The Natural Product Lepidiline A as an N-Heterocyclic Carbene Ligand Precursor in Complexes of the Type [Ir(cod)(NHC)PPh3)]X: Synthesis, Characterisation, and Application in Hydrogen Isotope Exchange Catalysis


 , , and
, , and 
Abstract
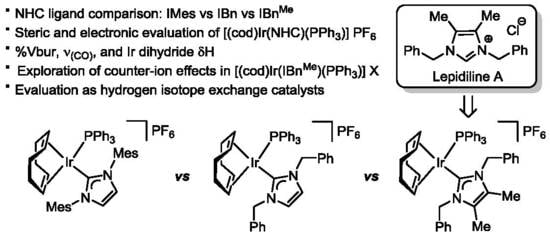
:
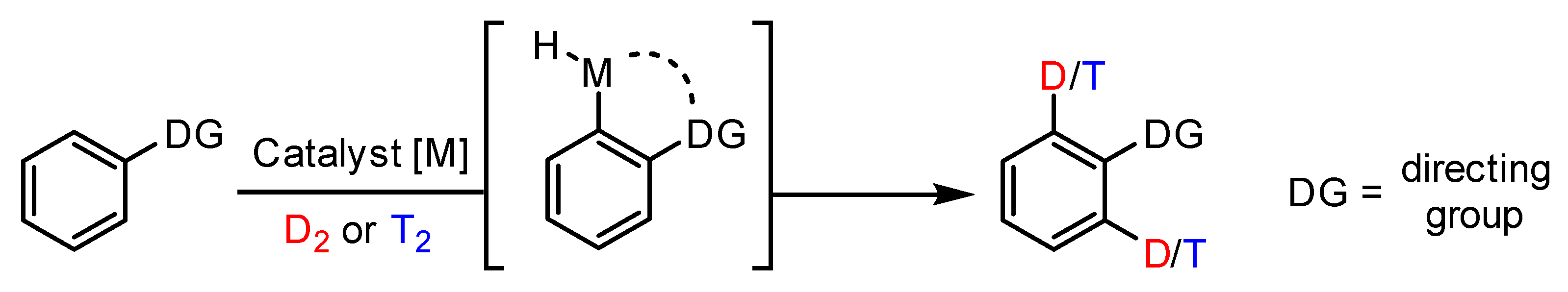
1. Introduction
2. Results and Discussion
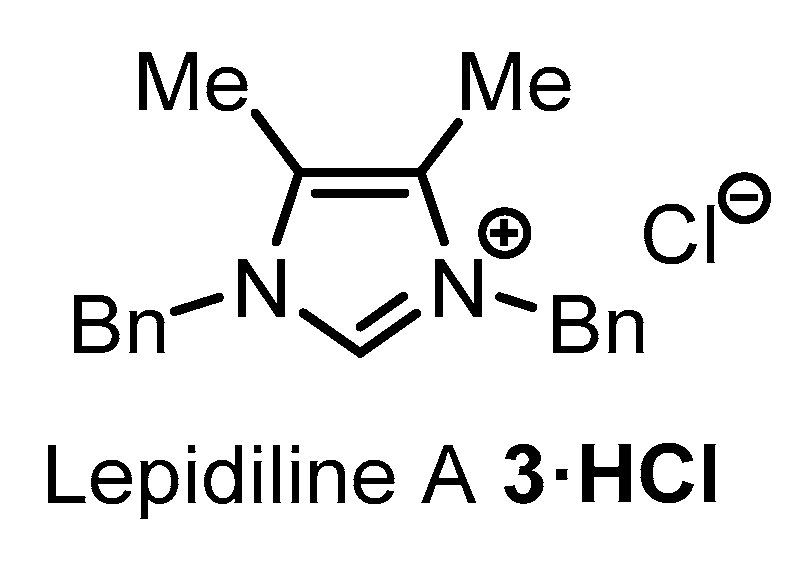
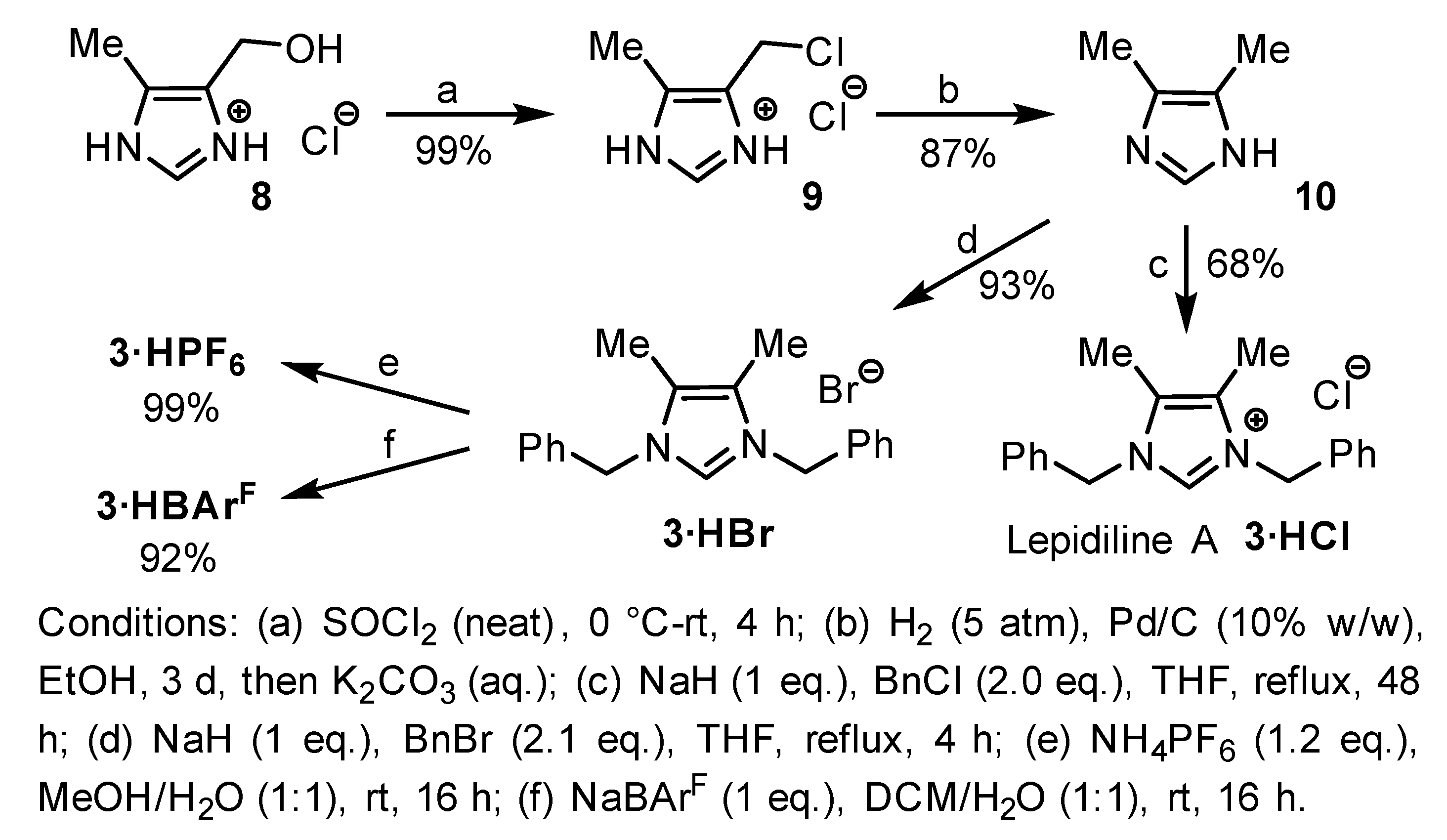
2.1. Synthesis of Lepidiline A
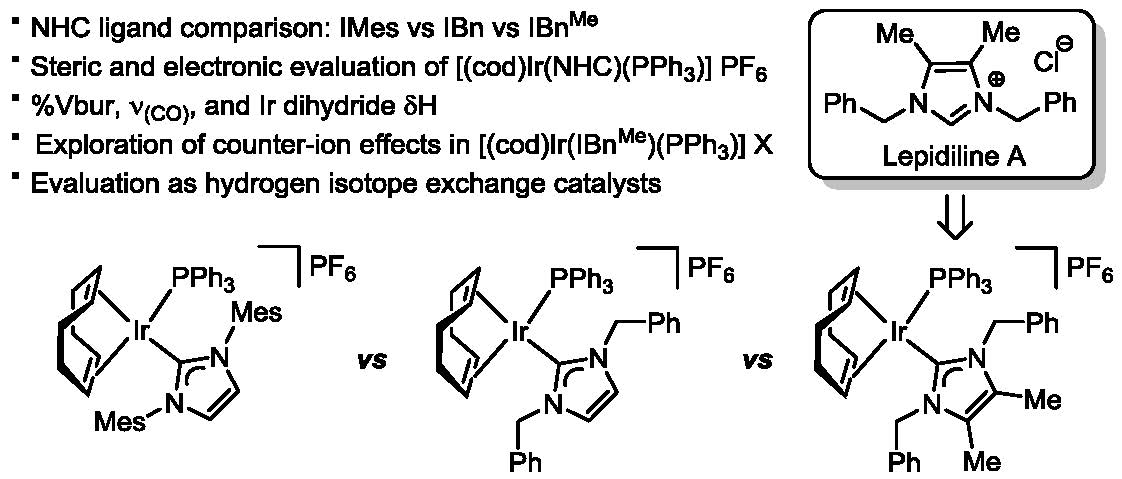
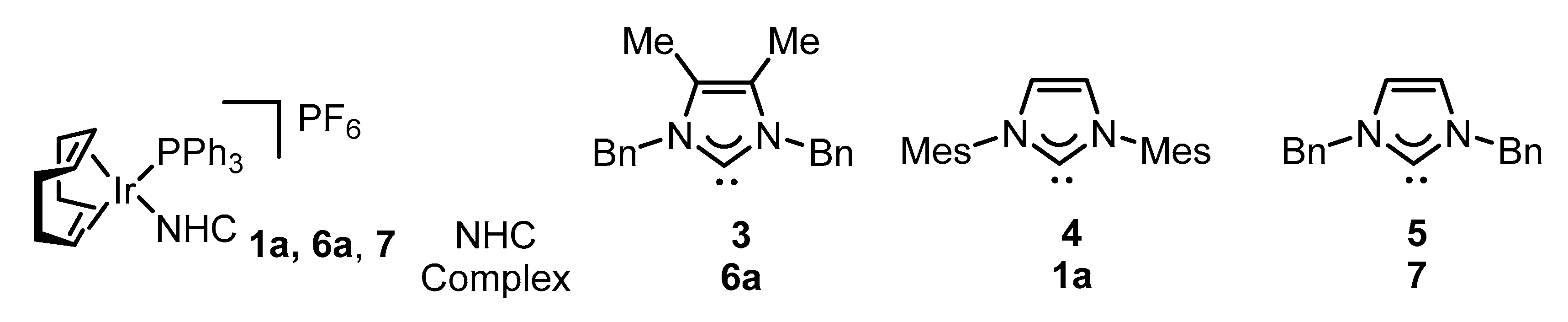
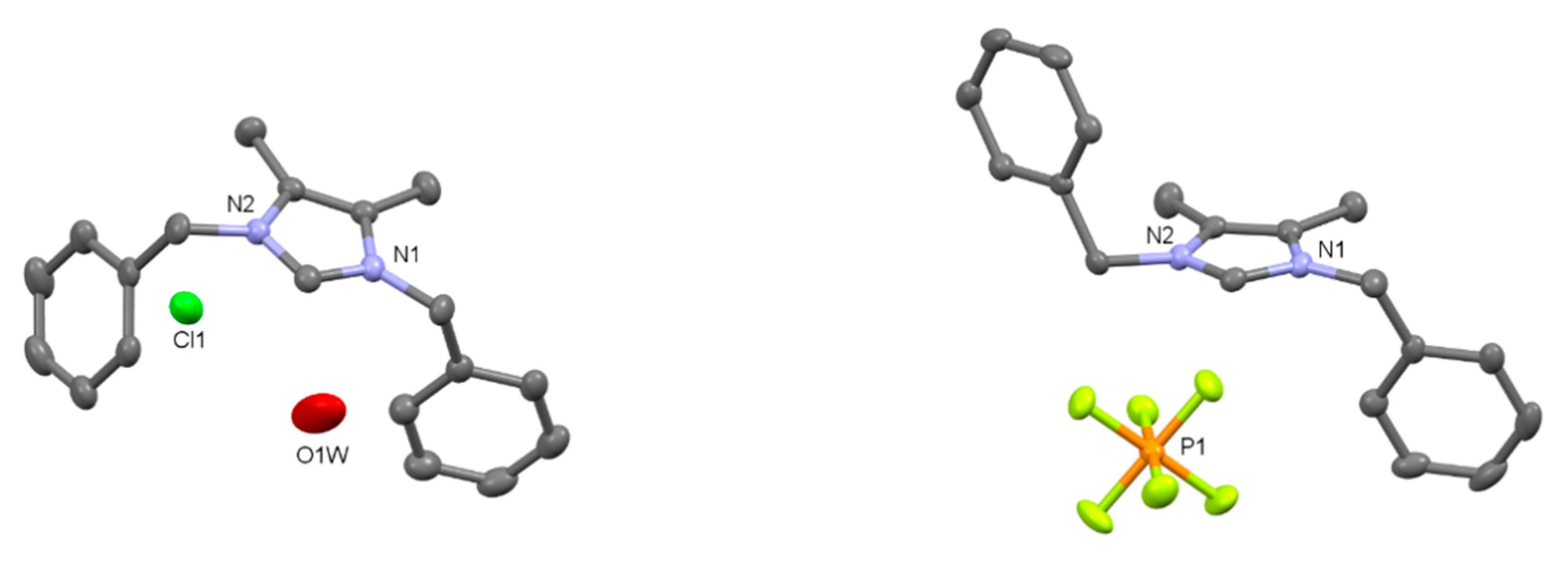
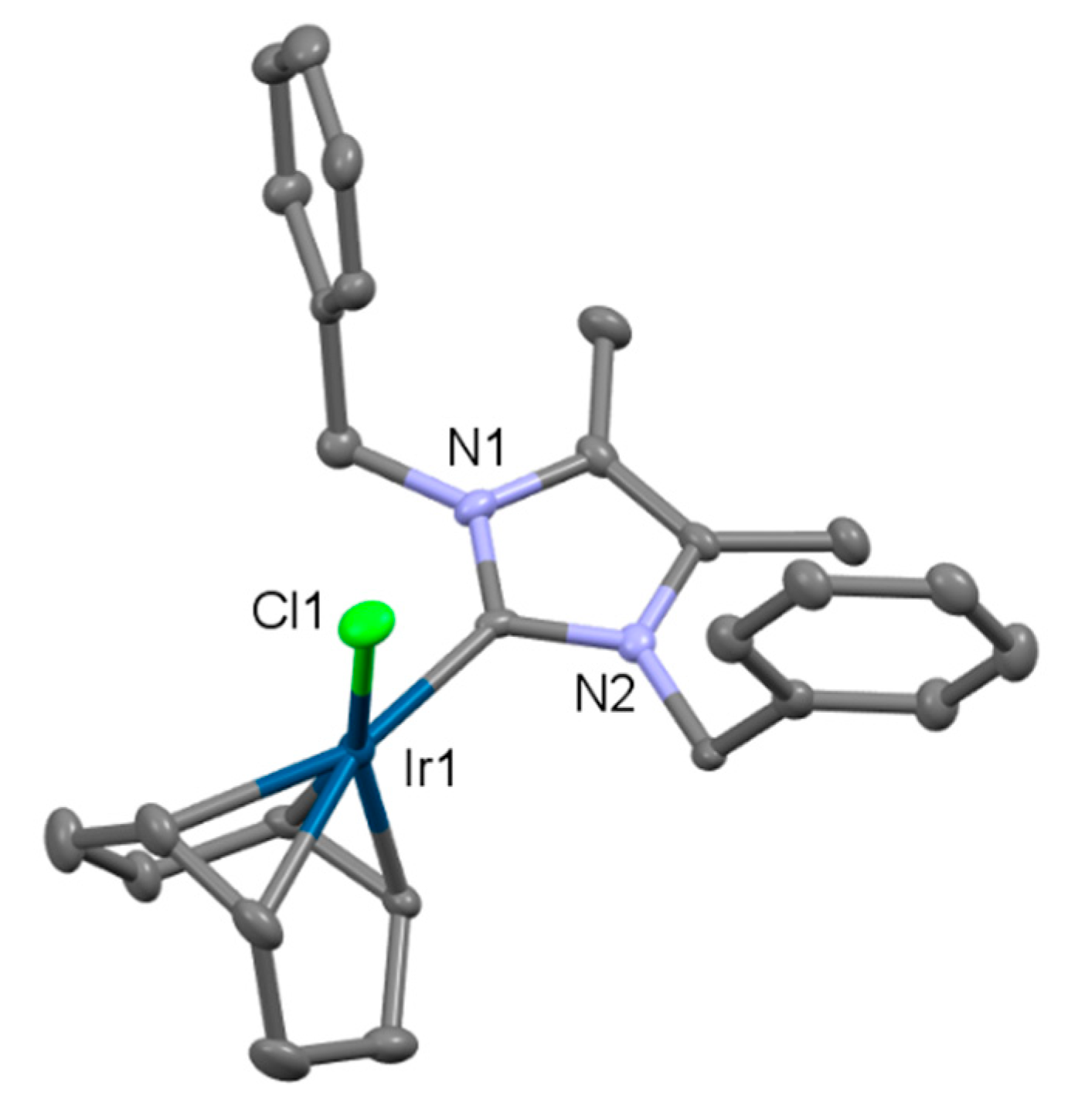
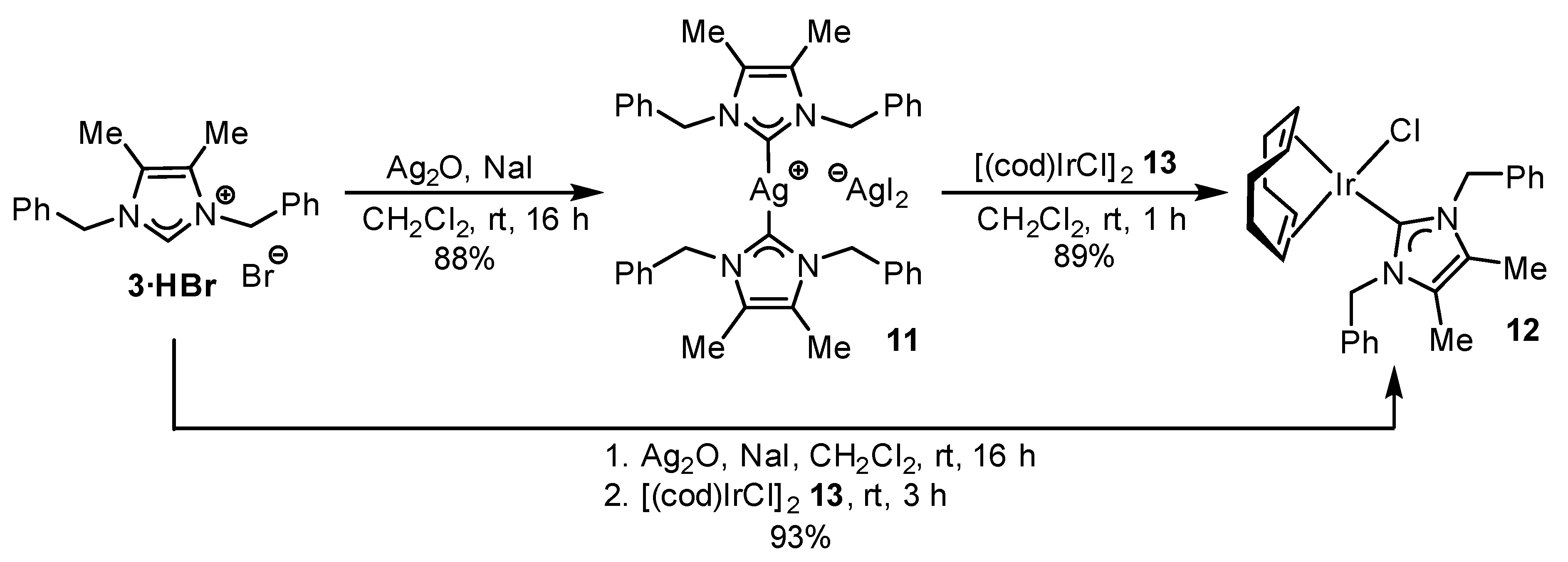
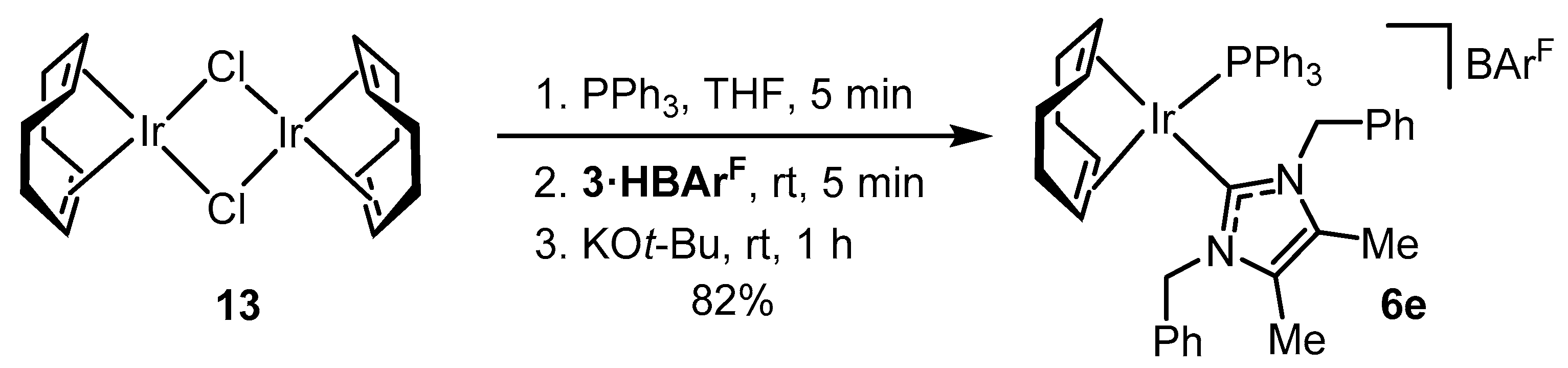
2.2. Preparation of Iridium Complexes Based on Lepidiline A 3
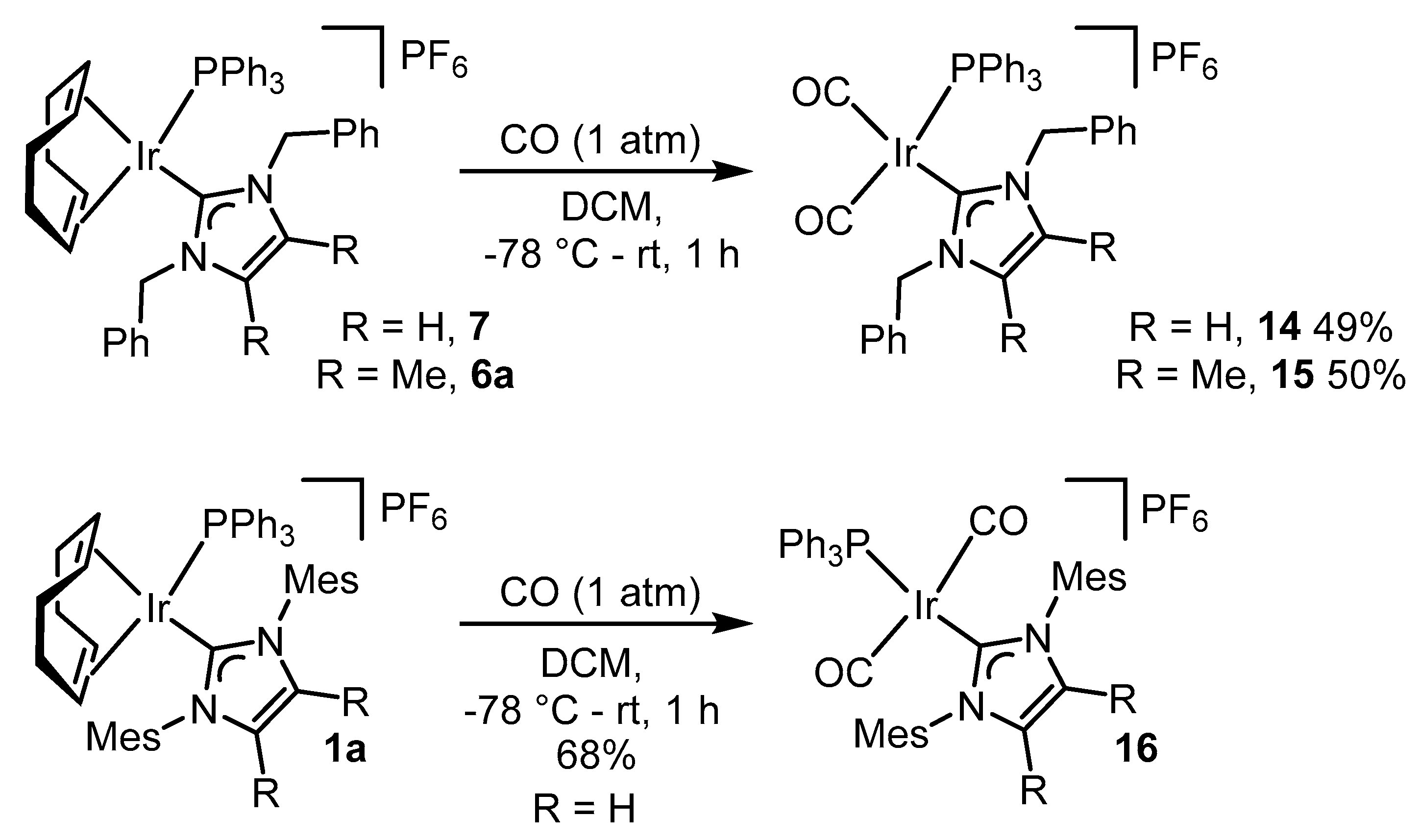
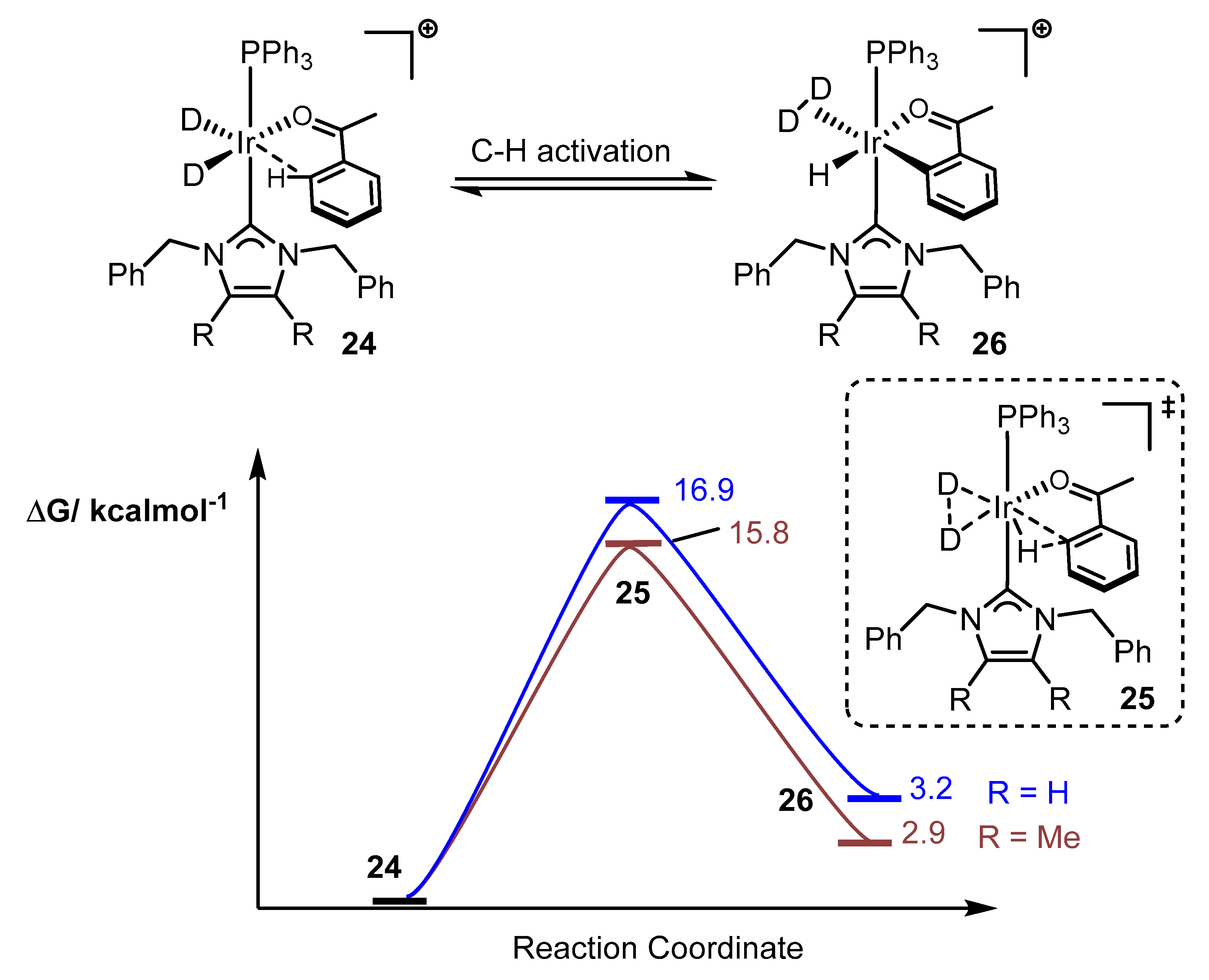
2.3. Steric and Electronic Characterisation of the Iridium NHC/Phosphine Complexes
2.4. Evaluation of Iridium Complex Catalytic Activity in HIE
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Meyer, M.P. New Applications of Isotope Effects in the Determination of Organic Reaction Mechanisms. In Advances in Physical Organic Chemistry; Williams, I., Williams, N., Eds.; Elsevier: London, UK, 2012; Volume 46, pp. 57–114. [Google Scholar]
