Abstract
The hydroamination reaction is a convenient alternative strategy for the formation of C–N bonds. Herein, we report a new versatile and convenient protocol for the hydroamination of arylacetylenes with anilines using palladium iodide in the absence of any added ligand as catalyst. Mild conditions, excellent regio- and stereoselectivity, and high functional group tolerance are the main features of this methodology. A subsequent reduction step gives access to a wide variety of secondary aromatic amines.
1. Introduction
Amines, imines, and enamines are useful building blocks in organic synthesis and ubiquitous motifs in biologically active molecules, as exemplified in Figure 1.
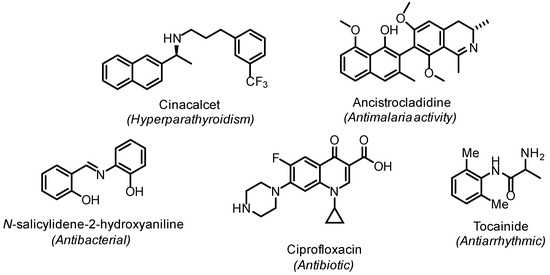
Figure 1.
Selected example of biologically active compounds containing amine, imine, or enamine functionalities.
One of the most attractive strategies for their preparation is the catalytic hydroamination of unsaturated compounds, such as alkenes or alkynes [1,2,3,4,5,6,7,8]. The hydroamination reaction, consisting of the addition of an N–H bond to a C–C multiple bond, features a 100% atom efficiency associated with the large availability of the starting materials (these are alkynes, alkenes, amines). Nevertheless, in order to promote the addition of the amino group on the multiple C–C bond, a high activation barrier, resulting from the repulsion between the high electron density of the nucleophile and the π-electrons of the multiple bond, needs to be overcome. In this context, metal catalysis can serve as a powerful tool to reduce the activation barrier either by coordinating the multiple bond and thus subtracting the electron density or by N-H metalation, resulting in a strongly nucleophilic metal amide moiety. Amide intermediates can be also generated by the oxidative addition of the N–H bond to the metal. Several metals have been found to catalyze hydroamination reactions. Early transition metals, such as Ti or Zr, as well as lanthanides, display high activity [9,10,11,12,13,14], although they are poorly stable, owing to their oxophilicity that renders them air and moisture sensitive. Late transition metals [3,15,16,17,18,19] feature a superior stability to moisture and air, together with broader functional group tolerance. Remarkably, cationic gold-based catalysts were highly active in the hydroamination of alkynes with amines [20,21]. The choice of the catalytic system is driven not only by the performance but also by its availability and cost. In this regard, palladium is clearly more attractive than rhodium, iridium, or gold.
Palladium-catalyzed hydroamination reactions have found several applications in the synthesis of biologically relevant heterocycles [22,23,24,25]. Alkynes [26], alkenes [27], dienes [28], and allenes [29] can serve as acceptor in combination with both aromatic and aliphatic amines. Despite the versatility of palladium-catalyzed hydroaminations, elaborate and expensive catalytic systems are often required. In particular, in the reaction between an alkyne and an aromatic amine, palladium has been employed in the presence of NHC [30] or NHCP [31] ligands. In both these cases, the catalytic precursor needs to be activated with a silver salt that acts as halogen scavenger. 3-Imino phosphine (3IP) ligands [32] also provide an efficient alternative but, similarly, a multistep synthesis for obtaining the catalytic active complex is required. Yamamoto and coworkers contributed significantly to the field by exploiting the possibility of using simple palladium salts such as palladium nitrate [33]. A dramatic rate enhancement effect was observed with 2-aminophenols, where the chelating OH group was crucial for the reaction outcome. Subsequently, the same research group was able to expand the reaction scope to variously substituted aryl amines resorting to an aqua palladium complex [Pd(dppe)(H2O)2](TfO)2 [34]. The Yamamoto’s group also reported the use of Pd(PPh3)4 in combination with benzoic acid for the hydroamination of internal alkynes to allyl amines [35].
Owing to its flexibility and synthetic attractiveness, hydroamination reactions are still of high interest, and we recently reported the hydroamidation of propargylic ureas to give imidazolidin-2-ones and imidazol-2-ones [36]. In this work, we report the use of a cheap and commercially available palladium catalyst (i.e., PdI2) for the hydroamination of arylacetylenes with anilines under mild reaction conditions and low catalyst loading.
