Manganese Catalysts with Tetradentate N-donor Pyridine-Appended Bipiperidine Ligands for Olefin Epoxidation Reactions: Ligand Electronic Effect and Mechanism

Abstract
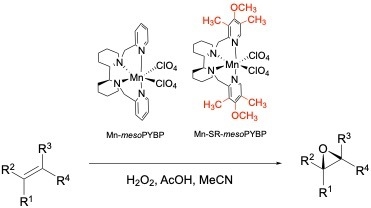
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
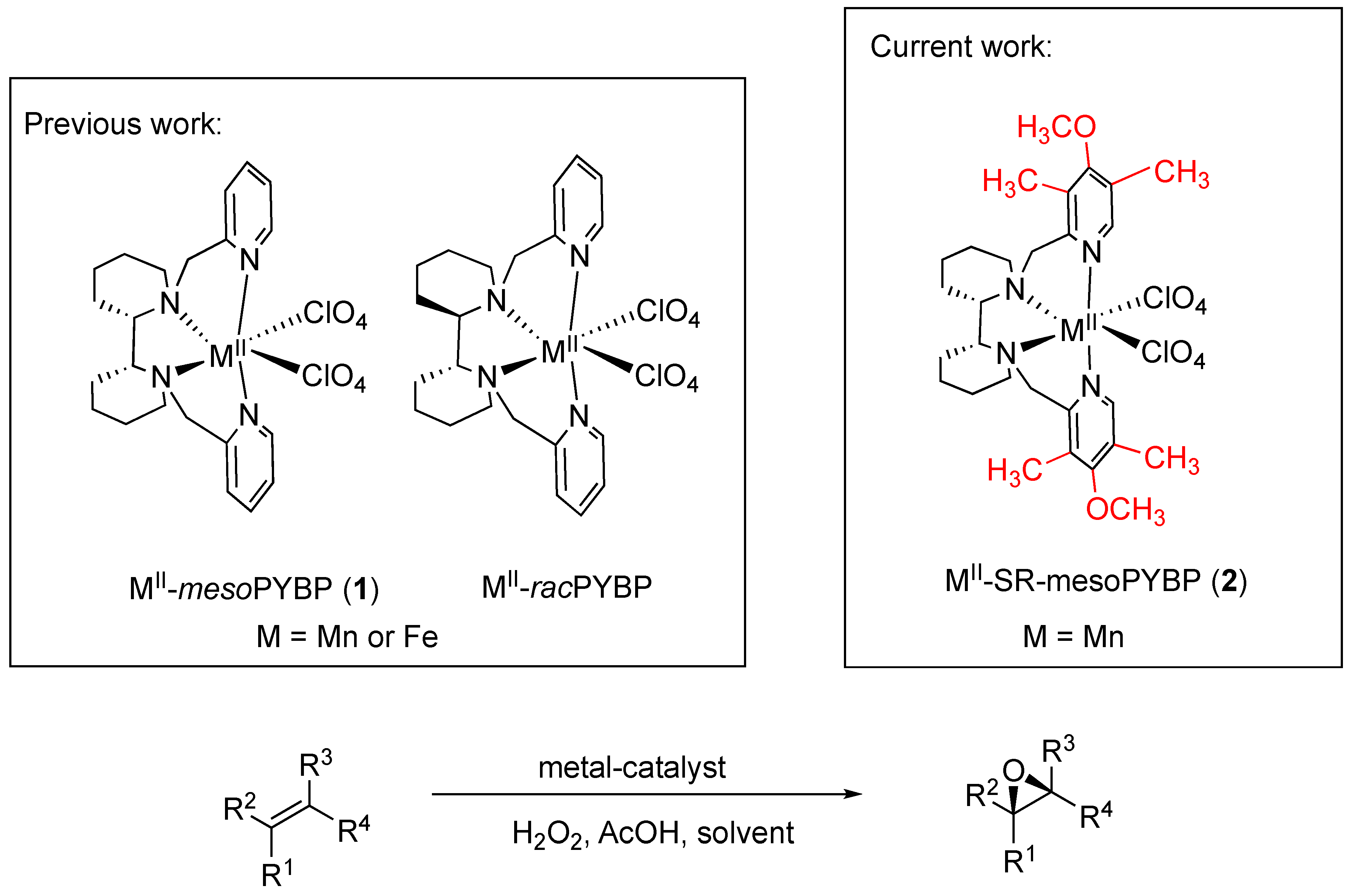
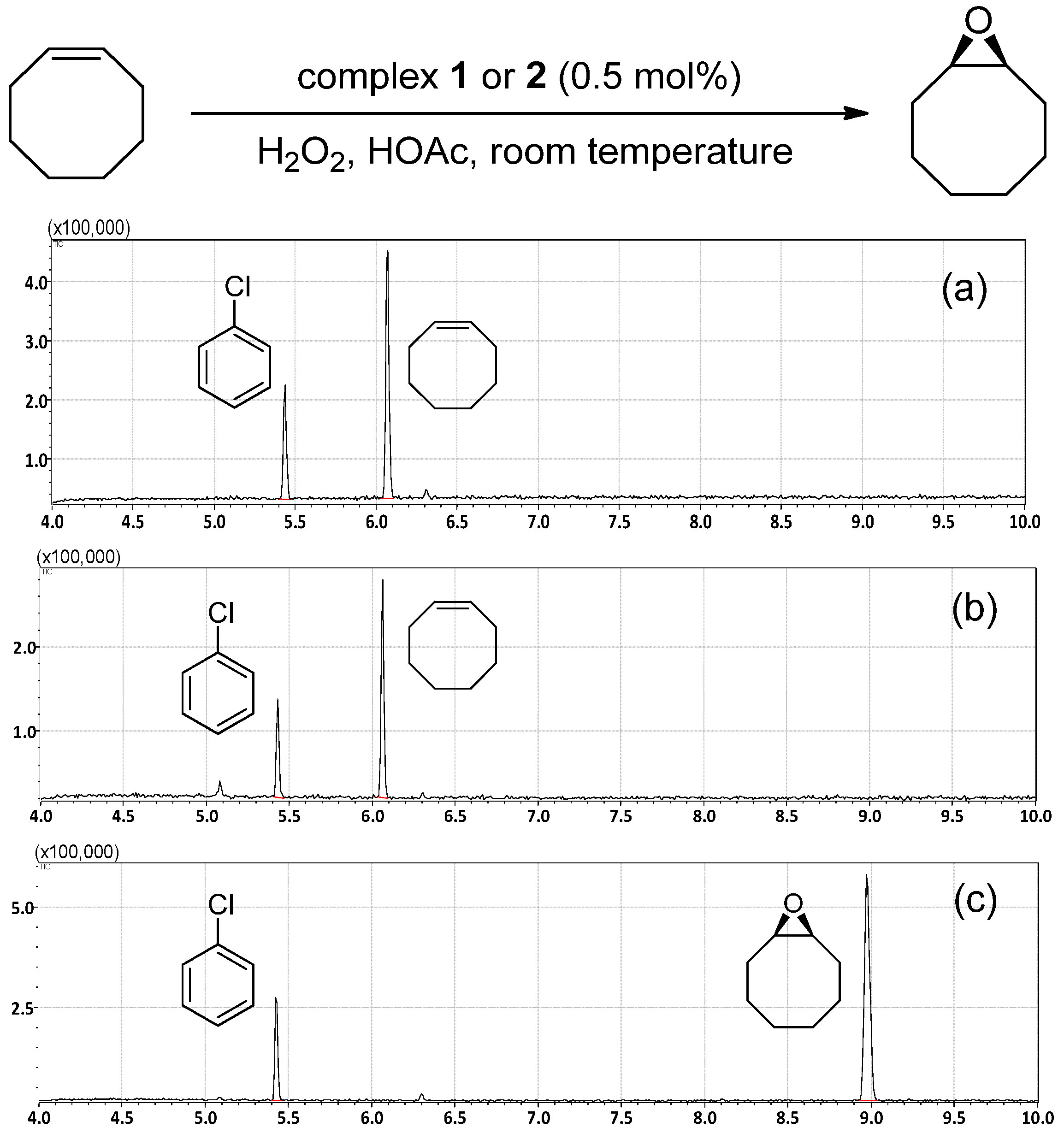
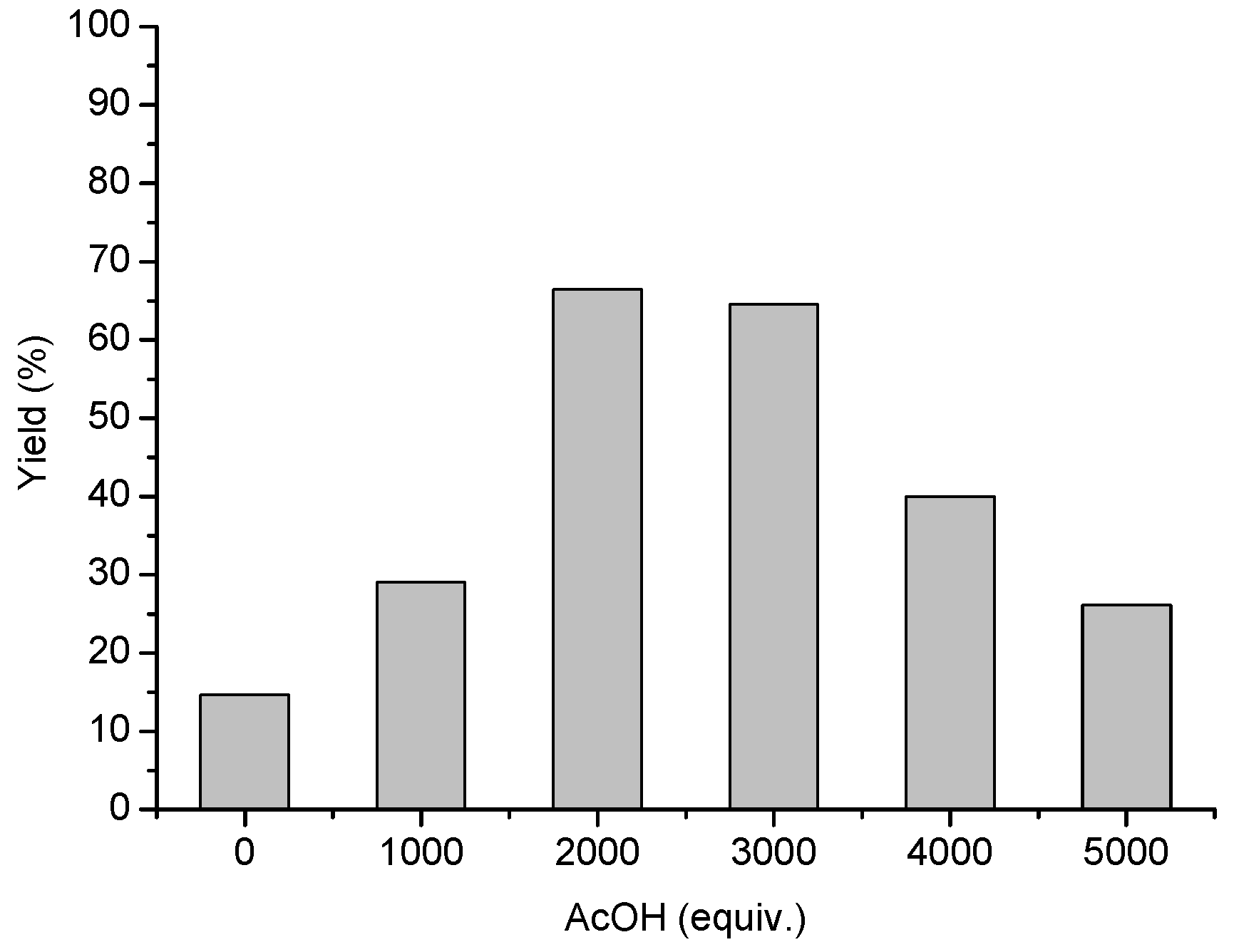
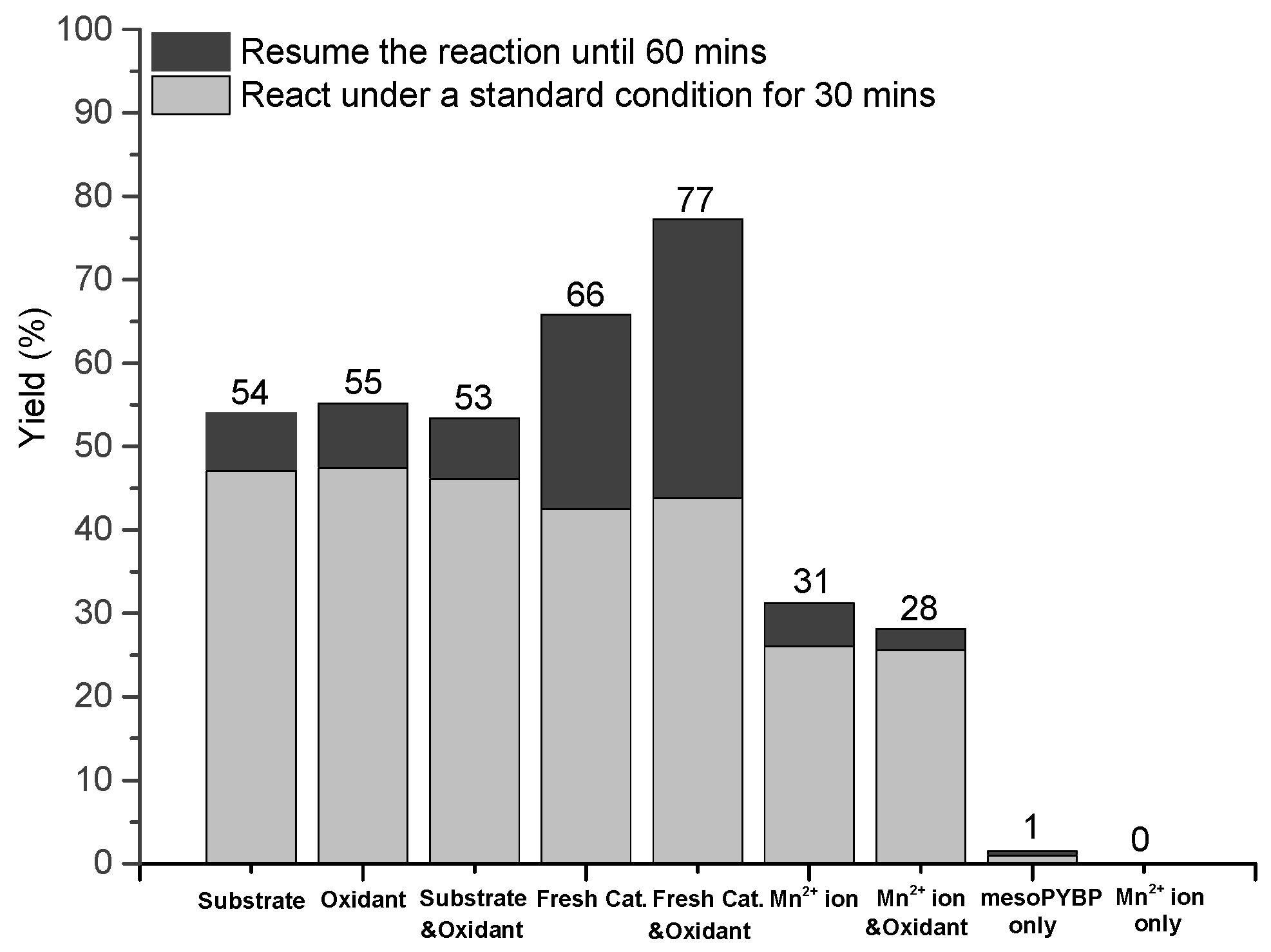
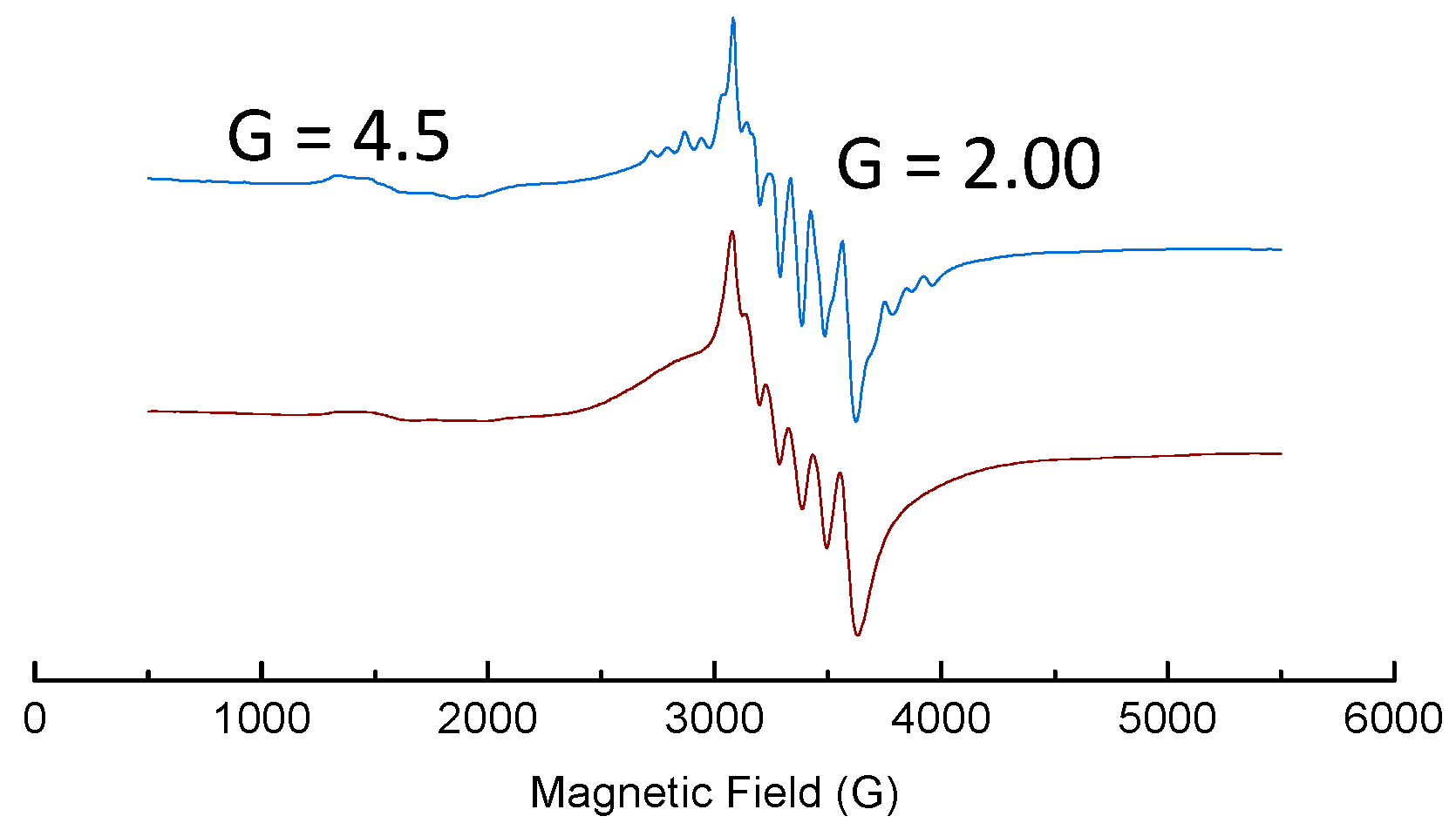
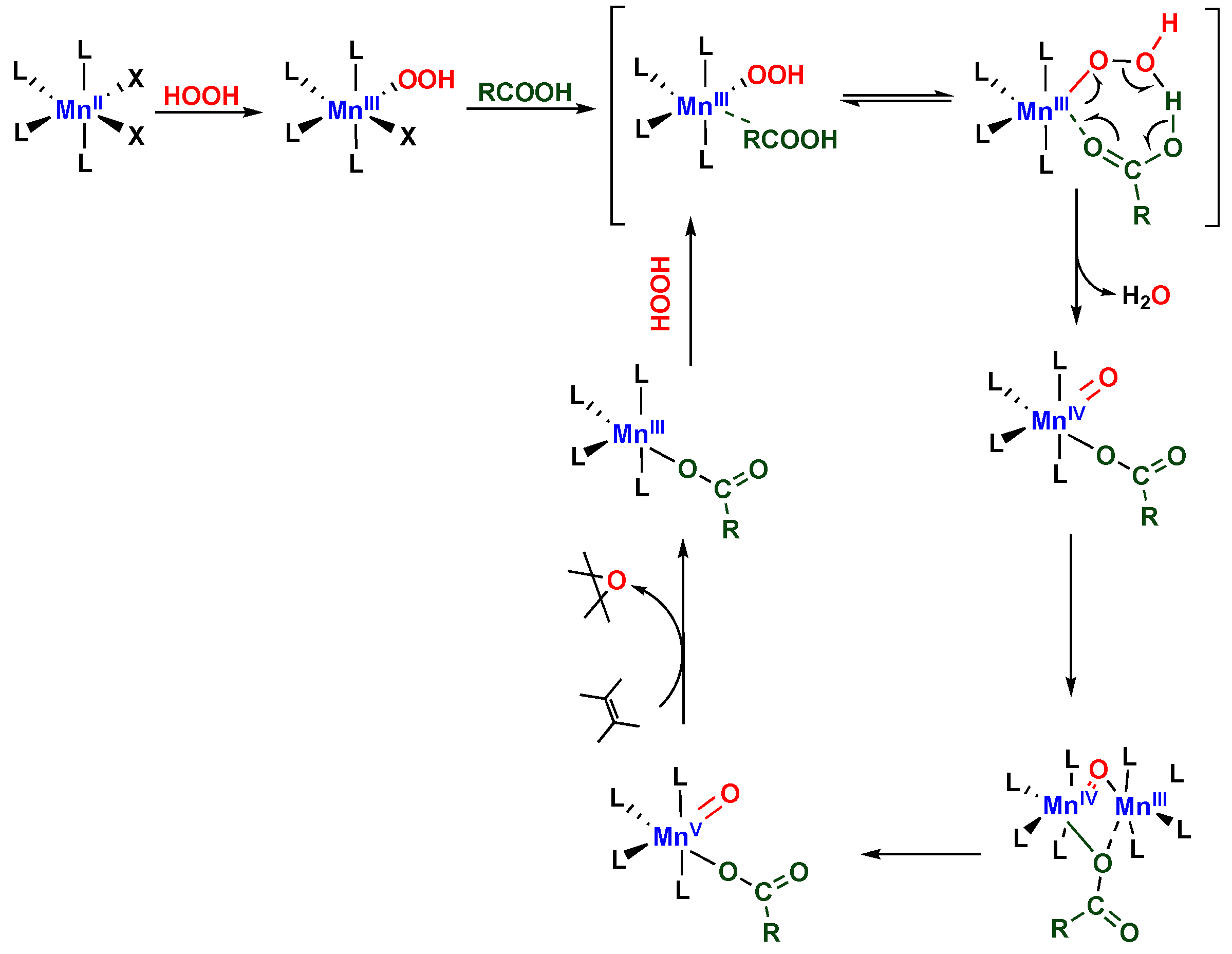
2. Results and Discussion
3. Materials and Methods
3.1. General
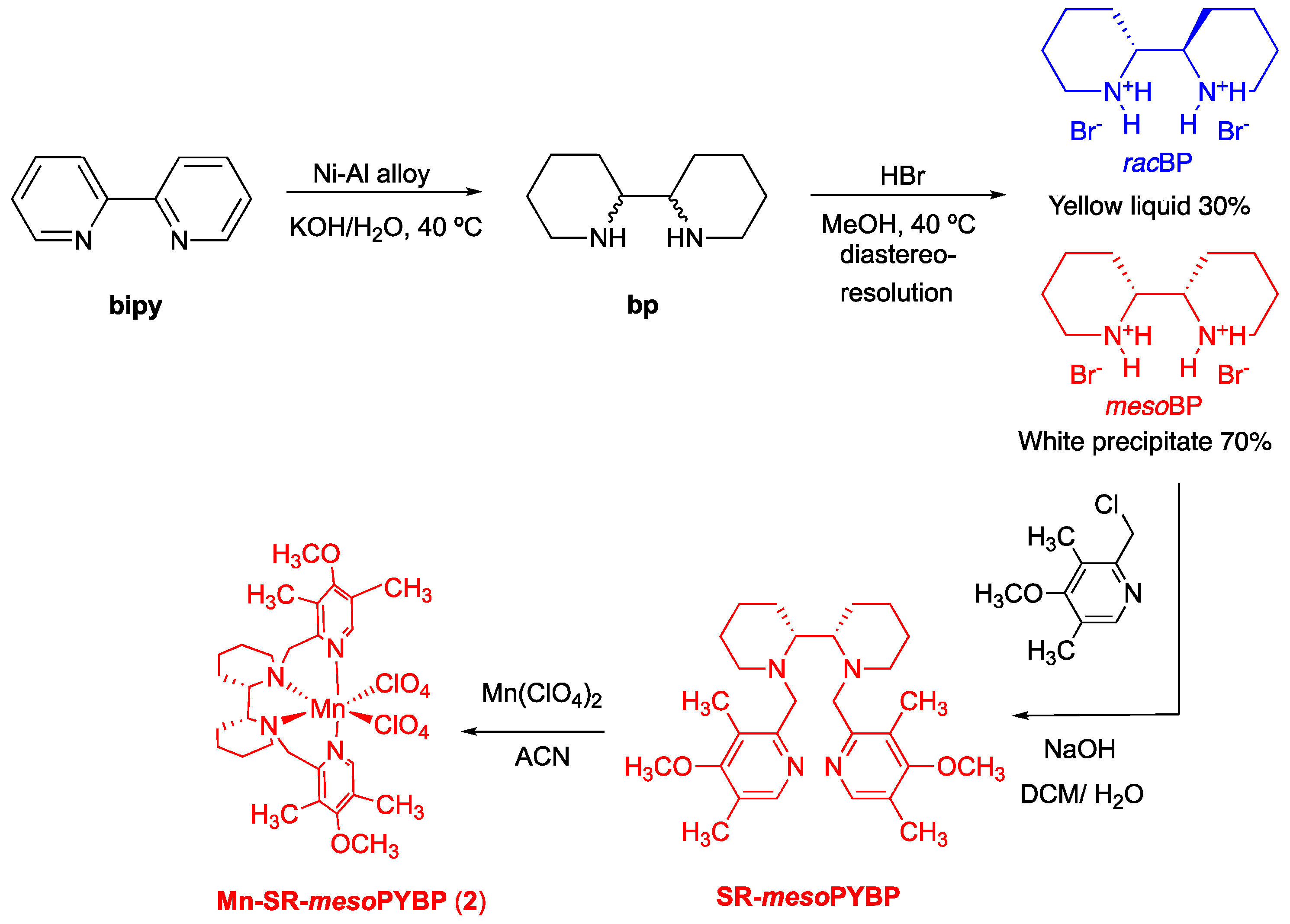
3.2. Synthesis of SR-MesoPYBP Ligand
3.3. Synthesis of Manganese SR-mesoPYBP Complex
3.4. General Catalysis Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mikhalyova, E.A.; Makhlynets, O.V.; Palluccio, T.D.; Filatov, A.S.; Rybak-Akimova, E.V. A new efficient iron catalyst for olefin epoxidation with hydrogen peroxide. Chem. Commun. 2012, 48, 687. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, E.; Wucher, B.; Rominger, F.; Törnroos, K.W.; Kunz, D. Selective rearrangement of terminal epoxides into methylketones catalysed by a nucleophilic rhodium–NHC–pincer complex. Chem. Commun. 