- Isin, E.M.; Elmore, C.S.; Nilsson, G.N.; Thompson, R.A.; Weidolf, L. Use of radiolabeled compounds in drug metabolism and pharmacokinetic studies. Chem. Res. Toxicol. 2012, 25, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Penner, N.; Xu, L.; Prakash, C. Radiolabeled absorption, distribution, metabolism, and excretion studies in drug development: why, when and how? Chem. Res. Toxicol. 2012, 25, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Atzrodt, J.; Derdau, V.; Kerr, W.J.; Reid, M. Deuterium- and tritium-labelled compounds: Applications in the life sciences. Angew. Chem. Int. Ed. 2018, 57, 1758–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schillerstrom, R. Heavy Drugs. Drug Discov. Dev. 2009, 12, 6–8. [Google Scholar]
- Katsnelson, A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 2013, 19, 656. [Google Scholar] [CrossRef]
- Atzrodt, J.; Derdau, V.; Kerr, W.J.; Reid, M. C-H functionalization for hydrogen isotope exchange. Angew. Chem. Int. Ed. 2018, 57, 3022–3047. [Google Scholar] [CrossRef]
- Hesk, D. Highlights of C(sp2)-H hydrogen isotope exchange reactions. J. Labelled Compd. Radiopharm. 2019. [Google Scholar] [CrossRef]
- Valero, M.; Derdau, V. Highlights of aliphatic C(sp3)-H hydrogen isotope exchange reactions. J. Labelled Compd. Radiopharm. 2019. [Google Scholar] [CrossRef]