2. Results and Discussion
The palladium-catalyzed intermolecular hydroamination reaction was initially investigated using aniline (1a, 1 equiv, 0.4 mmol) and phenylacetylene (2a, 1.2 equiv) as model substrates, in dioxane (0.2 M) as the solvent at 80 °C for 18 h, in the presence of catalytic amount of a palladium salt (2 mol%, Table 1). Palladium acetate did not provide any conversion, while palladium dichloride produced only traces of imine 3a (Table 1, Entries 1–2). Gratifyingly, palladium iodide resulted in 73% 1HNMR yield of the hydroamination product, which was identified as (E)-N,1-diphenylethan-1-imine 3a deriving from the Markovnikov addition to the triple bond followed by isomerization (Table 1, Entry 3). An excess of iodide anions (from added KI) caused a slight decrease in the catalytic activity of PdI2 (Table 1, Entry 4), and a similar outcome was found in the presence of K2PdI4 (Table 1, Entry 5). This result is in agreement with Brunet’s study, where PtBr2 was used in combination with bromide anions for the hydroamination of terminal alkynes [37]. The excess of iodide anions can in this case be detrimental to the yield, since it can affect the equilibrium of species I and II in the catalytic cycle (see below). We then investigated the effect of concentration, temperature, catalyst loading, and solvent. At 0.4 M, compound 3a was obtained with 81% yield (Table 1, entries 6), while a further increase of the concentration to 0.8 M led to a less satisfactory result (Table 1, entries 7). As expected, temperature also plays a fundamental role in this transformation. In fact, when the reaction was performed at 100 °C, we observed a slightly decrease in the yield of 3a (Table 1, entry 8, 77%). Synthetically useful yields were still observed when reducing the catalyst loading to 1.0 mol% (Table 1, entry 9) and to 0.5 mol% (Table 1, entry 10) with yields of 3a of 63% and 47%, respectively.

Table 1.
Optimization study for the hydroamination of phenylacetylene with anilinea.
Lastly, different solvents were evaluated. Dimethylformamide (DMF) proved to be ineffective under these reaction conditions (entry 11), while acetonitrile and toluene allowed the formation of the desired imine 3a even though in lower yields (entries 12 and 13).
Once the best reaction conditions (Table 1, entry 6) were determined, we then investigated the scope for variously substituted anilines (1) and arylacetylenes (2) (Scheme 1). In general, the yields attained using PdI2 as catalyst were comparable with the ones achieved with the previously employed more complex catalytic systems [30]. For example, different alkyl chains at the para position of the aniline provided good yields, irrespective of the length of the chain (yields of 3b, 67%; 3c, 79%; 3e, 67%) or the steric hindrance (3d, 80%). Interestingly, an isopropyl group at the ortho position of the aniline also allowed an excellent product yield (3f, 83%). Fluoro-containing groups were well tolerated under these reaction conditions, regardless of their position or electronic properties. With a CF3 group at the meta position, the expected imine 3g was formed in 78% yield, and the same result was observed starting from the aniline ring bearing an OCF3 substituent para to the NH2 group (3h, 78%). para-Substituted anilines with fluoro, chloro, or bromo smoothly underwent the hydroamination, resulting in 75% (3i), 70% (3j), and 60% (3k) yields of the desired imines, respectively. Even a disubstituted aniline, such as 2-methyl-3-chloroaniline, behaved nicely, providing the corresponding product 3l with 85% yield. The OMe substituent in para position on the aniline ring led to 95% yield of 3m, while the presence of two OMe groups caused a significant decrease of the yield (3n, 29%).
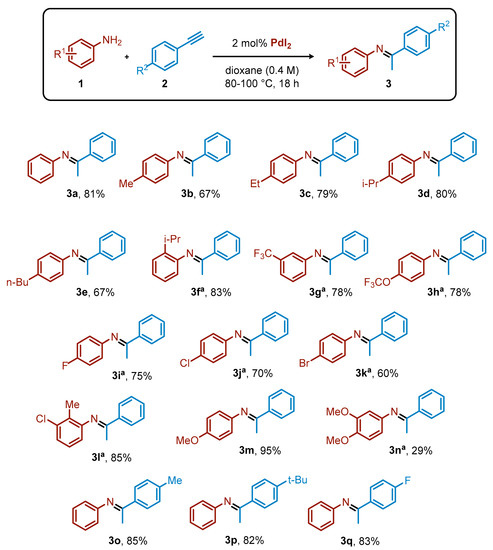
Scheme 1.
Scope for the hydroamination of arylacetylene derivatives with anilines. Reaction conditions: 1 (1 equiv, 0.8 mmol), 2 (1.2 equiv, 1 mmol), PdI2 (2 mol%, 0.016 mmol), dioxane (2 mL), 80 °C, 18 h. Yield calculated by 1H NMR using dimethyl maleate as internal standard. a Reaction performed at 100 °C.
The versatility of the process with respect to the nature of the arylacetylene partner was also considered. Both electron-withdrawing and electron-donating groups were highly compatible with the catalytic protocol, providing excellent yields of the corresponding imines (3o–q, 82–85%).
Other substrates were less tolerated (Scheme 2). For instance, 4-iodoaniline gave only 26% yield of the corresponding imine (3r). The iodide on the ring was highly reactive under palladium catalysis, leading to a complex mixture of byproducts. For the same reason, 2-bromoaniline in combination with phenyl acetylene gave only 30% of the expected 3s. The presence of a strongly electron-withdrawing group, such as the nitro group on the aniline ring, completely inhibited the process. Alkylacetylenes were considerably less reactive than arylacetylenes, as exemplified by the formation of 3v in 20% yield starting from 1-decyne. Primary alkylamines were not reactive under the optimized conditions, likely due to their pronounced coordinating nature.
