Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
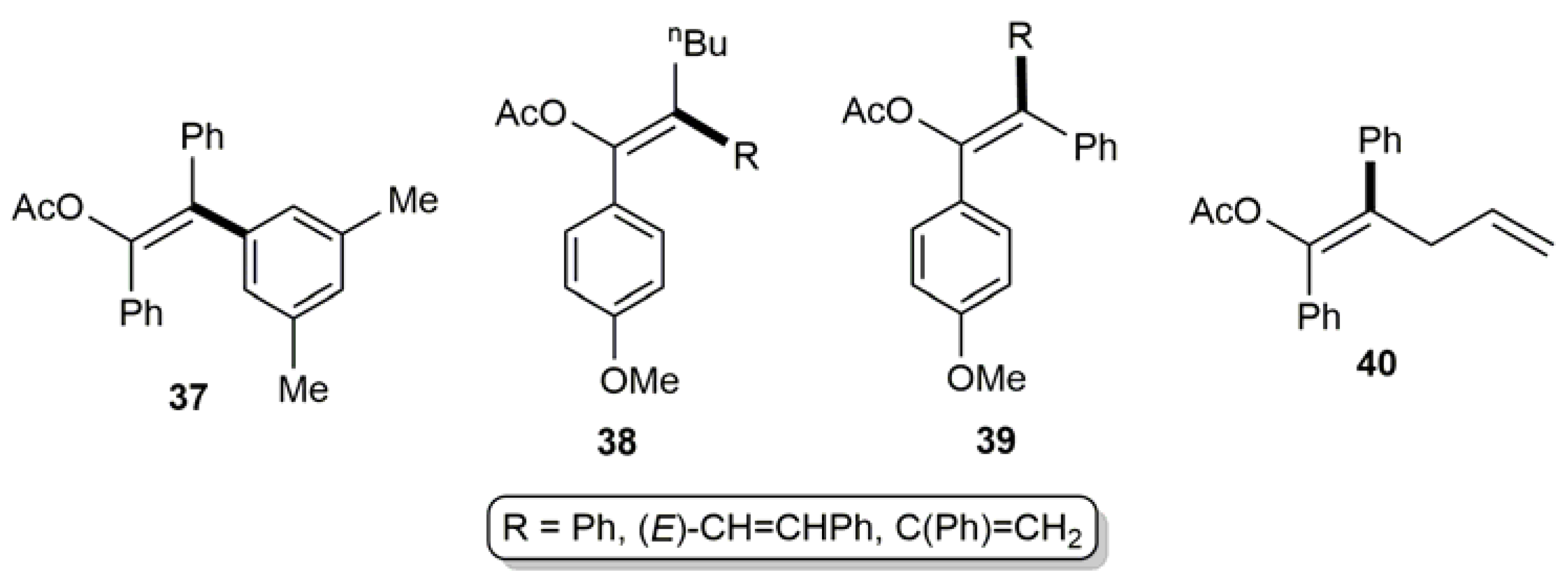{kind=link}
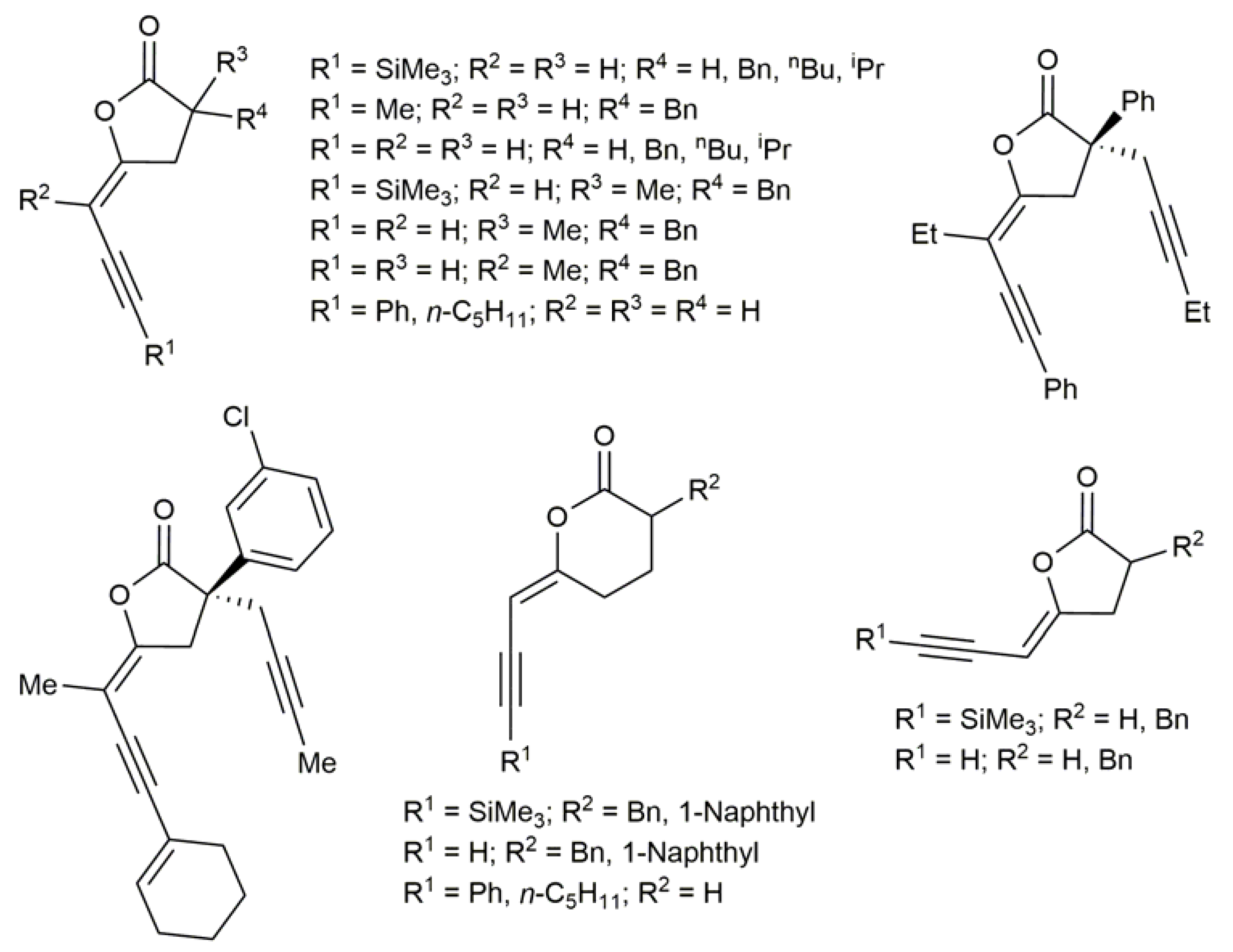