- Kerr, W.J.; Knox, G.J.; Paterson, L.C. Recent Advances in Iridium(I) Catalysis towards Directed Hydrogen Isotope Exchange. J. Labelled Compd. Radiopharm. 2019. [Google Scholar] [CrossRef]
- Brown, J.A.; Irvine, S.; Kennedy, A.R.; Kerr, W.J.; Andersson, S.; Nilsson, G.N. Highly Active Iridium(I) Complexes for Catalytic Hydrogen Isotope Exchange. Chem. Commun. 2008, 1115–1117. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Cochrane, A.R.; Irvine, S.; Kerr, W.J.; Mondal, B.; Parkinson, J.A.; Paterson, L.C.; Reid, M.; Tuttle, T.; Andersson, S.; et al. The Synthesis of Highly Active Iridium(I) Complexes and Their Application in Catalytic Hydrogen Isotope Exchange. Adv. Synth. Catal. 2014, 356, 3551–3562. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.R.; Kerr, W.J.; Moir, R.; Reid, M. Anion Effects to Deliver Enhanced Iridium Catalysts for Hydrogen Isotope Exchange Processes. Org. Biomol. Chem. 2014, 12, 7927–7931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, W.J.; Mudd, R.J.; Paterson, L.C.; Brown, J.A. Iridium(I)-Catalyzed Regioselective C-H Activation and Hydrogen-Isotope Exchange of Non-Aromatic Unsaturated Functionality. Chem. Eur. J. 2014, 20, 