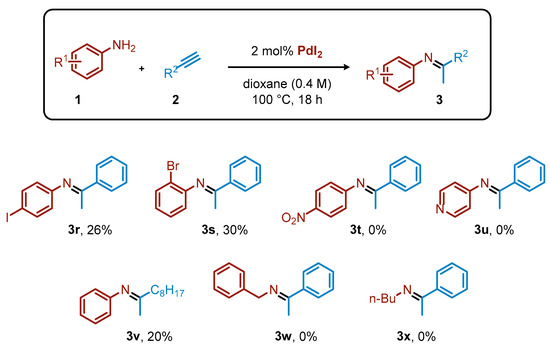
Scheme 2.
Limitations for the PdI2-catalyzed hydroamination of acetylene derivatives. Yield calculated by 1H NMR.
Not surprisingly, imine products 3 were unstable under acidic conditions and the isolated yields after column chromatography were very low. We then decided to reduce compounds 3 to the corresponding, more stable secondary amines 4 (See Supplementary Materials for NMR spectra of 4), using simple and inexpensive NaBH4. The overall isolated yields for the two-step synthesis of secondary amines 4 (I step: PdI2-catalyzed hydroamination and II step: reduction) starting from anilines and terminal arylacetylenes, reported in Scheme 3, ranged from 17% to 90%. In some cases, the two-step yield was about 20% lower than the corresponding NMR hydroamination yield (4o-q). This was mainly due to a non-optimized reduction procedure, which can be improved with the use of alternative reduction reagents, such as NaBH3CN.
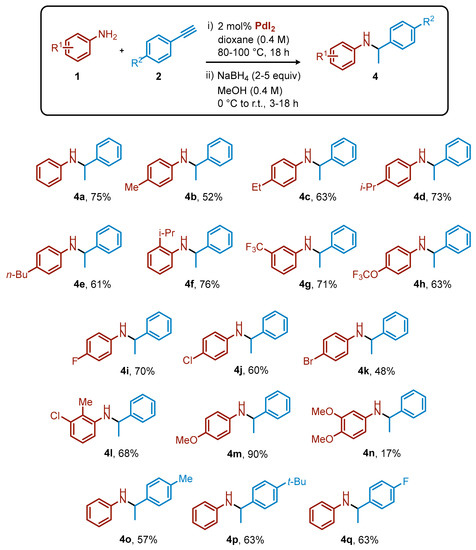
Scheme 3.
Overall isolated yields for secondary amines 4 after two steps (hydroamination and reduction).
From a mechanistic point of view, coordination of the triple bond of 2 to PdI2 generates the π-complex I (Scheme 4). This type of coordination makes the triple bond susceptible to undergo nucleophilic attack of the nitrogen atom of amine 1 resulting in the vinylpalladium iodide intermediate II. Then, protonolysis of II regenerates PdI2 and leads to enamine intermediate III, which readily tautomerizes to imine 3. The presence of the iodide counteranion in the process is crucial and we believe that its role may be connected, on one hand, to its higher ability to act as the leaving group in the nucleophilic attack step and, on the other hand, to its higher electron-releasing power, which tends to favor the protonolysis step [38]. Moreover, iodide anions display a superior ability to stabilize palladium(II) species, compared to other counteranions, such as acetates [39]. Efforts at understanding the effect of iodide anions in this transformation are underway in our laboratories [40].
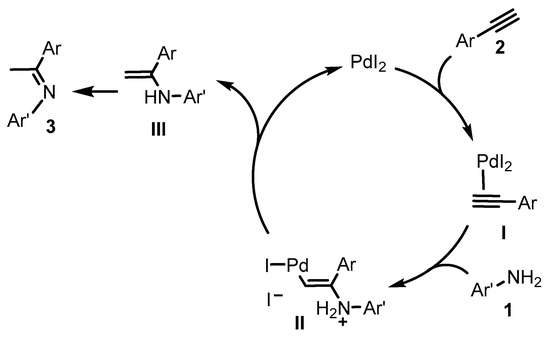
Scheme 4.
Proposed mechanism for PdI2-catalyzed hydroamination of terminal alkynes 2 with anilines 1 leading to imines 3 through the formation of vinylamines III as intermediates.
3. Materials and Methods
3.1. Materials
All chemicals were purchased from commercial sources and used as received. Dioxane and methanol were dried and stored over molecular sieves previously activated in an oven at 300 °C overnight. Catalytic reactions were carried out under nitrogen using the standard Schlenk technique. Reaction mixtures were analyzed using a GC Agilent Tenchnologies 7820A GC System equipped with FID detector and column Agilent Technologies 19091J 413 (30 m × 0.32mm) and a gas chromatograph Agilent Technologies 6890N Network GC System equipped with quadrupole Agilent Technologies 5973 Network Mass Selective Detector and column Fused Silica Capillary Column (30 m × 0.25 mm) for GC-MS analyses. 1H NMR and 13C NMR spectra were recorded at 300 K on a Bruker 400 MHz using the solvent as internal standard (7.26 ppm for 1H NMR and 77.16 ppm for 13C NMR for CDCl3). The terms m, s, d, t, q and quint represent multiplet, singlet, doublet, triplet, quadruplet, and quintuplet respectively, and the term br means a broad signal.