{kind=link}
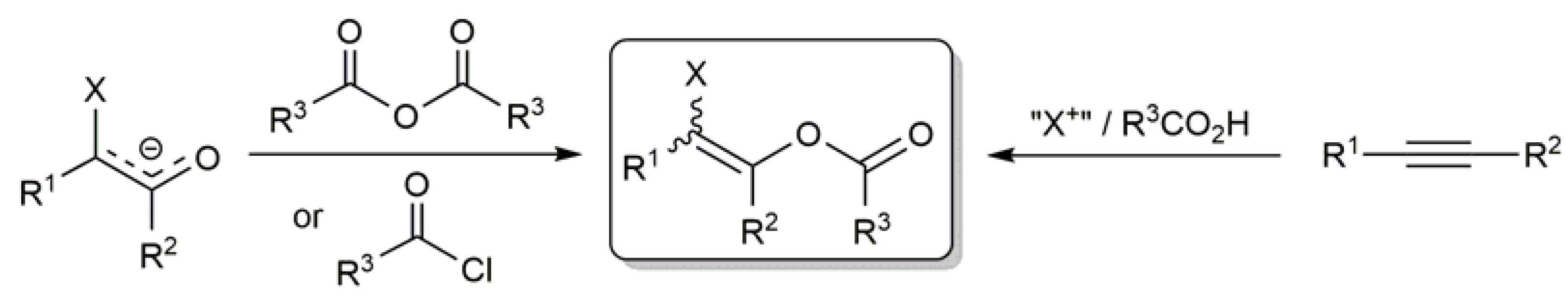{kind=link}
{kind=link}
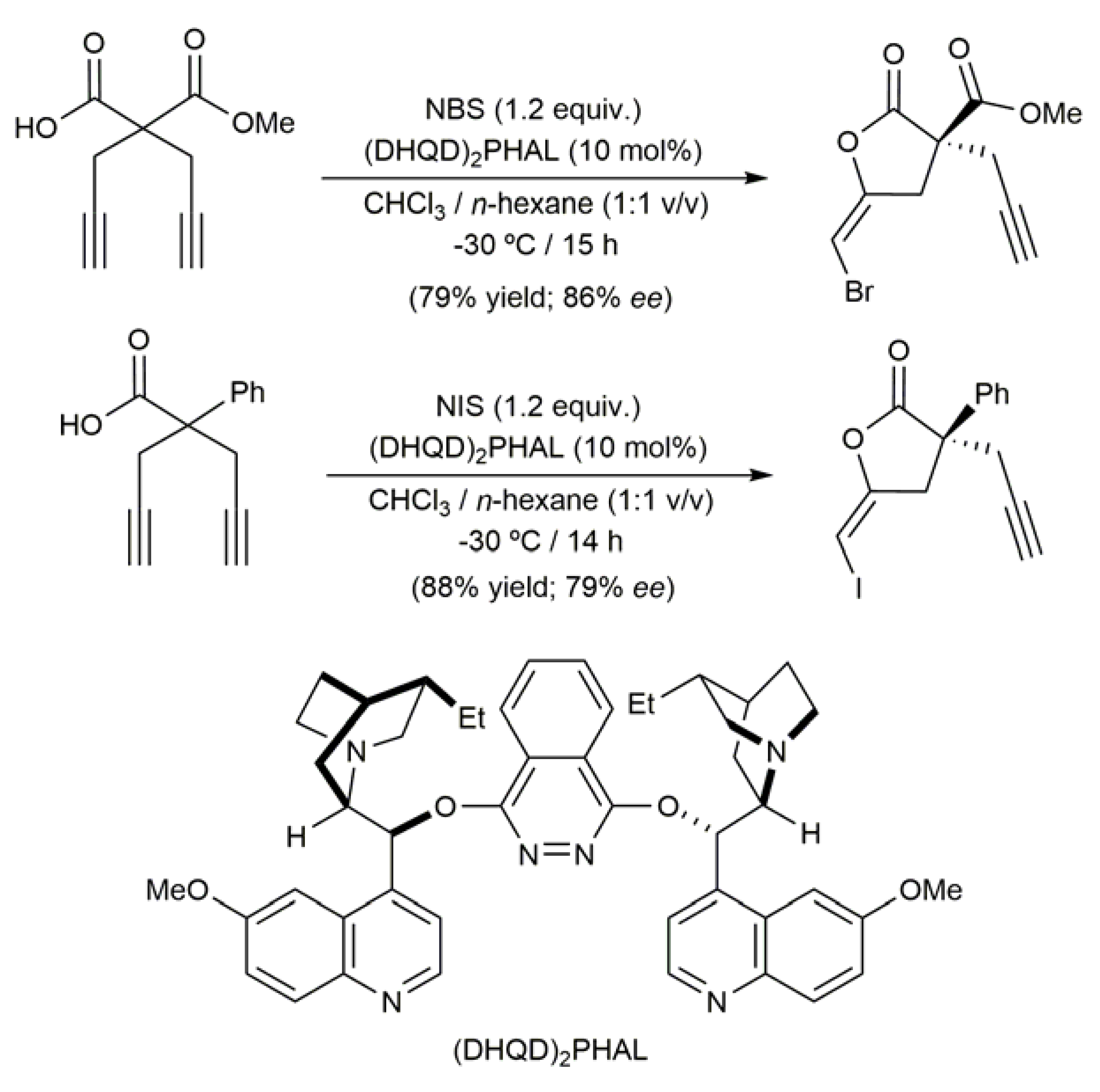{