14604–14607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atzrodt, J.; Derdau, V.; Kerr, W.J.; Reid, M.; Rojahn, P.; Weck, R. Expanded Applicability of Iridium(I) NHC/Phosphine Catalysts in Hydrogen Isotope Exchange Processes with Pharmaceutically-Relevant Heterocycles. Tetrahedron 2015, 71, 1924–1929. [Google Scholar] [CrossRef] [Green Version]
- Kerr, W.J.; Lindsay, D.M.; Reid, M.; Atzrodt, J.; Derdau, V.; Rojahn, P.; Weck, R. Iridium-Catalysed Ortho-H/D and -H/T Exchange under Basic Conditions: C–H Activation of Unprotected Tetrazoles. Chem. Commun. 2016, 52, 6669–6672. [Google Scholar] [CrossRef] [Green Version]
- Kerr, W.J.; Mudd, R.J.; Owens, P.K.; Reid, M.; Brown, J.A.; Campos, S. Hydrogen Isotope Exchange with Highly Active Iridium(I) NHC/Phosphine Complexes: A Comparative Counterion Study. J. Labelled Compd. Radiopharm. 2016, 59, 601–603. [Google Scholar] [CrossRef] [Green Version]
- Kerr, W.J.; Lindsay, D.M.; Owens, P.K.; Reid, M.; Tuttle, T.; Campos, S. Site-Selective Deuteration of N-Heterocycles via Iridium-Catalyzed Hydrogen Isotope Exchange. ACS Catal. 2017, 7, 7182–7186. [Google Scholar] [CrossRef] [Green Version]
- Kerr, W.J.; Reid, M.; Tuttle, T. Iridium-Catalyzed C-H Activation and Deuteration of Primary Sulfonamides: An Experimental and Computational Study. ACS Catal. 2015, 5, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Kerr, W.J.; Reid, M.; Tuttle, T. Iridium-Catalyzed Formyl-Selective Deuteration of Aldehydes. Angew. Chem. Int. Ed. 2017, 56, 7808–7812. [Google Scholar] [CrossRef]