3.2. General Procedures
3.2.1. Hydroamination of Terminal Alkynes with Anilines
A flame-dried test tube of 10 mL was charged with palladium iodide (0.016 mmol, 2 mol%), aniline 1 (0.8 mmol, 1 equiv) and alkyne 2 (1 mmol, 1.2 equiv). The test tube was sealed with a rubber cup, filled with nitrogen, evacuated, and backfilled with nitrogen three times. Then, dry 1,4-dioxane (2 mL) was added. The tube was placed in an oil bath at 80 or 100 °C and stirred for 18 hours. The reaction crude was cooled to room temperature, filtered through celite, and dried under reduced pressure. The 1H NMR analysis on the reaction crude using dimethyl maleate as the internal standard was immediately acquired to determine the yield of imine 3.
3.2.2. Reduction of Imine 3 to Secondary Amine 4
The hydroamination crude was transferred in a two-neck round-bottom flask and dissolved in dry methanol (2 mL). The flask was placed in an ice bath and sodium borohydride (2–5 equiv) was added portion wise. The mixture was stirred at 0 °C until a complete conversion of 3, detected by TLC analysis. The mixture was quenched with 2 mL of KOH (1M). The crude was diluted with 20 mL of EtOAc, washed with water and brine and dried over MgSO4. The pure amine 4 was obtained after flash column chromatography using a mixture of hexane and EtOAc as eluent.
3.3. Product Characterization
N-(1-Phenylethyl)aniline (4a) was synthesized from aniline (73 μL) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4a (118 mg, 75%) as pale-yellow oil. The spectroscopic data of 4a are consistent with literature values [41]. 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.6 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.2 Hz, 1H), 7.18 (t, J = 7.9 Hz, 2H), 6.75 (t, J = 7.3 Hz, 1H), 6.62 (d, J = 7.8 Hz, 2H), 4.57 (q, J = 6.7 Hz, 1H), 4.38 (br s, 1H), 1.60 (d, J = 6.7 Hz, 3H).
4-Methyl-N-(1-phenylethyl)aniline (4b) was synthesized from p-toluidine (86 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Four equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4b (88 mg, 52%) as yellow oil. The spectroscopic data of 4b are consistent with literature values [41]. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.7 Hz, 2H), 7.49 (t, J = 7.1 Hz, 2H), 7.43–7.37 (m, 1H), 7.10 (d, J = 7.7 Hz, 2H), 6.63 (d, J = 7.8 Hz, 2H), 4.64 (q, J = 6.8 Hz, 1H), 4.05 (br s, 1H), 2.39 (s, 3H), 1.68 (d, J = 6.9 Hz, 3H).
4-Ethyl-N-(1-phenylethyl)aniline (4c) was synthesized from 4-ethylaniline (99 μL) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4c (113 mg, 63%) as yellow oil. The spectroscopic data of 4c are consistent with literature values [42]. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 7.6 Hz, 2H), 7.43 (t, J = 7.5 Hz, 2H), 7.34 (t, J = 7.2 Hz, 1H), 7.06 (d, J = 8.5 Hz, 2H), 6.60 (d, J = 8.4 Hz, 2H), 4.58 (q, J = 6.7 Hz, 1H), 4.19 (br s, 1H), 2.63 (q, J = 7.6 Hz, 2H), 1.62 (d, J = 6.7 Hz, 3H), 1.28 (t, J = 7.6 Hz, 3H).
4-Isopropyl-N-(1-phenylethyl)aniline (4d) was synthesized from 4-isopropylaniline (108 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4d (140 mg, 73%) as orange viscous oil. The spectroscopic data of 4d are consistent with literature values [43]. 1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 7.5 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.29 (t, J = 7.2 Hz, 1H), 7.04 (d, J = 8.2 Hz, 2H), 6.56 (d, J = 8.2 Hz, 2H), 4.52 (q, J = 6.7 Hz, 1H), 4.32 (br s, 1H), 2.83 (hept, J = 6.9 Hz, 1H), 1.57 (d, J = 6.7 Hz, 3H), 1.24 (d, J = 6.9, 6H).
4-Butyl-N-(1-phenylethyl)aniline (4e) was synthesized from 4-butylaniline (119 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4e (124 mg, 61%) as dark orange oil. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.5 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.28 (t, J = 7.2 Hz, 1H), 6.98 (d, J = 8.4 Hz, 2H), 6.54 (d, J = 8.4 Hz, 2H), 4.52 (q, J = 6.7 Hz, 1H), 4.29 (br s, 1H), 2.52 (t, J = 7.7 Hz, 1H), 1.64–1.51 (m, 5H), 1.42-1.34 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 145.3, 144.9, 132.1, 129.1 (2C), 128.7 (2C), 127.0, 126.1 (2C), 113.7 (2C), 54.1, 34.8, 34.0, 25.0, 22.4, 14.1.
