Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction


Abstract
1. Introduction
2. Results and Discussion
2.1. Polymerizations
2.2. Optical Properties and Microstructures
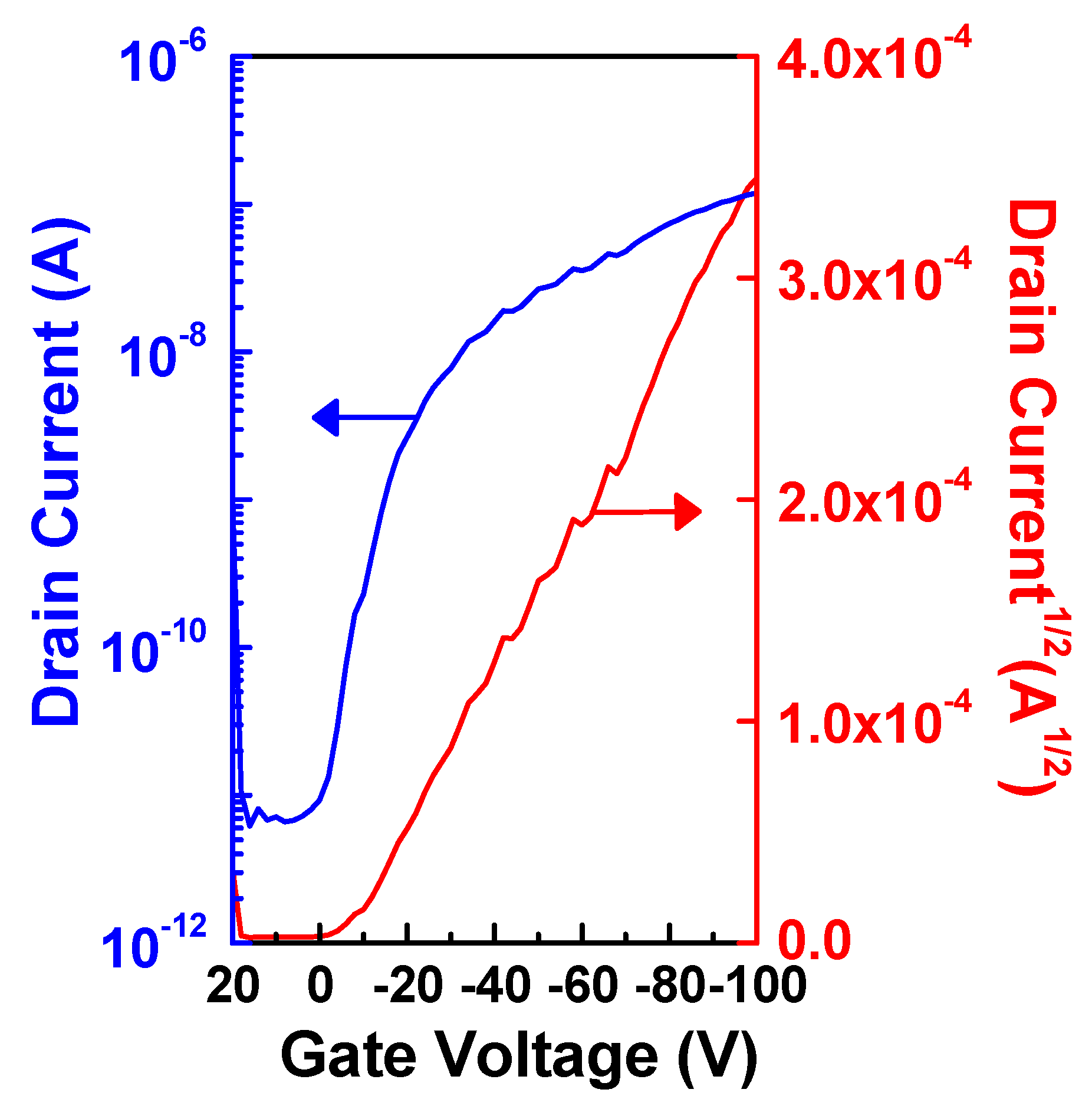
2.3. OFET Performances
3. Experimental
3.1. Materials
3.2. Synthesis of OTV
3.2.1. 1-Bromo-2-hexyldecane (1)
3.2.2. 6-Bromo-(N-2-hexyldecyl)isatin (2)
3.2.3. 6-Bromo-(N-2-hexyldecyl)oxindole (3)
3.2.4. 5-Iodothiophene-2-carbaldehyde (4)
3.2.5. 5-(Trimetyltin)thiophene-2-carbaldehyde (5)
3.2.6. N-(2-hexyldecyl)oxindolidene thienylene vinylene (OTV)
3.3. Synthesis of POTV
3.4. Measurements and Characterization
3.5. OFET Fabrication
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boudreault, P.L.T.; Najari, A.; Leclerc, M. Processable low-bandgap polymers for photovoltaic applications. Chem. Mater. 2011, 23, 456–469. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Fréchet, J.M.J. Molecular design and ordering effects in π-functional materials for transistor and solar cell applications. J. Am. Chem. Soc. 2011, 133, 20009–20029. [Google Scholar] [CrossRef]
- Dou, L.; Liu, Y.; Hong, Z.; Li, G.; Yang, Y. Low-bandgap near-IR conjugated polymers/molecules for organic electronics. Chem. Rev. 2015, 115, 12633–12665. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent advances in bulk heterojunction polymer solar cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef] [PubMed]
- Holliday, S.; Li, Y.; Luscombe, C.K. Recent advances in high performance donor-acceptor polymers for organic photovoltaics. Prog. Polym. Sci. 2017, 70, 34–51. [Google Scholar] [CrossRef]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J.P., Jr. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Negishi, E.; King, A.O.; Okukado, N. Selective carbon-carbon bond formation via transition metal catalysis. 3. A highly selective synthesis of unsymmetrical biaryls and diarylmethanes by the nickel- or palladium-catalyzed reaction of aryl- and benzylzinc derivatives with aryl halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar] [CrossRef]
- Kosugi, M.; Sasazawa, K.; Shimizu, Y.; Migita, T. Reactions of allyltin compounds III. Allylation of aromatic halides with allyltributyltin in the presence of tetrakis(triphenylphosphine)palladium(0). Chem. Lett. 1977, 6, 301–302. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J.K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc. Chem. Commun. 1979, 866–867. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef]
- Carsten, B.; He, F.; Son, H.J.; Xu, T.; Yu, L. Stille polycondensation for synthesis of functional materials. Chem. Rev. 2011, 111, 1493–1528. [Google Scholar] [CrossRef]