- Cui, B.; Zheng, B.; He, K.; Zheng, Q. Imidazole Alkaloids from Lepidium meyenii. J. Nat. Prod. 2003, 66, 1101–1103. [Google Scholar] [CrossRef]
- Curran, D.; Dada, O.; Müller-Bunz, H.; Rothemund, M.; Sánchez-Sanz, G.; Schobert, R.; Zhu, X.; Tacke, M. Synthesis and Cytotoxic Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules 2018, 23, 2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See the Supplementary Materials for further details.
- Cochrane, A.R.; Irvine, S.; Kerr, W.J.; Reid, M.; Andersson, S.; Nilsson, G.N. Application of neutral iridium(I) N-heterocyclic carbene complexes in ortho-directed hydrogen isotope exchange. J. Labelled Compd. Radiopharm. 2013, 46, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.M.J.; Lin, I.J.B. Facile Synthesis of Silver(I)−Carbene Complexes. Useful Carbene Transfer Agents. Organometallics 1998, 17, 972–975. [Google Scholar] [CrossRef]
- Chang, Y.-H.; Fu, C.-F.; Liu, Y.-H.; Peng, S.-M.; Chen, J.-T.; Liu, S.-T. Synthesis, characterization and catalytic activity of saturated and unsaturated N-heterocyclic carbene iridium(I) complexes. Dalton Trans. 