef]
- Halskov, K.S.; Næsborg, L.; Tur, F.; Jørgensen, K.A. Asymmetric [3+2] cycloaddition of vinylcyclopropanes and α,β-unsaturated aldehydes by synergistic palladium and organocatalysis. Org. Lett. 2016, 18, 2220–2223. [Google Scholar] [CrossRef]
- Meazza, M.; Rios, R. Synergistic catalysis: Enantioselective ring expansion of vinyl cyclopropanes combining four catalytic cycles for the synthesis of highly substituted spirocyclopentanes bearing up to four stereocenters. Chem. Eur. J. 2016, 22, 9923–9928. [Google Scholar] [CrossRef]
- Laugeois, M.; Ponra, S.; Ratovelomanana-Vidal, V.; Michelet, V.; Vitale, M.R. Asymmetric preparation of polysubstituted cyclopentanes by synergistic Pd(0)/amine catalyzed formal [3+2] cycloadditions of vinyl cyclopropanes with enals. Chem. Commun. 2016, 52, 5332–5335. [Google Scholar] [CrossRef]
- Zhang, K.; Meazza, M.; Izaga, A.; Contamine, C.; Gimeno, M.C.; Herrera, R.P.; Rios, R. Synergistic catalysis: Asymmetric synthesis of cyclopentanes bearing four stereogenic centers. Synthesis 2017, 49, 167–174. [Google Scholar]
- Kamlar, M.; Franc, M.; Císařová, I.; Gyepes, R.; Vesely, J. Formal [3+2] cycloaddition of vinylcyclopropane azlactones to enals using synergistic catalysis. Chem. Commun. 2019, 55, 3829–3832. [Google Scholar] [CrossRef]
- Afewerki, S.; Córdova, A. Combinations of aminocatalysts and metal catalysts: A powerful cooperative approach in selective organic synthesis. Chem. Rev. 2016, 116, 13512–13570. [Google Scholar] [CrossRef]
- Olmstead, W.N.; Bordwell, F.G. Ion-pair association constants in dimethyl sulfoxide. J. Org. Chem. 1980, 45, 3299–3305. [Google Scholar] [CrossRef]
- Christ, P.; Lindsay, A.G.; Vormittag, S.S.; Neudörfl, J.-M.; Berkessel, A.; O’Donoghue, A.C. pKa Values of chiral Brønsted acid catalysts: Phosphoric acids/amides, sulfonyl/sulfuryl imides, and perfluorinated TADDOLs (TEFDDOLs). Chem. Eur. J. 2011, 17, 8524–8528. [Google Scholar] [CrossRef] [PubMed]
- McCallum, C.; Pethybridge, A.D. Conductance of acids in dimethyl-sulphoxide—II. Conductance of some strong acids in DMSO at 25 °C. Electrochim. Acta 1975, 20, 815–818. [Google Scholar] [CrossRef]
- Spielmann, K.; van der Lee, A.; de Figueiredo, R.M.; Campagne, J.-M. Diastereoselective palladium-catalyzed (3+2)-cycloadditions from cyclic imines and vinyl aziridines. Org. Lett. 2008, 20, 1444–1447. [Google Scholar] [CrossRef]
- Trost, B.M.; Toste, F.D. Regio- and enantioselective allylic alkylation of an unsymmetrical substrate: a working model. J. Am. Chem. Soc. 1999, 121, 4545–4554. [Google Scholar] [CrossRef]
- Trost, B.M.; Morris, P.J.; Sprague, S.J. Palladium-catalyzed diastereo- and enantioselective formal [3+2]-cycloadditions of substituted vinylcyclopropanes. J. Am. Chem. Soc. 2012, 134, 17823–17831. [Google Scholar] [CrossRef] [Green Version]
- Laugeois, M.; Ling, J.; Férard, C.; Michelet, V.; Ratovelomanana-Vidal, V.; Vitale, M.R. Palladium(0)-catalyzed dearomative [3+2] cycloaddition of 3-nitroindoles with vinylcyclopropanes: An entry to stereodefined 2,3-fused cyclopentannulated indoline derivatives. Org. Lett. 2017, 19, 2266–2269. [Google Scholar] [CrossRef]