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
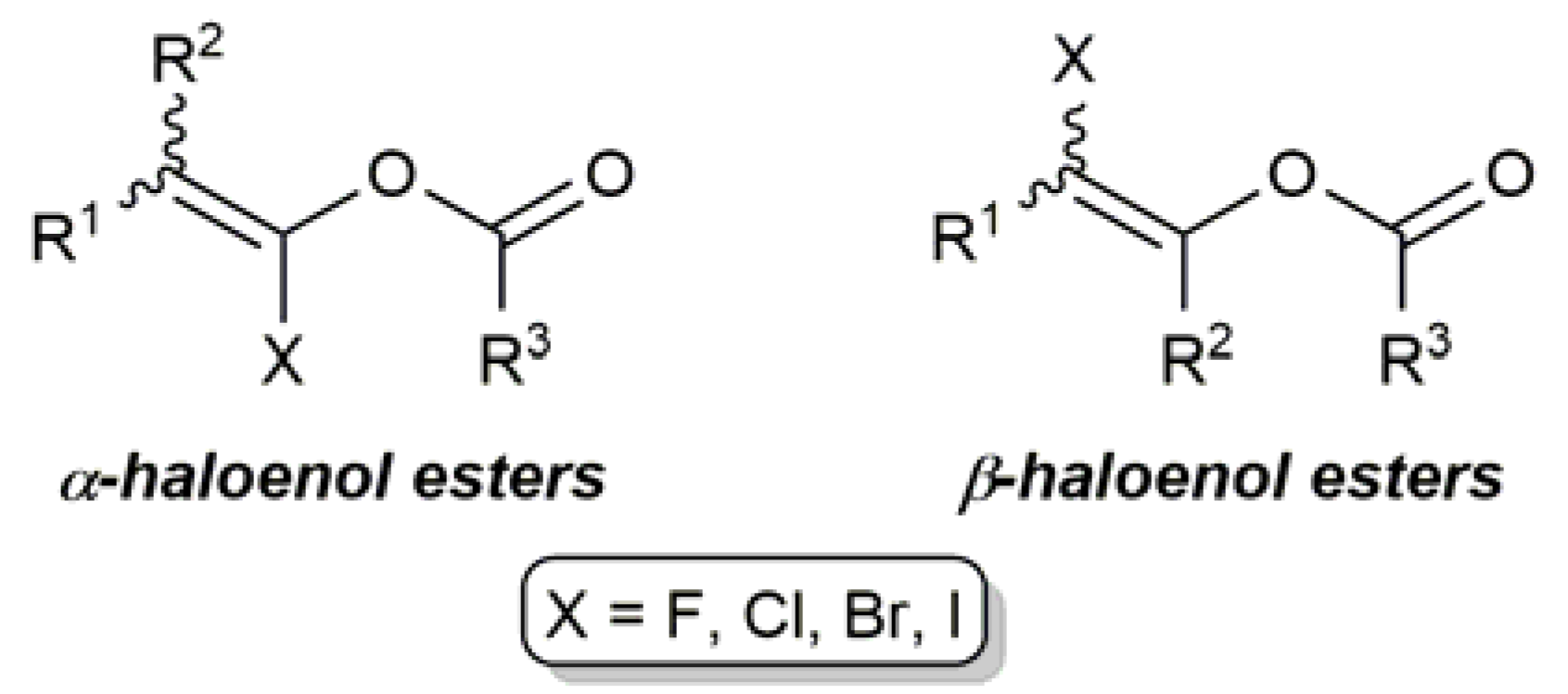
1. Introduction
2. Metal-Catalyzed Synthesis