2009, 861–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolman, C.R. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Mantilli, L.; Gérard, D.; Besnard, C.; Mazet, C. Structure-Activity Relationship in the Iridium-Catalyzed Isomerization of Primary Allylic Alcohols. Eur. J. Inorg. Chem. 2012, 2012, 3320–3330. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. Stereoelectronic parameters associated with N-heterocyclic carbene (NHC) liagnds: A quest for understanding. Coord. Chem. Rev. 2007, 251, 874–883. [Google Scholar]
- Nelson, D.J.; Truscott, B.J.; Slawin, A.M.Z.; Nolan, S.P. Synthesis and Reactivity of New Bis(N-heterocyclic carbene) Iridium(I) Complexes. Inorg. Chem. 2013, 52, 12674–12681. [Google Scholar] [CrossRef]
- Torres, O.; Martín, M.; Sola, E. Labile N-Heterocyclic Carbene Complexes of Irdium. Organometallics 2009, 28, 863–870. [Google Scholar] [CrossRef]
- Smidt, S.P.; Zimmermann, N.; Studer, M.; Pfaltz, A. Enantioselective Hydrogenation of Alkenes with Iridium–PHOX Catalysts: A Kinetic Study of Anion Effects. Chem. Eur. J. 2004, 10, 4685–4693. [Google Scholar] [CrossRef]












{kind=link}
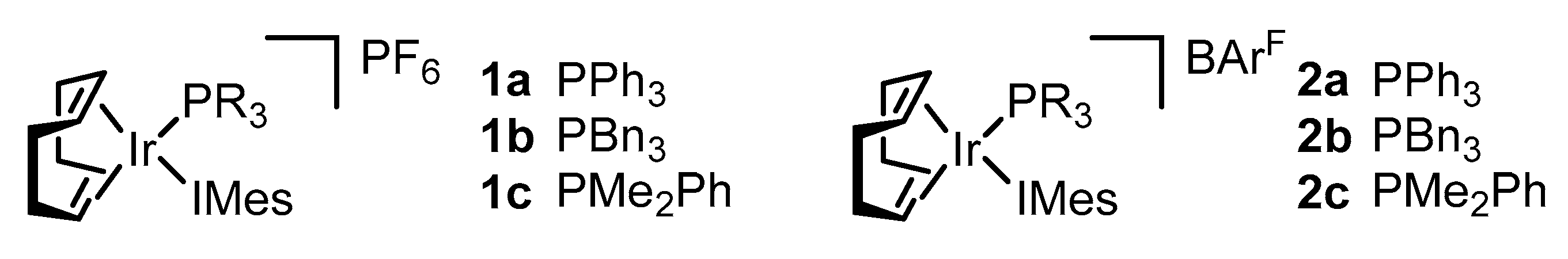{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Complex | X | Yield |
|---|---|---|---|
| 1 | 6a | PF6 | 75 |