2-Isopropyl-N-(1-phenylethyl)aniline (4f) was synthesized from 2-isopropylaniline (108 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4f (145 mg, 76%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 7.7 Hz, 2H), 7.34 (t, J = 7.4 Hz, 2H), 7.28–7.21 (m, 1H), 7.18 (d, J = 7.6 Hz, 1H), 6.97 (t, J = 7.7 Hz, 1H), 6.73 (br s, 1H), 6.45 (br s, 1H), 4.55 (q, J = 6.7 Hz, 1H), 4.07 (br s, 1H), 3.02 (br s, 1H), 1.61 (br s, 3H), 1.35 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 145.5, 143.9, 131.8, 128.8, 126.9, 126.7, 125.9, 124.9, 117.2, 111.8, 53.5, 27.5, 25.5, 22.5, 22.4.
N-(1-Phenylethyl)-3-(trifluoromethyl)aniline (4g) was synthesized from 3-trifluoromethylaniline (100 μL) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4g (151 mg, 71%) as dark yellow oil. The spectroscopic data of 4g are consistent with literature values [44]. 1H NMR (400 MHz, CDCl3) δ 7.39–7.31 (m, 4H), 7.29–7.23 (m, 1H), 7.17 (t, J = 7.9 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 6.80 (s, 1H), 6.65 (d, J = 8.2 Hz, 1H), 4.51 (q, J = 6.7 Hz, 1H), 1.57 (d, J = 6.7 Hz, 3H).
N-(1-Phenylethyl)-4-(trifluoromethoxy)aniline (4h) was synthesized from 4-trifluoromethoxyaniline (108 μL) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4h (142 mg, 63%) as dark orange oil. 1H NMR (400 MHz, CDCl3) δ 7.44–7.34 (m, 4H), 7.32–7.24 (m, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.55–6.42 (m, 2H), 4.47 (q, J = 6.7 Hz, 1H), 4.14 (br s, 1H), 1.55 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 146.1, 144.8, 140.3 (q, J = 1.7 Hz), 128.8 (2C), 127.1, 125.8 (2C), 122.3 (2C), 120.7 (q, J = 255.1 Hz), 113.5 (2C), 53.8, 25.1.
4-Fluoro-N-(1-phenylethyl)aniline (4i) was synthesized from 4-fluoraniline (76 μL) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (95:5) as eluent to give 4i (120 mg, 70%) as dark orange oil. The spectroscopic data of 4i are consistent with literature values [41]. 1H NMR (400 MHz, CDCl3) δ 7.44–7.33 (m, 4H), 7.32–7.24 (m, 1H), 6.90–6.79 (m, 2H), 6.51–6.43 (m, 2H), 4.46 (q, J = 6.7 Hz, 1H), 3.96 (br s, 1H), 1.55 (d, J = 6.7 Hz, 3H).
4-Chloro-N-(1-phenylethyl)aniline (4j) was synthesized from 4-chloroaniline (102 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Five equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (95:5) as eluent to give 4j (111 mg, 60%) as orange solid. The spectroscopic data of 4i are consistent with literature values [41]. 1H NMR (400 MHz, CDCl3) δ 7.46–7.38 (m, 4H), 7.37–7.29 (m, 1H), 7.17–7.09 (m, 2H), 6.55–6.47 (m, 2H), 4.52 (q, J = 6.7 Hz, 1H), 4.12 (s, 1H), 1.58 (d, J = 6.7 Hz, 3H).
4-Bromo-N-(1-phenylethyl)aniline (4k) was synthesized from 4-bromoaniline (138 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4k (106 mg, 48%) as red solid. The spectroscopic data of 4k are consistent with literature values [42]. 1H NMR (400 MHz, CDCl3) δ 7.38–7.31 (m, J = 7.3, 6.1 Hz, 4H), 7.30–7.23 (m, 1H), 7.21–7.14 (m, 2H), 6.40 (d, J = 8.8 Hz, 2H), 4.46 (q, J = 6.7 Hz, 1H), 4.10 (br s, 1H), 1.53 (d, J = 6.7 Hz, 3H).
3-Chloro-2-methyl-N-(1-phenylethyl)aniline (4l) was synthesized from 3-chloro-2-methylaniline (119 μL) and phenylacetylene (105 μL) following the general hydroamination procedure. Three equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate/CH2Cl2 (89:2:9) as eluent to give 4l (134 mg, 68%) as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.41–7.34 (m, 4H), 7.32–7.25 (m, 1H), 6.89 (t, J = 8.0 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H), 6.33 (d, J = 8.1 Hz, 1H), 4.57 (q, J = 6.7 Hz, 1H), 4.04 (br s, 1H), 2.36 (s, 3H), 1.62 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 146.3, 144.8, 134.5, 128.8 (2C), 127.2, 127.1, 125.8 (2C), 119.4, 118.0, 109.7, 53.7, 25.3, 13.8.
4-Methoxy-N-(1-phenylethyl)aniline (4m) was synthesized from 4-methoxyaniline (98 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Two equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4m (181 mg, 90%) as orange crystals. The spectroscopic data of 4m are consistent with literature values [41]. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 7.44–7.33 (m, 4H), 7.30–7.25 (m, 1H), 6.77–6.71 (m, 2H), 6.55–6.50 (m, 2H), 4.46 (q, J = 6.6 Hz, 1H), 4.00–3.60 (two signals: br s, 1H and s, 3H centred at 3.74 ppm), 1.54 (d, J = 6.7 Hz, 3H).