- Kuwabara, J.; Yasuda, T.; Takase, N.; Kanbara, T. Effects of the terminal structure, purity, and molecular weight of an amorphous conjugated polymer on its photovoltaic characteristics. ACS Appl. Mater. Interfaces 2016, 8, 1752–1758. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Y.; Liu, Y.; Liu, J.; Lu, H.; Qiu, L.; Cho, K. Facile green synthesis of isoindigo-based conjugated polymers using aldol polycondensation. Polym. Chem. 2017, 8, 3448–3456. [Google Scholar] [CrossRef]
- Onwubiko, A.; Yue, W.; Jellett, C.; Xiao, M.; Chen, H.Y.; Ravva, M.K.; Hanifi, D.A.; Knall, A.C.; Purushothaman, B.; Nikolka, M.; et al. Fused electron deficient semiconducting polymers for air stable electron transport. Nat. Commun. 2018, 9, 416. [Google Scholar] [CrossRef]
- Himmelberger, S.; Vandewal, K.; Fei, Z.; Heeney, M.; Salleo, A. Role of molecular weight distribution on charge transport in semiconducting polymers. Macromolecules 2014, 47, 7151–7157. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, T.; Xu, T.; Zhao, D.; Yu, L. Mechanistic studies of effect of dispersity on the photovoltaic performance of PTB7 polymer solar cells. Chem. Mater. 2015, 27, 537–543. [Google Scholar] [CrossRef]
- Yokoyama, A.; Miyakoshi, R.; Yokozawa, T. Chain-growth polymerization for poly(3-hexylthiophene) with a defined molecular weight and a low polydispersity. Macromolecules 2004, 37, 1169–1171. [Google Scholar] [CrossRef]
- Iovu, M.C.; Sheina, E.E.; Gil, R.R.; McCullough, R.D. Experimental evidence for the quasi-“living” nature of the grignard metathesis method for the synthesis of regioregular poly(3-alkylthiophenes). Macromolecules 2005, 38, 8649–8656. [Google Scholar] [CrossRef]
- Sheina, E.E.; Liu, J.; Lovu, M.C.; Laird, D.W.; McCullough, R.D. Chain growth mechanism for regioregular nickel-initiated cross-coupling polymerizations. Macromolecules 2004, 37, 3526–3528. [Google Scholar] [CrossRef]
- Higashihara, T.; Goto, E.; Ueda, M. Purification-free and protection-free synthesis of regioregular poly(3-hexylthiophene) and poly(3-(6-hydroxyhexyl)thiophene) using a zincate complex of tBu4ZnLi2. ACS Macro Lett. 2012, 1, 167–170. [Google Scholar] [CrossRef]
- Higashihara, T.; Goto, E. Controlled synthesis of low-polydisperse regioregular poly(3- hexylthiophene) and related materials by zincate-complex metathesis polymerization. Polym. J. 2014, 46, 381–390. [Google Scholar] [CrossRef]
- Goto, E.; Mori, H.; Ueda, M.; Higashihara, T. Controlled polymerization of electron-deficient naphthalene-diimide containing monomer by negishi-type catalyst-transfer polymerization. J. Photopolym. Sci. Technol. 2015, 28, 279–283. [Google Scholar] [CrossRef]