of β-Haloenol Esters
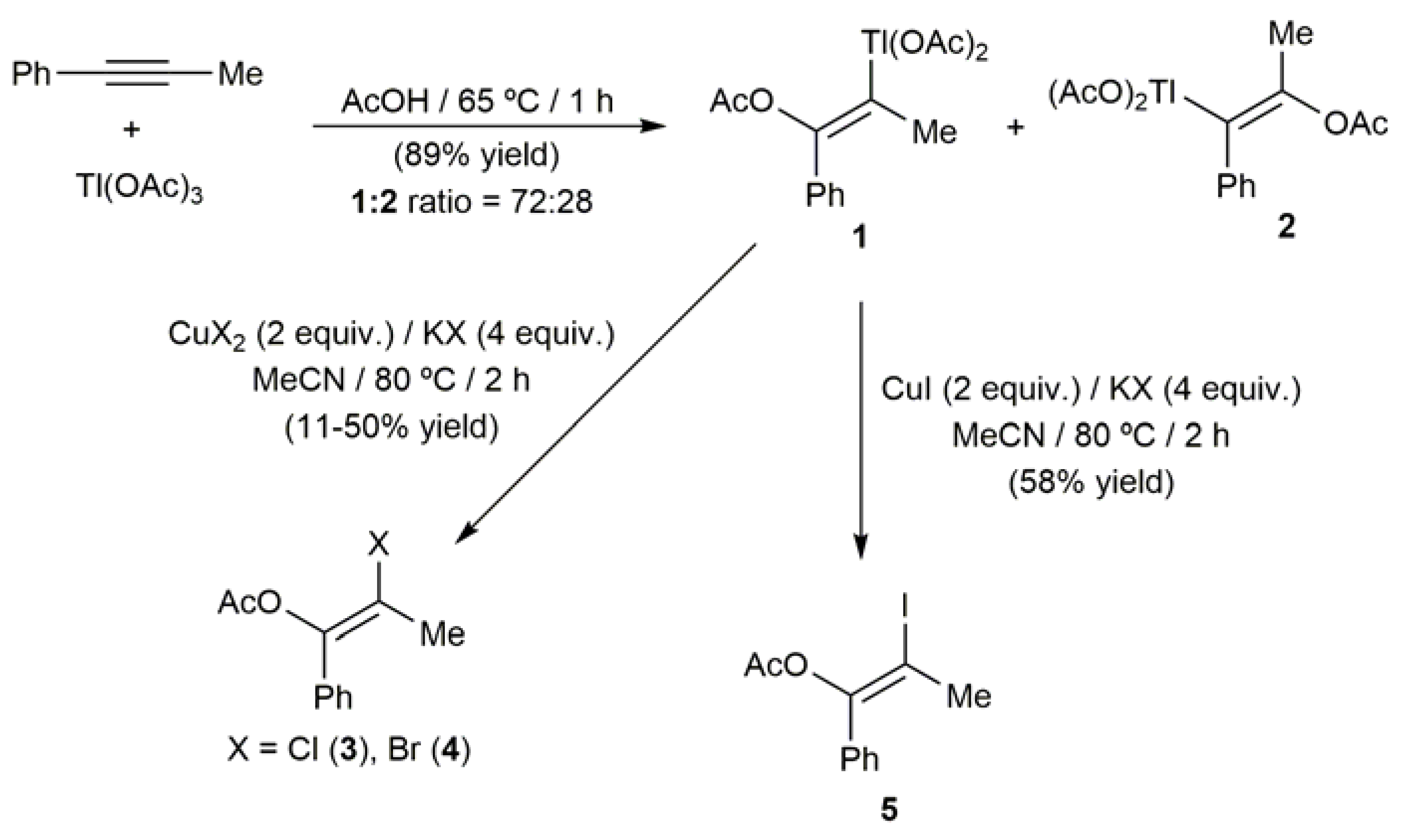
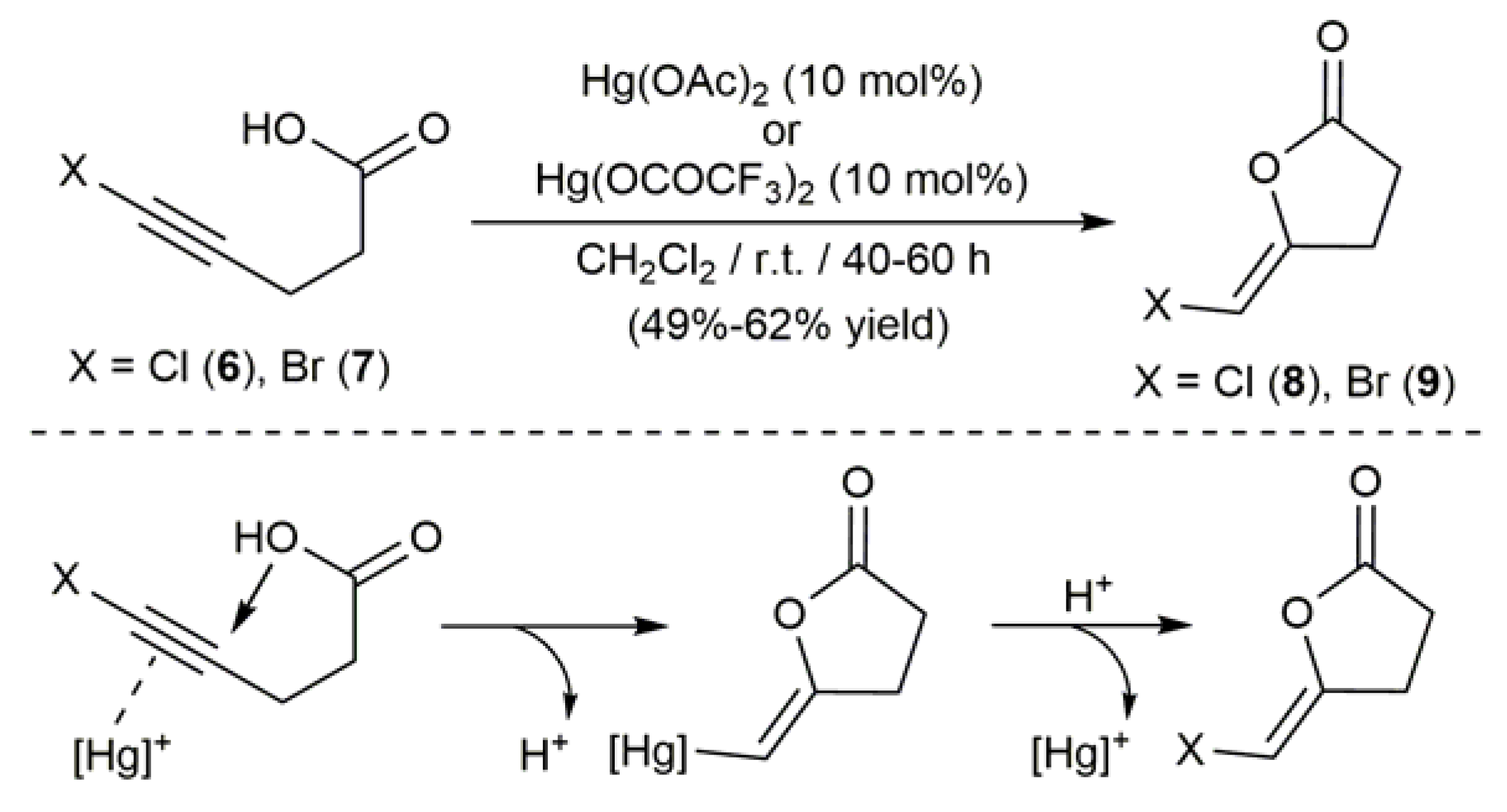
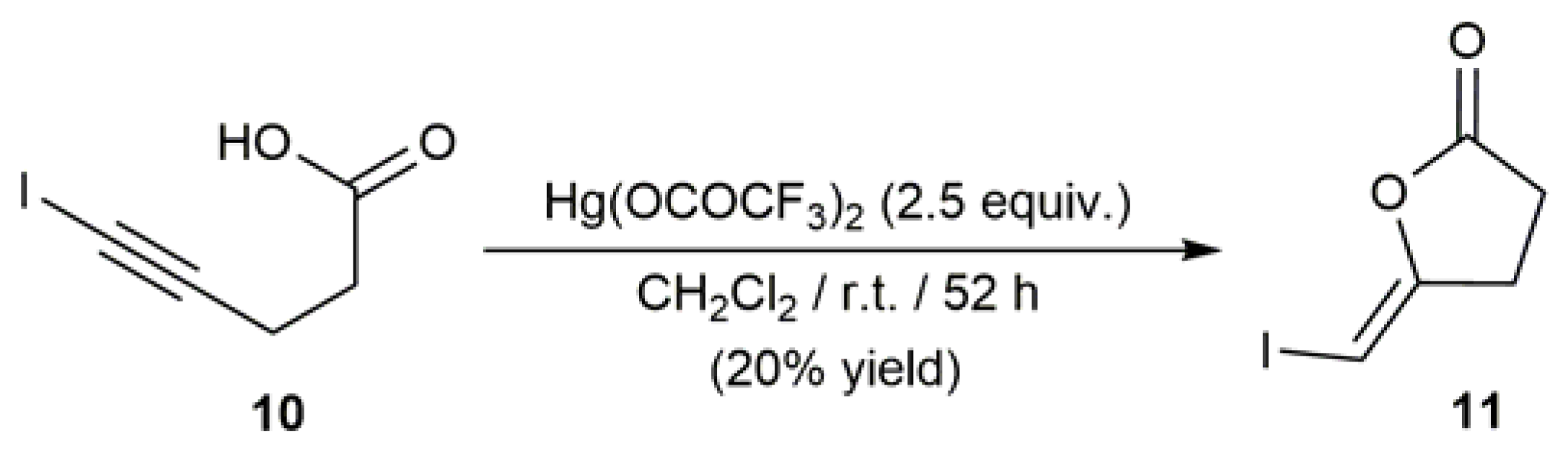