3,4-Dimethoxy-N-(1-phenylethyl)aniline (4n) was synthesized from 3,4-dimethoxyaniline (123 mg) and phenylacetylene (105 μL) following the general hydroamination procedure. Five equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate/CH2Cl2 (89:2:9) as eluent to give 4n (35 mg, 17%) as dark red oil. 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 7.6 Hz, 2H), 7.32 (t, J = 7.7 Hz, 2H), 7.27–7.19 (m, 1H), 6.65 (d, J = 8.6 Hz, 1H), 6.20 (s, 1H), 6.05 (d, J = 8.6 Hz, 1H), 4.43 (q, J = 6.7 Hz, 1H), 3.76 (s, 3H), 3.73 (s, 3H), 1.53 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 149.7, 144.9, 141.7, 128.7, 127.0, 126.0, 112.8, 104.9, 99.8, 56.5, 55.6, 54.9, 24.8.
N-(1-(p-Tolyl)ethyl)aniline (4o) was synthesized from aniline (72 μL) and 4-ethynyltoluene (122 μL) following the general hydroamination procedure. Three equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4o (96 mg, 57%) as yellow oil. The spectroscopic data of 4o are consistent with literature values [43]. 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 8.3 Hz, 2H), 7.29–7.17 (m, 4H), 6.82–6.73 (m, 1H), 6.64 (d, J = 6.0 Hz, 2H), 4.60–4.54 (m, 1H), 3.97 (br s, 1H), 2.44 (s, 3H), 1.60 (d, J = 6.7 Hz, 3H).
N-(1-(4-(tert-Butyl)phenyl)ethyl)aniline (4p) was synthesized from aniline (72 μL) and 4-tert-butylphenylacetylene (179 μL) following the general hydroamination procedure. Three equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4p (128 mg, 63%) as orange solid. The spectroscopic data of 4p are consistent with literature values [45]. 1H NMR (300 MHz, CDCl3) δ 7.40–7.27 (m, 4H), 7.16–7.07 (m, 2H), 6.66 (t further split, J = 7.3, 1H), 6.55 (d, J = 8.5 Hz, 2H), 4.50 (q, J = 6.7 Hz, 1H), 4.03 (br s, 1H), 1.53 (d further split, J = 6.7, 3H), 1.35–1.30 (m, 9H).
N-(1-(4-Fluorophenyl)ethyl)aniline (4q) was synthesized from aniline (72 μL) and 4-fluorophenylacetylene (110 μL) following the general hydroamination procedure. Three equiv of NaBH4 were employed during the reduction step. The reaction crude was purified by flash column chromatography using hexane/ethyl acetate (98:2) as eluent to give 4q (108 mg, 63%) as dark orange oil. The spectroscopic data of 4q are consistent with literature values [41]. 1H NMR (400 MHz, CDCl3) δ 7.43–7.34 (m, 2H), 7.21–7.13 (m, 2H), 7.06 (t, J = 8.7 Hz, 2H), 6.74 (t, J = 7.3 Hz, 1H), 6.57 (d, J = 7.6 Hz, 2H), 4.52 (q, J = 6.7 Hz, 1H), 4.24 (br s, 1H), 1.55 (d, J = 6.8 Hz, 3H).
4. Conclusions
In conclusion, we developed a simple and versatile palladium-catalyzed protocol for the hydroamination of terminal arylacetylenes with anilines, in the absence of organic ligands or additives. This novel methodology features high functional group tolerance and offers a valuable alternative to reported methods for the synthesis of secondary aromatic amines. The presence of iodide anions was crucial to achieve good to excellent yields of the corresponding imine products. We envisage wide diffusion of this versatile methodology within the organic chemistry community.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/2/176/s1, Copies of NMR spectra.
Author Contributions
A.C. co-supervised the project and wrote the original draft of the paper; A.V. carried out most of the NMR analyses; D.G.F. carried out the catalytic experiments and the chromatographic separations; R.M., B.G., and E.M. were involved in writing and editing the manuscript; N.D.C. supervised the project and revised the paper. All authors have read and agreed to the published version of the manuscript.
Funding
We wish to thank the University of Parma and MIUR (Ministry of Education, University and Research, FFABR 2017) for financial support and CIM (Interdepartmental Measurements Centre) for NMR facilities.
Acknowledgments
This work has benefited from the equipment and framework of the COMP-HUB Initiative, funded by the ‘Departments of Excellence’ program of the Italian Ministry for Education, University and Research (MIUR, 2018-2022).