- Goto, E.; Ochiai, Y.; Ueda, M.; Higashihara, T. Transition-metal-free and halogen-free controlled synthesis of poly(3-alkylthienylene vinylene): Via the Horner-Wadsworth-Emmons condensation reaction. Polym. Chem. 2018, 9, 1996–2001. [Google Scholar] [CrossRef]
- Yokozawa, T.; Muroya, D.; Sugi, R.; Yokoyama, A. Convenient method of chain-growth polycondensation for well-defined aromatic polyamides. Macromol. Rapid Commun. 2005, 26, 979–981. [Google Scholar] [CrossRef]
- Shao, M.; Keum, J.; Chen, J.; He, Y.; Chen, W.; Browning, J.F.; Jakowski, J.; Sumpter, B.G.; Ivanov, I.N.; Ma, Y.-Z.; et al. The isotopic effects of deuteration on optoelectronic properties of conducting polymers. Nat. Commum. 2014, 5, 3180. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run a | Base | Solvent | Temp. | Mnb | ÐM | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | sodium hexamethyldisilazide (NaHMDS)/15-crown-5 | Tetrahydrofuran (THF) | −40 °C | 1300 | 1.17 | 71 c |
| 2 | NaHMDS/15-crown-5 | THF | r. t. | 2100 | 1.97 | 58 c |
| 3 | NaHMDS/15-crown-5 | Toluene/THF f | r. t. | 2600 | 1.46 | ~100 d |
| 4 | lithium hexamethyldisilazide (LiHMDS)/12-crown-4 | Toluene/THF g | r. t. | 2500 | 1.37 | 66 c |
| 5 e | LiHMDS/12-crown-4 | Toluene/THF g | r. t. | 3000 | 1.45 | 73 c |
| 6 e | LiHMDS/12-crown-4 | Toluene/THF g | 30 °C | 3700 (5300) h | 1.47 (1.57) h | 65 c (40) h |
| Conditions | λmax (nm) | ε (L/mol/cm) | λonset (nm) | Eg (eV) |
|---|---|---|---|---|
| Solution | 486 | 2.47 × 104 | 677 | 1.82 |
| As-cast | 433, 577 | - | 740 | 1.66 |
| Annealed at 200 °C | 431, 588 | - | 748 | 1.64 |
| ION/IOFF | Max mobility (cm V−1 s−1) | Threshold voltage (V) | W/L | |
|---|---|---|---|---|
| POTV (Run 6) | 1.92 × 104 | 2.24 × 10−4 | −21.6 | 25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, S.; Lin, P.-S.; Wu, W.-N.; Liu, C.-L.; Higashihara, T. Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction. Catalysts 2020, 10, 364. https://doi.org/10.3390/catal10040364
Sato S, Lin P-S, Wu W-N, Liu C-L, Higashihara T. Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction. Catalysts. 2020; 10(4):364. https://doi.org/10.3390/catal10040364
Chicago/Turabian StyleSato, Shingo, Po-Shen Lin, Wei-Ni Wu, Cheng-Liang Liu, and Tomoya Higashihara. 2020. "Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction" Catalysts 10, no. 4: 364. https://doi.org/10.3390/catal10040364
APA StyleSato, S., Lin, P.-S., Wu, W.-N., Liu, C.-L., & Higashihara, T. (2020). Atom-economical Synthesis and Characterization of Poly(oxindolidene thienylene vinylene) Based on Aldol Polycondensation Reaction. Catalysts, 10(4), 364. https://doi.org/10.3390/catal10040364