2015, 51, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, K.K.; Thomas, A.M.; Sindhu, K.S.; Anilkumar, G. Recent advances and perspectives in the manganese-catalysed epoxidation reactions. Tetrahedron 2016, 72, 1. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Enantioselective Bioinspired Epoxidation of Electron-Deficient Olefins with H2O2 on Aminopyridine Mn Catalysts. ACS Catal. 2014, 4, 1599. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Bioinspired Mn-aminopyridine catalyzed epoxidations of olefins with various oxidants: Enantioselectivity and mechanism. Catal. Today 2016, 278, 30. [Google Scholar] [CrossRef]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Direct selective oxidative functionalization of C–H bonds with H2O2: Mn-Aminopyridine complexes challenge the dominance of non-heme Fe catalysts. Molecules 2016, 21, 1454. [Google Scholar] [CrossRef] [Green Version]
- Milan, M.; Bietti, M.; Costas, M. Aliphatic C–H Bond Oxidation with Hydrogen Peroxide Catalyzed by Manganese Complexes: Directing Selectivity through Torsional Effects. Org. Lett. 2018, 20, 2720. [Google Scholar] [CrossRef]
- White, M.C.; Doyle, A.G.; Jacobsen, E.N. A Synthetically Useful, Self-Assembling MMO Mimic System for Catalytic Alkene Epoxidation with Aqueous H2O2. J. Am. Chem. Soc. 2001, 123, 7194. [Google Scholar] [CrossRef]
- Costas, M.; Tipton, A.K.; Chen, K.; Jo, D.H.; Que, J. Modeling Rieske Dioxygenases: The First Example of Iron-Catalyzed Asymmetric cis-Dihydroxylation of Olefins. J. Am. Chem. Soc. 2001, 123, 6722. [Google Scholar] [CrossRef]
- Murphy, A.; Dubois, G.; Stack, T.D.P. Efficient epoxidation of electron-deficient olefins with a cationic manganese complex. J. Am. Chem. Soc. 2003, 125, 5250. [Google Scholar] [CrossRef] [PubMed]
- Makhlynets, O.V.; Oloo, W.N.; Moroz, Y.S.; Belaya, I.G.; Palluccio, T.D.; Filatov, A.S.; Müller, P.; Cranswick, M.A.; Que, L.; Rybak-Akimova, E.V. H 2 O 2 activation with biomimetic non-haem iron complexes and AcOH: Connecting the g = 2.7 EPR signal with a visible chromophore. Chem. Commun. 2014, 50, 645. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.J.; Yang, X.N.; Lee, Y.M.; Nam, W.; Sun, L.C. A nonheme manganese (IV)–Oxo species generated in photocatalytic reaction using water as an oxygen source. Chem. Commun. 2015, 51, 4013. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Non-Heme Manganese Complexes Catalyzed Asymmetric Epoxidation of Olefins by Peracetic Acid and Hydrogen Peroxide. Adv. Synth. Catal. 2011, 353, 885. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Dramatic effect of carboxylic acid on the electronic structure of the active species in Fe (PDP)-catalyzed asymmetric epoxidation. ACS Catal. 2012, 2, 1196. [Google Scholar] [CrossRef]
- Cussó, O.; Garcia-Bosch, I.; Font, D.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Highly Stereoselective Epoxidation with H2O2 Catalyzed by Electron-Rich Aminopyridine Manganese Catalysts. Org. Lett. 2013, 15, 6158. [Google Scholar] [CrossRef]
- Cussó, O.; Garcia-Bosch, I.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Asymmetric Epoxidation with H2O2 by Manipulating the Electronic Properties of Non-heme Iron Catalysts. J. Am. Chem. Soc. 2013, 135, 14871. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Iron-Catalyzed Enantioselective Epoxidations with Various Oxidants: Evidence for Different Active Species and Epoxidation Mechanisms. ACS Catal. 2017, 7, 60–69. [Google Scholar] [CrossRef]
- Cussó, O.; Serrano-Plana, J.; Costas, M. Evidence of a Sole Oxygen Atom Transfer Agent in Asymmetric Epoxidations with Fe-pdp Catalysts. ACS Catal. 2017, 7, 5046. [Google Scholar] [CrossRef]
- Hölzl, S.M.; Altmann, P.J.; Kück, J.W.; Kühn, F.E. Speciation in iron epoxidation catalysis: A perspective on the discovery and role of non-heme iron (III)-hydroperoxo species in iron-catalyzed oxidation reactions. Coord. Chem. Rev. 2017, 352, 517. [Google Scholar] [CrossRef]
- Yazerski, V.A.; Spannring, P.; Gatineau, D.; Woerde, C.H.M.; Wieclawska, S.M.; Lutz, M.; Kleijn, H.; Klein Gebbink, R.J.M. Making Fe (BPBP)-catalyzed C–H and C [double bond, length as m-dash] C oxidations more affordable. Org. Biomol. Chem. 2014, 12, 2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Rybak-Akimova, E.V.; Campana, C. {1, 1′-Bis [(pyridin-2-yl) methyl]-2, 2′-bipiperidyl}(perchlorato) copper (II) perchlorate. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, 73, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G. Bioinspired mononuclear transition-metal aminopyridine complexes for olefin epoxidation and molecular oxygen activation: Synthesis, characterization, catalysis and mechanistic studies. Ph.D. Thesis, Tufts University, Medford, MA, USA, 2017. [Google Scholar]