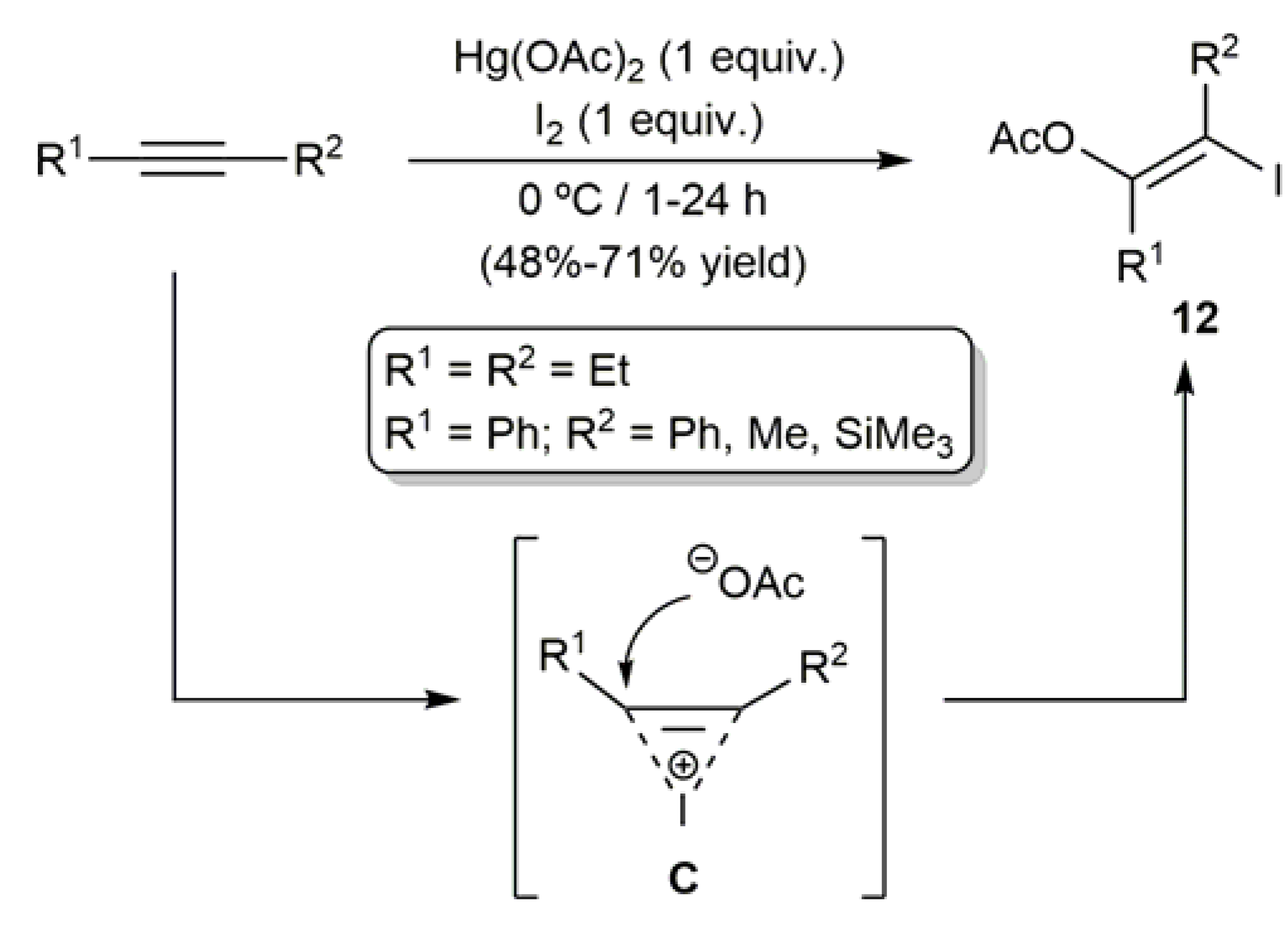
2.1. Hg-Catalyzed Synthesis of β-Haloenol Esters
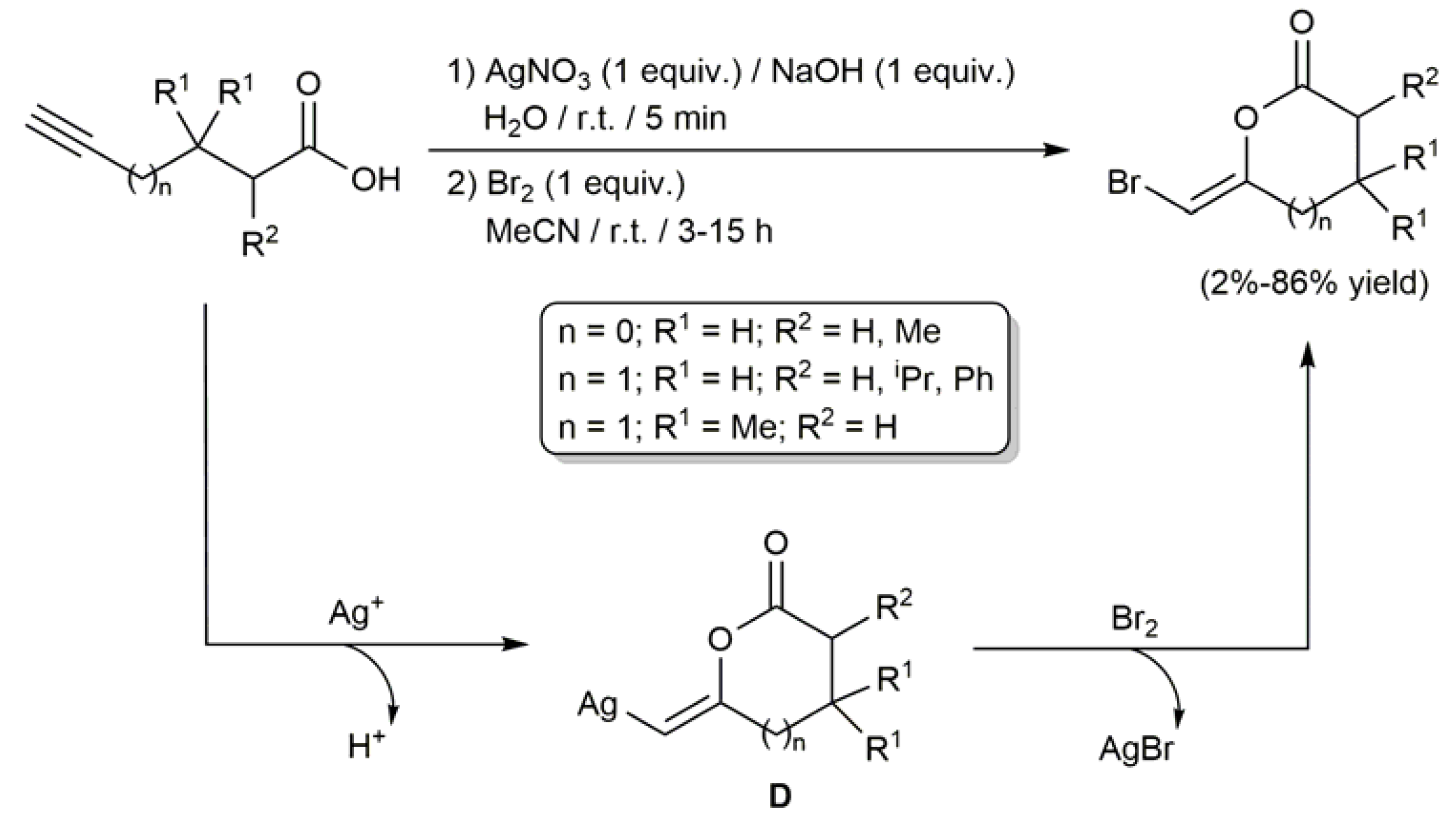