| 2 | 6b | BF4 | 87 |
| 3 | 6c | SbF6 | 81 |
| 4 | 6d | OTf | 88 |
| Entry | Complex | N-Substituent | R | Yield | %Vbur (PPh3) 2 | %Vbur (NHC) 2 | Σ (%Vbur) | θ (°) 1 |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 | Bn | H | 49% | 26.9 | 28.9 | 55.8 | 93.2 |
| 2 | 15 | Bn | Me | 50% | 27.0 | 29.2 | 56.2 | 89.4 3 |
| 92.6 3 | ||||||||
| 3 | 16 | Mes | H | 68% | 27.9 | 32.6 | 60.5 | 160.0 |

| Entry | R | R’ | Cl/CO Complex | ν(CO)DCM | Dihydride Complex | δH |
|---|---|---|---|---|---|---|
| 1 | Bn | H | 17 | 1950 cm−1 | 20 | −21.35 ppm |
| 2 | Bn | Me | 18 | 1948 cm−1 | 21 | −21.40 ppm |
| 3 | Mes | H | 19 | 1942 cm−1 | 22 | −21.58 ppm |

| Entry | Complex | NHC | kobs |
|---|---|---|---|
| 1 | 6a | IBnMe | 0.0224 s−1 |
| 2 | 7 | IBn | 0.0145 s−1 |

| Entry | Substrate | %D | ||||
|---|---|---|---|---|---|---|
| 6a, X = PF6 | 6b, X = BF4 | 6c, X = SbF6 | 6d, X = OTf | 6e, X = BArF | ||
| 1 | 23 | 96 | 96 | 96 | 79 | 97 |
| 2 | 27 | 91 | 87 | 93 | 29 | 97 |
| 3 | 28 | 98 | 87 | 93 | 43 | 99 |
| 4 | 29 | 82 | 89 | 90 | 85 | 87 |
| 5 | 30 | 5 | 4 | 5 | 4 | 16 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cochrane, A.R.; Kennedy, A.R.; Kerr, W.J.; Lindsay, D.M.; Reid, M.; Tuttle, T. The Natural Product Lepidiline A as an N-Heterocyclic Carbene Ligand Precursor in Complexes of the Type [Ir(cod)(NHC)PPh3)]X: Synthesis, Characterisation, and Application in Hydrogen Isotope Exchange Catalysis. Catalysts 2020, 10, 161. https://doi.org/10.3390/catal10020161