- Costas, M.; Que, L., Jr. Ligand topology tuning of iron-catalyzed hydrocarbon oxidations. Angew. Chem. Int. Ed. 2002, 41, 2179. [Google Scholar] [CrossRef]
- Saravanan, N.; Sankaralingam, M.; Palaniandavar, M. Manganese(ii) complexes of tetradentate 4N ligands with diazepane backbones for catalytic olefin epoxidation: Effect of nucleophilicity of peroxo complexes on reactivity. RSC Adv. 2014, 4, 12000. [Google Scholar] [CrossRef]
- Hubin, T.J.; McCormick, J.M.; Collinson, S.R.; Buchalova, M.; Perkins, C.M.; Alcock, N.W.; Kahol, P.K.; Raghunathan, A.; Busch, D.H. New iron (II) and manganese (II) complexes of two ultra-rigid, cross-bridged tetraazamacrocycles for catalysis and biomimicry. J. Am. Chem. Soc. 2000, 122, 2512. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. J Electronic effects in (salen)Mn-based epoxidation catalysts. J. Org. Chem. 2003, 68, 6202. [Google Scholar] [CrossRef]
- Goldsmith, C.R.; Jiang, W. Alkene epoxidation catalyzed by manganese complexes with electronically modified 1,10-phenanthroline ligands. Inorg. Chim. Acta. 2012, 384, 340. [Google Scholar] [CrossRef]