- Leth, L.A.; Glaus, F.; Meazza, M.; Fu, L.; Thøgersen, M.K.; Bitsch, E.A.; Jørgensen, K.A. Decarboxylative [4+2] cycloaddition by synergistic palladium and organocatalysis. Angew. Chem. Int. Ed. 2016, 55, 15272–15726. [Google Scholar] [CrossRef]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen Bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Caruana, L.; Fochi, M.; Ranieri, S.; Mazzanti, A.; Bernardi, L. Catalytic highly enantioselective vinylogous Povarov reaction. Chem. Commun. 2013, 49, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Ren, C.-L.; Wang, D.; Liu, L. Highly enantioselective [3+2] cycloaddition of vinylcyclopropane with nitroalkenes catalyzed by palladium(0) with a chiral bis(tert-amine) ligand. Chem. Eur. J. 2015, 21, 2335–2338. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry 1 | [Pd] (mol%) | 3 (mol%) | Solvent | Additive | t (h) | Y 2 (%) | d.r. 3 |
| 1 | Pd(PPh3)4 [10] | 3a [10] | toluene | - | 1 | 92 | 2.0:1 |
| 2 | - | 3a [10] | toluene | - | 24 | <5 | - |
| 3 | Pd(PPh3)4 [10] | - | toluene | - | 24 | 49 | 5.1:1 |
| 4 | Pd(PPh3)4 [10] | 3b [10] | toluene | - | 3 | 47 | 1.7:1 |
| 5 | Pd(PPh3)4 [10] | 3c [10] | toluene | - | 1 | 96 | 2.0:1 |
| 6 | Pd(PPh3)4 [2.5] | 3a [10] | toluene | - | 1 | 96 | 5.3:1 |
| 7 | Pd(PPh3)4 [2.5] | 3a [2.5] | toluene | - | 1 | 98 | 4.5:1 |
| 8 | Pd(PPh3)4 [10] | 3a [5] | toluene | - | 1 | >95 4 | 1.9:1 |
| 9 | Pd(dba)2 [5] | 3a [10] | toluene | - | 6 | 36 4 | 3.9:1 |
| 10 | Pd(PPh3)4 [2.5] | 3a [10] | CH2 Cl2 | - | 1 | 88 | 1.5:1 |
| 11 | Pd(PPh3)4 [2.5] | 3a [10] | THF | - | 1 | 76 | 4.2:1 |
| 12 | Pd(PPh3)4 [2.5] | 3a [10] | CH3 CN | - | 1 | 97 | 1.2:1 |
| 13 | Pd(PPh3)4 [2.5] | 3a [10] | toluene | TBACl 5 | 30 | 83 | 6.4:1 |
| 14 | Pd(PPh3)4 [2.5] | 3a [10] | toluene | TBABr 5 | 30 | 61 | 11.8:1 |
| 15 | Pd(PPh3)4 [2.5] | 3a [10] | toluene | TBAI 5 | 21 | 21 | 18.0:1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corti, V.; Marcantonio, E.; Mamone, M.; Giungi, A.; Fochi, M.; Bernardi, L. Synergistic Palladium-Phosphoric Acid Catalysis in (3 + 2) Cycloaddition Reactions between Vinylcyclopropanes and Imines. Catalysts 2020, 10, 150. https://doi.org/10.3390/catal10020150
Corti V, Marcantonio E, Mamone M, Giungi A, Fochi M, Bernardi L. Synergistic Palladium-Phosphoric Acid Catalysis in (3 + 2) Cycloaddition Reactions between Vinylcyclopropanes and Imines. Catalysts. 2020; 10(2):150. https://doi.org/10.3390/catal10020150
Chicago/Turabian StyleCorti, Vasco, Enrico Marcantonio, Martina Mamone, Alessandro Giungi, Mariafrancesca Fochi, and Luca Bernardi. 2020. "Synergistic Palladium-Phosphoric Acid Catalysis in (3 + 2) Cycloaddition Reactions between Vinylcyclopropanes and Imines" Catalysts 10, no. 2: 150. https://doi.org/10.3390/catal10020150
APA StyleCorti, V., Marcantonio, E., Mamone, M., Giungi, A., Fochi, M., & Bernardi, L. (2020). Synergistic Palladium-Phosphoric Acid Catalysis in (3 + 2) Cycloaddition Reactions between Vinylcyclopropanes and Imines. Catalysts, 10(2), 150. https://doi.org/10.3390/catal10020150