Cochrane AR, Kennedy AR, Kerr WJ, Lindsay DM, Reid M, Tuttle T. The Natural Product Lepidiline A as an N-Heterocyclic Carbene Ligand Precursor in Complexes of the Type [Ir(cod)(NHC)PPh3)]X: Synthesis, Characterisation, and Application in Hydrogen Isotope Exchange Catalysis. Catalysts. 2020; 10(2):161. https://doi.org/10.3390/catal10020161
Chicago/Turabian StyleCochrane, Alison R., Alan R. Kennedy, William J. Kerr, David M. Lindsay, Marc Reid, and Tell Tuttle. 2020. "The Natural Product Lepidiline A as an N-Heterocyclic Carbene Ligand Precursor in Complexes of the Type [Ir(cod)(NHC)PPh3)]X: Synthesis, Characterisation, and Application in Hydrogen Isotope Exchange Catalysis" Catalysts 10, no. 2: 161. https://doi.org/10.3390/catal10020161
APA StyleCochrane, A. R., Kennedy, A. R., Kerr, W. J., Lindsay, D. M., Reid, M., & Tuttle, T. (2020). The Natural Product Lepidiline A as an N-Heterocyclic Carbene Ligand Precursor in Complexes of the Type [Ir(cod)(NHC)PPh3)]X: Synthesis, Characterisation, and Application in Hydrogen Isotope Exchange Catalysis. Catalysts, 10(2), 161. https://doi.org/10.3390/catal10020161