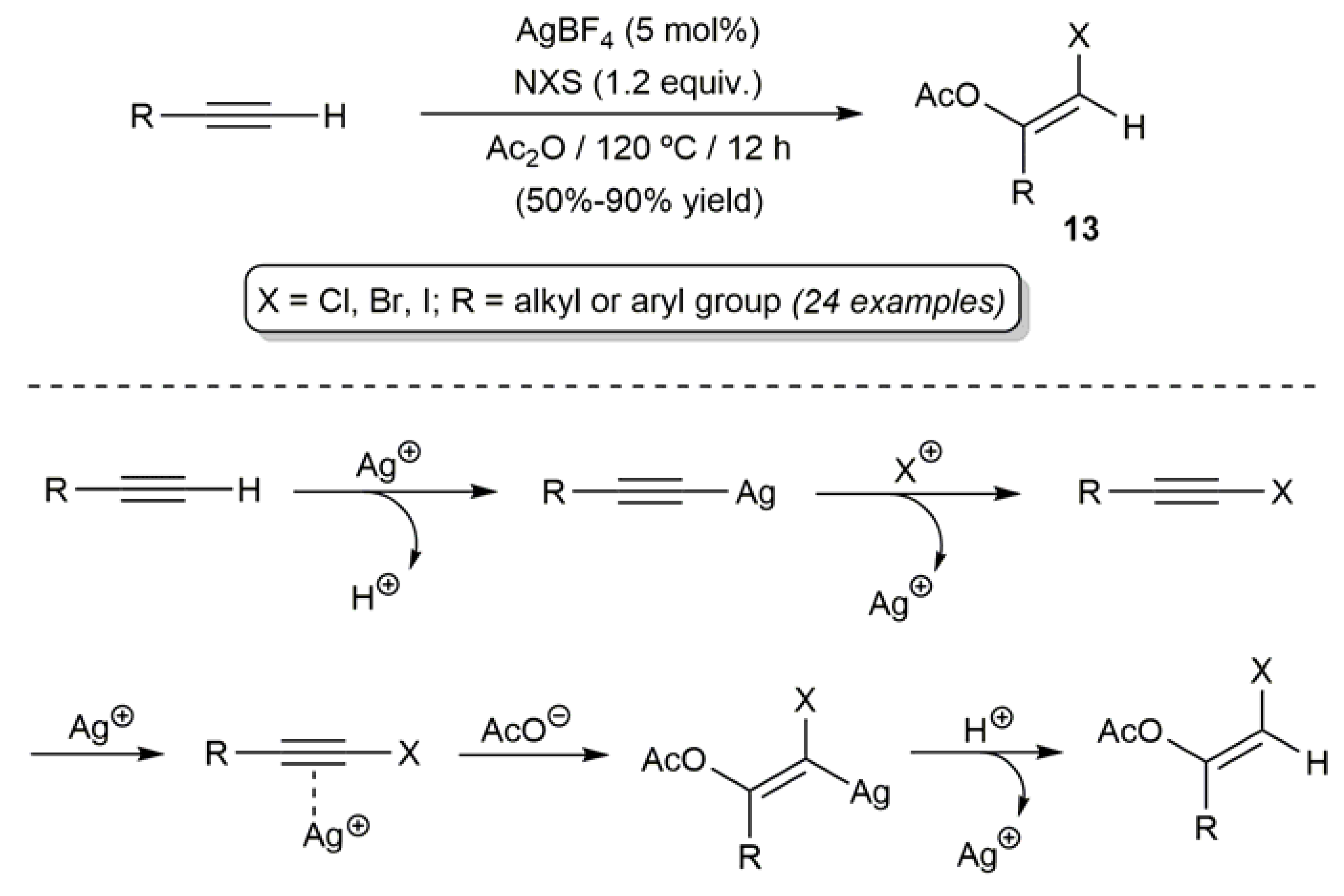
2.2. Ag-Catalyzed Synthesis of β-Haloenol Esters
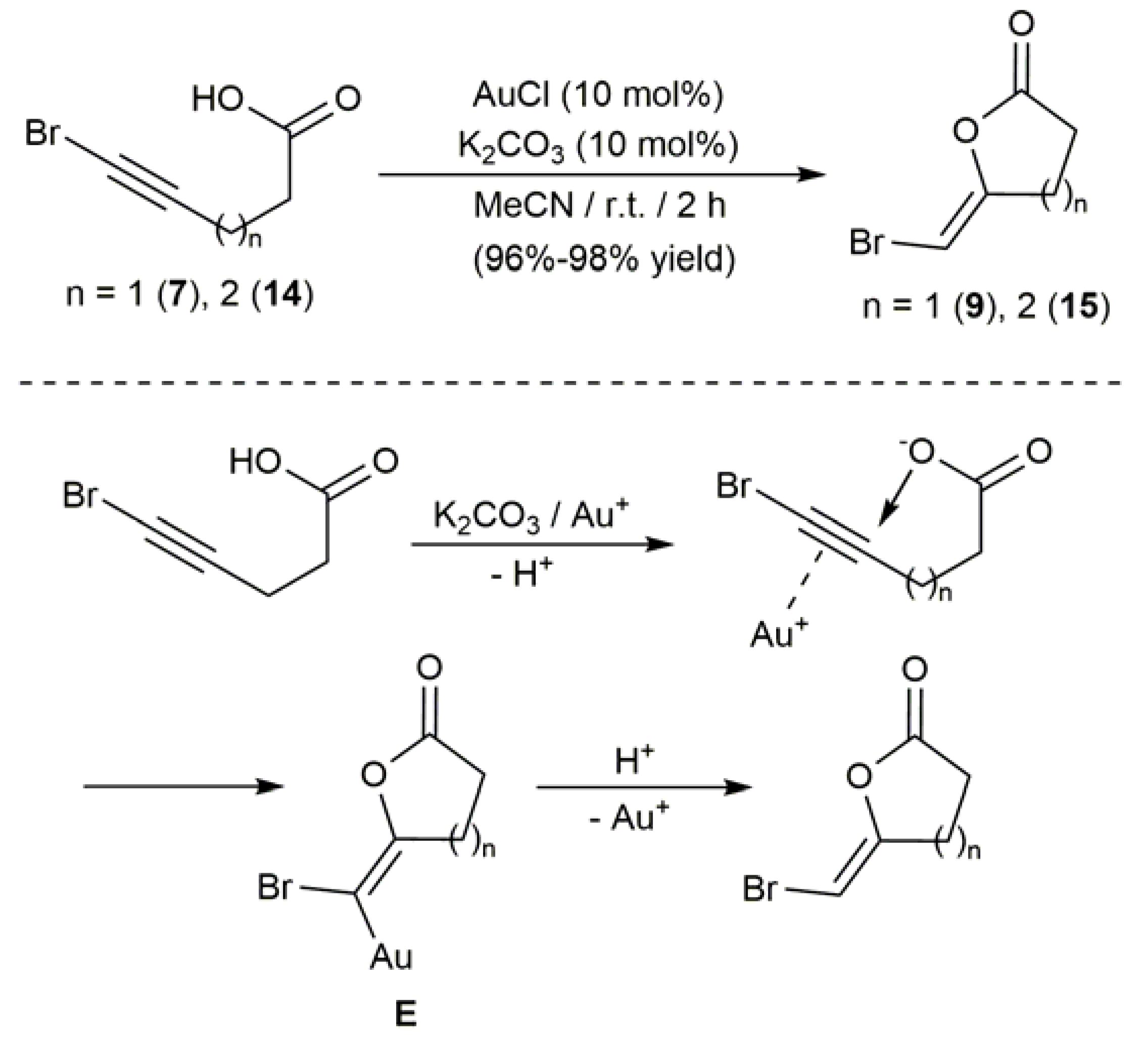