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Huo, J.; He, G.; Chen, W.; Hu, X.; Deng, Q.; Chen, D. A minireview of hydroamination catalysis: Alkene and alkyne substrate selective, metal complex design. BMC Chem. 2019, 13, 89–101. [Google Scholar] [CrossRef]
- Patel, M.; Saunthwal, R.K.; Verma, A.K. Base-Mediated Hydroamination of Alkynes. Acc. Chem. Res. 2017, 50, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L.J. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Carbon–heteroatom bond formation catalysed by organometallic complexes. Nature 2008, 455, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct addition of amines to alkenes and alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef]
- Severin, R.; Doye, S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 2007, 36, 1407–1420. [Google Scholar] [CrossRef]
- Pohlki, F.; Doye, S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 2003, 32, 104–114. [Google Scholar] [CrossRef]
- Müller, T.E.; Beller, M. Metal-initiated amination of alkenes and alkynes. Chem. Rev. 1998, 98, 675–704. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Z. Asymmetric hydrofunctionalization of minimally functionalized alkenes via earth abundant transition metal catalysis. Org. Chem. Front. 2018, 5, 260–272. [Google Scholar] [CrossRef]
- Griffin, S.E.; Pacheco, J.; Schafer, L.L. Reversible C–N Bond Formation in the Zirconium-Catalyzed Intermolecular Hydroamination of 2-Vinylpyridine. Organometallics 2019, 38, 1011–1016. [Google Scholar] [CrossRef]
- Eedugurala, N.; Hovey, M.; Ho, H.-A.; Jana, B.; Lampland, N.L.; Ellern, A.; Sadow, A.D. Cyclopentadienyl-bis(oxazoline) Magnesium and Zirconium Complexes in Aminoalkene Hydroaminations. Organometallics 2015, 34, 5566–5575. [Google Scholar] [CrossRef]
- Reznichenko, A.L.; Hultzsch, K.C. Early Transition Metal (Group 3–5, Lanthanides and Actinides) and Main Group Metal (Group 1, 2, and 13) Catalyzed Hydroamination. In Hydrofunctionalization; Ananikov, V.P., Tanaka, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 43, pp. 51–114. [Google Scholar]
- Reznichenko, A.L.; Nguyen, H.N.; Hultzsch, K.C. Asymmetric intermolecular hydroamination of unactivated alkenes with simple amines. Angew. Chem. Int. Ed. 2010, 49, 8984–8987. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Marks, T.J. Organolanthanide-Catalyzed Hydroamination. Acc. Chem. Res. 2004, 37, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Yahata, K.; Kaneko, Y.; Akai, S. Cobalt-Catalyzed Intermolecular Markovnikov Hydroamination of Nonactivated Olefins: N 2 -Selective Alkylation of Benzotriazole. Org. Lett. 2020, 22, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-H.; Lu, A.; Dong, V.M. Intermolecular Hydroamination of 1,3-Dienes To Generate Homoallylic Amines. J. Am. Chem. Soc. 2017, 139, 14049–14052. [Google Scholar] [CrossRef] [PubMed]
- Gurak, J.A.; Yang, K.S.; Liu, Z.; Engle, K.M. Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc. 2016, 138, 5805–5808. [Google Scholar] [CrossRef] [PubMed]
- Sevov, C.S.; Zhou, J.; Hartwig, J.F. Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc. 2014, 136, 3200–3207. [Google Scholar] [CrossRef]
- Cooke, M.L.; Xu, K.; Breit, B. Enantioselective Rhodium-Catalyzed Synthesis of Branched Allylic Amines by Intermolecular Hydroamination of Terminal Allenes. Angew. Chem. Int. Ed. 2012, 51, 10876–10879. [Google Scholar] [CrossRef]
- Malhotra, D.; Mashuta, M.S.; Hammond, G.B.; Xu, B. A Highly Efficient and Broadly Applicable Cationic Gold Catalyst. Angew. Chem. Int. Ed. 2014, 53, 4456–4459. [Google Scholar] [CrossRef]
- Lavallo, V.; Wright, J.H.; Tham, F.S.; Quinlivan, S. Perhalogenated Carba-closo-dodecaborate Anions as Ligand Substituents: Applications in Gold Catalysis. Angew. Chem. Int. Ed. 2013, 52, 3172–3176. [Google Scholar] [CrossRef]
- Platon, M.; Amardeil, R.; Djakovitch, L.; Hierso, J.C. Progress in palladium-based catalytic systems for the sustainable synthesis of annulated heterocycles: A focus on indole backbones. Chem. Soc. Rev. 2012, 41, 3929–3968. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Junge, K.; Beller, M. A General Catalytic Hydroamidation of 1, 3-Dienes: Atom-Efficient Synthesis of N-Allyl Heterocycles, Amides, and Sulfonamides. Angew. Chem. Int. Ed. 2014, 53, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Lutete, L.M.; Kadota, I.; Yamamoto, Y. Palladium-catalyzed intramolecular asymmetric hydroamination of alkynes. J. Am. Chem. Soc. 2004, 126, 1622–1623. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, B.; Salerno, G.; Fazio, A. General and regioselective synthesis of substituted pyrroles by Metal-Catalyzed or spontaneous cycloisomerization of (Z)-(2-En-4-ynyl) amines. J. Org. Chem. 2003, 68, 7853–7861. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition metal-catalyzed addition of heteroatom−hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Michael, F.E.; Cochran, B.M. Room temperature palladium-catalyzed intramolecular hydroamination of unactivated alkenes. J. Am. Chem. Soc. 2006, 128, 4246–4247. [Google Scholar] [CrossRef]