- Odom, B.; Hanneke, D.; D’urso, B.; Gabrielse, G. New measurement of the electron magnetic moment using a one-electron quantum cyclotron. Phys. Rev. Lett. 2006, 97, 6. [Google Scholar] [CrossRef] [Green Version]
- Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Nonheme manganese-catalyzed asymmetric oxidation. A Lewis acid activation versus oxygen rebound mechanism: Evidence for the “Third Oxidant”. Inorg. Chem. 2010, 49, 8620. [Google Scholar] [CrossRef]
- Larrow, J.F.; Jacobsen, E.N. (R, R)-N, N′-BIS (3, 5-DI-TERT-BUTYLSALICYLIDENE)-1, 2-CYCLOHEXANEDIAMINO MANGANESE (III) CHLORIDE, A HIGHLY ENANTIOSELECTIVE EPOXIDATION CATALYST:(MANG ANESE, CHLORO 2, 2′-1, 2-CYCLOHEXANEDIYLBIS (NITRILOMETHYLIDYNE)-BIS4, 6-BIS (1, 1-DIMETHYLETHYL) PHENALATO (2-)-N, N′, O, O′-, SP-5-1 3-(1 R-TRANS--). Org. Synth. 1998, 75, 1. [Google Scholar]
- Mas-Balleste, R.; Que, L. Iron-catalyzed olefin epoxidation in the presence of acetic acid: Insights into the nature of the metal-based oxidant. J. Am. Chem. Soc. 2007, 129, 15964. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Miao, C.; Xia, C.; Lee, Y.M.; Nam, W.; Sun, W. Mechanistic Insights into the Enantioselective Epoxidation of Olefins by Bioinspired Manganese Complexes: Role of Carboxylic Acid and Nature of Active Oxidant. ACS Catal. 