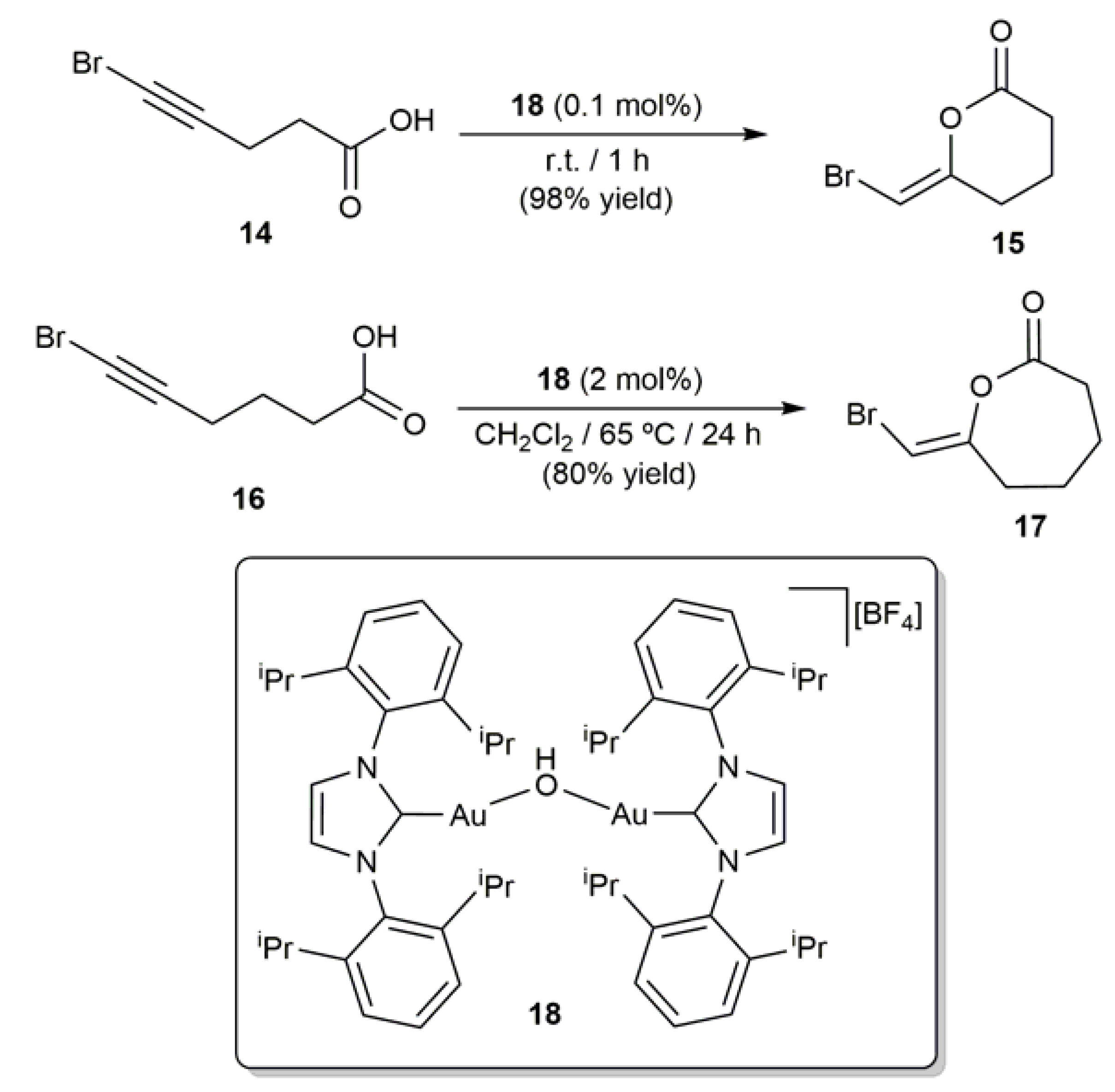
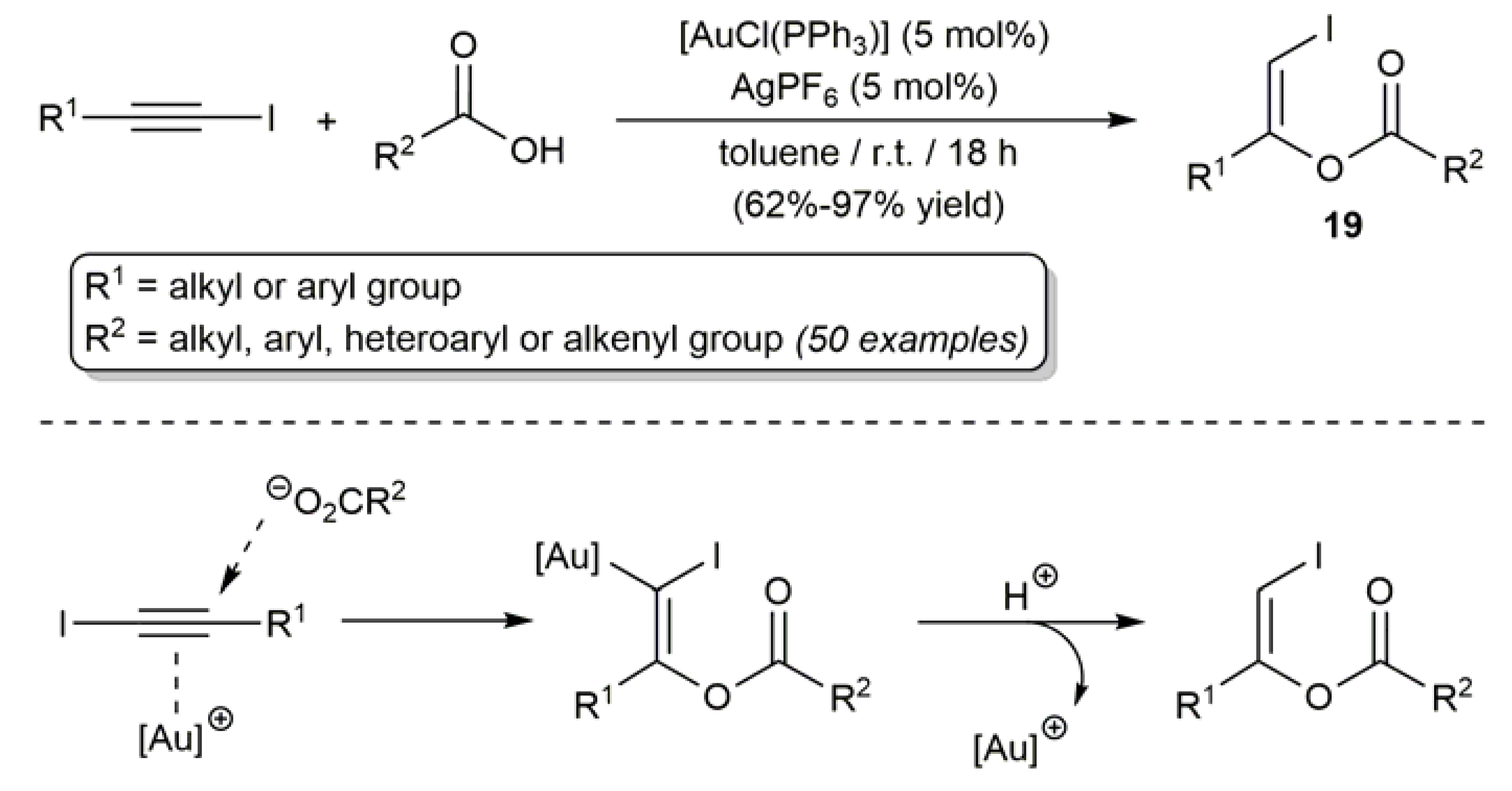
2.3. Au-Catalyzed Synthesis of β-Haloenol Esters