- Löber, O.; Kawatsura, M.; Hartwig, J.F. Palladium-catalyzed hydroamination of 1, 3-dienes: A colorimetric assay and enantioselective additions. J. Am. Chem. Soc. 2001, 123, 4366–4367. [Google Scholar] [CrossRef]
- Besson, L.; Goré, J.; Cazes, B. Synthesis of allylic amines through the palladium-catalyzed hydroamination of allenes. Tetrahedron Lett. 1995, 36, 3857–3860. [Google Scholar] [CrossRef]
- Chen, Q.; Lv, L.; Yu, M.; Shi, Y.; Li, Y.; Pang, G.; Cao, C. Simple, efficient and reusable Pd–NHC catalysts for hydroamination. RSC Adv. 2013, 3, 18359–18366. [Google Scholar] [CrossRef]
- Franco, D.; Marchenko, A.; Koidan, G.; Hurieva, A.N.; Kostyuk, A.; Biffis, A. Palladium (II) Complexes with N-Phosphanyl-N-heterocyclic Carbenes as Catalysts for Intermolecular Alkyne Hydroaminations. ACS Omega 2018, 3, 17888–17894. [Google Scholar] [CrossRef]
- Shaffer, A.R.; Schmidt, J.A. Palladium (II) 3-iminophosphine complexes as intermolecular hydroamination catalysts for the formation of imines and enamines. Organometallics 2008, 27, 1259–1266. [Google Scholar] [CrossRef]
- Shimada, T.; Yamamoto, Y. Palladium-catalyzed intermolecular hydroamination of alkynes: A dramatic rate-enhancement effect of o-aminophenol. J. Am. Chem. Soc. 2002, 124, 12670–12671. [Google Scholar] [CrossRef]
- Shimada, T.; Bajracharya, G.B.; Yamamoto, Y. Aquapalladium complex: A stable and convenient catalyst for the intermolecular hydroamination of alkynes. Eur. J. Org. Chem. 2005, 2005, 59–62. [Google Scholar] [CrossRef]
- Kadota, I.; Shibuya, A.; Lutete, L.M.; Yamamoto, Y. Palladium/benzoic acid catalyzed hydroamination of alkynes. J. Org. Chem. 1999, 64, 4570–4571. [Google Scholar] [CrossRef] [PubMed]
- Casnati, A.; Perrone, A.; Mazzeo, P.P.; Bacchi, A.; Mancuso, R.; Gabriele, B.; Maggi, R.; Maestri, G.; Motti, E.; Stirling, A.; et al. Synthesis of Imidazolidin-2-ones and Imidazol-2-ones via Base-Catalyzed Intramolecular Hydroamidation of Propargylic Ureas under Ambient Conditions. J. Org. Chem. 2019, 84, 3477–3490. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.J.; Chu, N.C.; Diallo, O.; Vincendeau, S. Platinum-catalyzed intermolecular hydroamination of terminal alkynes. J. Mol. Catal. A Chem. 2005, 240, 245–248. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Costa, M. PdI2-Catalyzed Synthesis of Heterocycles. Synlett 2004, 2004, 2468–2483. [Google Scholar] [CrossRef]
- Maitlis, P.M.; Haynes, A.; James, B.R.; Catellani, M.; Chiusoli, G.P. Iodide effects in transition metal catalyzed reactions. Dalton Trans. 2004, 21, 3409–3419. [Google Scholar] [CrossRef]
- For a recent Pd-catalyzed transformation, involving a C-N bond formation, promoted by iodide anions, see: Casnati, A.; Fontana, M.; Coruzzi, G.; Aresta, B.M.; Corriero, N.; Maggi, R.; Maestri, G.; Motti, E.; Della Ca’, N. Enhancing Reactivity and Selectivity of Aryl Bromides: A Complementary Approach to Dibenzo [b,f] azepine Derivatives. ChemCatChem 2018, 10, 4346–4352. [Google Scholar] [CrossRef]
- Liang, S.; Hammond, L.; Xu, B.; Hammond, G.B. Commercial Supported Gold Nanoparticles Catalyzed Alkyne Hydroamination and Indole Synthesis. Adv. Synth. Catal. 2016, 358, 3313–3318. [Google Scholar] [CrossRef]
- Wallach, D.R.; Stege, P.C.; Shah, J.P.; Chisholm, J.D. Brønsted acid catalyzed monoalkylation of anilines with trichloroacetimidates. J. Org. Chem. 2015, 80, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Fleischer, S.; Jiao, H.; Junge, K.; Beller, M. Cooperative catalysis with iron and a chiral brønsted acid for asymmetric reductive amination of ketones. Adv. Synth. Catal. 2014, 356, 3451–3455. [Google Scholar] [CrossRef]
- Babu, N.S.; Reddy, K.M.; Prasad, P.S.; Suryanarayana, I.; Lingaiah, N. Intermolecular hydroamination of vinyl arenes using tungstophosphoric acid as a simple and efficient catalyst. Tetrahedron Lett. 2007, 48, 7642–7645. [Google Scholar] [CrossRef]
- Schroeter, F.; Lerch, S.; Kaliner, M.; Strassner, T. Cobalt-catalyzed hydroarylations and hydroaminations of alkenes in tunable aryl alkyl ionic liquids. Org. Lett. 2018, 20, 6215–6219. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).