2018, 8, 4528. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Chiral Manganese Aminopyridine Complexes: The Versatile Catalysts of Chemo- and Stereoselective Oxidations with H2O2. Chem. Rec. 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, F.; Yang, G.; Zoll, A.J.; Rybak-Akimova, E.V.; Zhu, X. Manganese Catalysts with Tetradentate N-donor Pyridine-Appended Bipiperidine Ligands for Olefin Epoxidation Reactions: Ligand Electronic Effect and Mechanism. Catalysts 2020, 10, 285. https://doi.org/10.3390/catal10030285
Zhu F, Yang G, Zoll AJ, Rybak-Akimova EV, Zhu X. Manganese Catalysts with Tetradentate N-donor Pyridine-Appended Bipiperidine Ligands for Olefin Epoxidation Reactions: Ligand Electronic Effect and Mechanism. Catalysts. 2020; 10(3):285. https://doi.org/10.3390/catal10030285
Chicago/Turabian StyleZhu, Fengfan, Guang Yang, Adam J. Zoll, Elena V. Rybak-Akimova, and Xinbao Zhu. 2020. "Manganese Catalysts with Tetradentate N-donor Pyridine-Appended Bipiperidine Ligands for Olefin Epoxidation Reactions: Ligand Electronic Effect and Mechanism" Catalysts 10, no. 3: 285. https://doi.org/10.3390/catal10030285