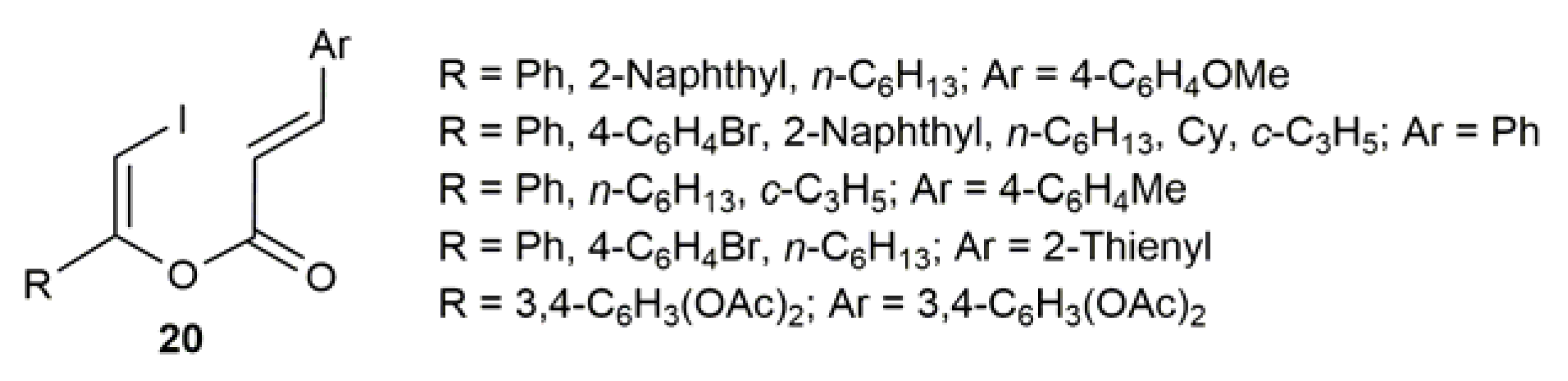
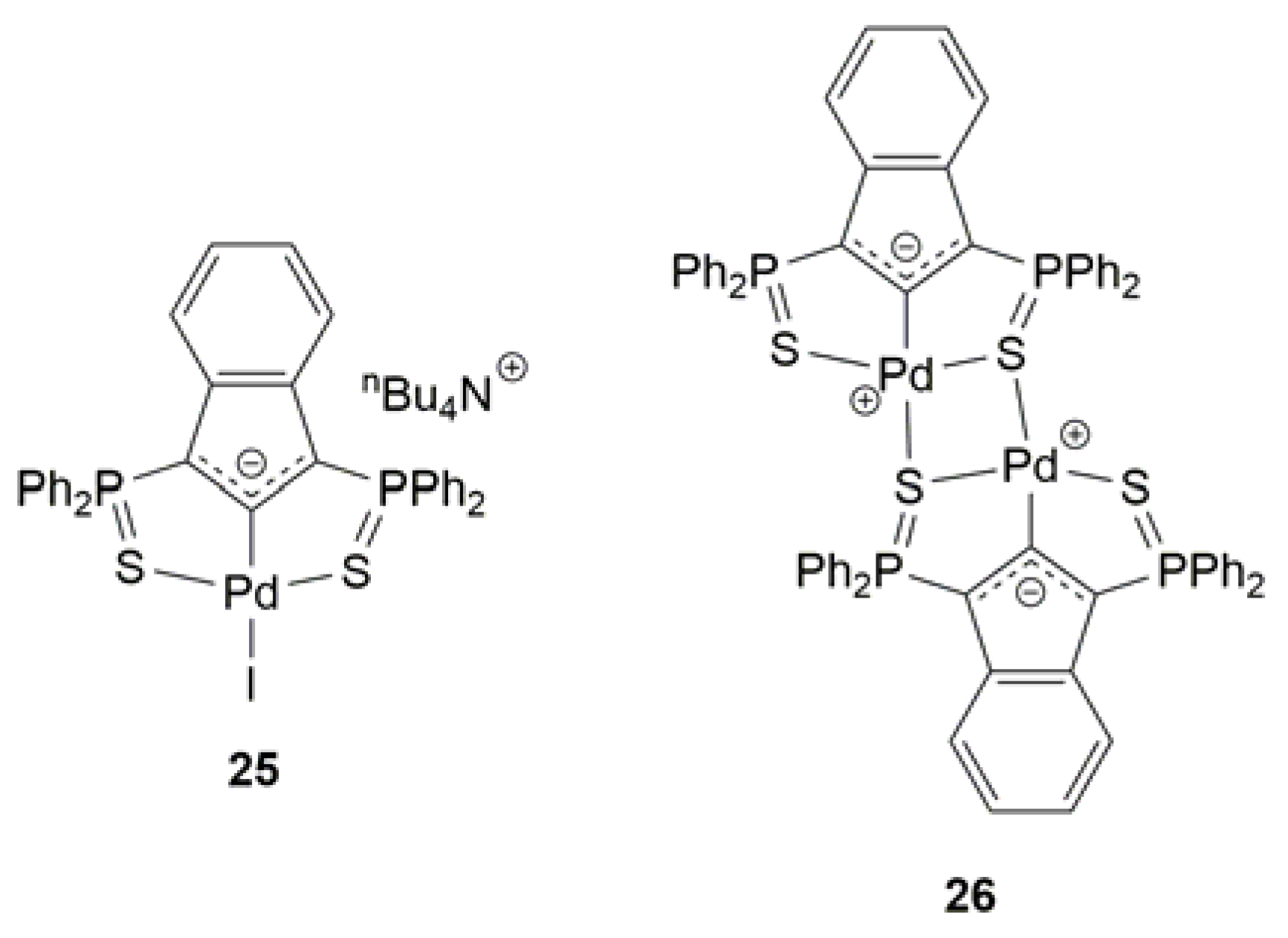
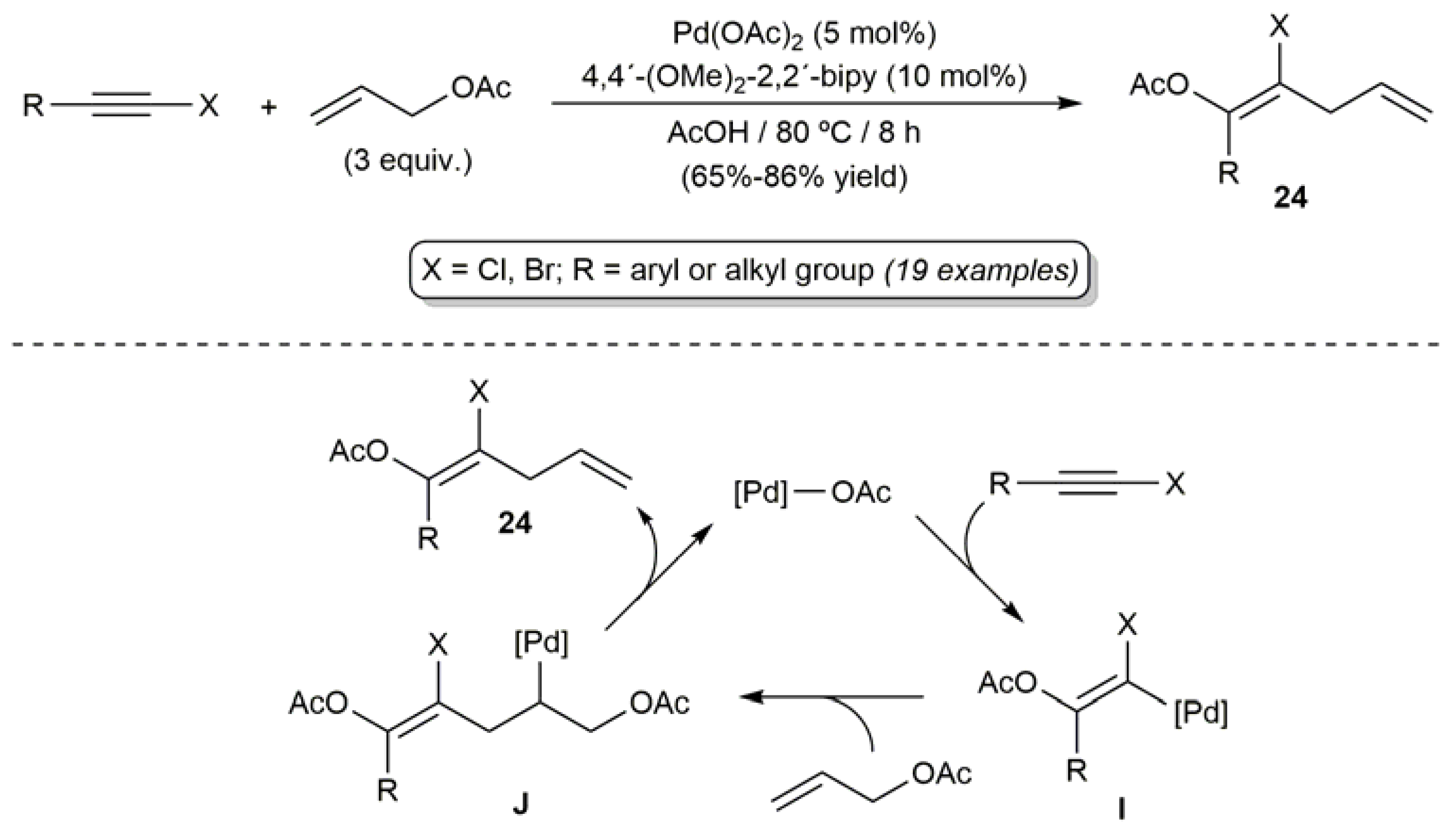
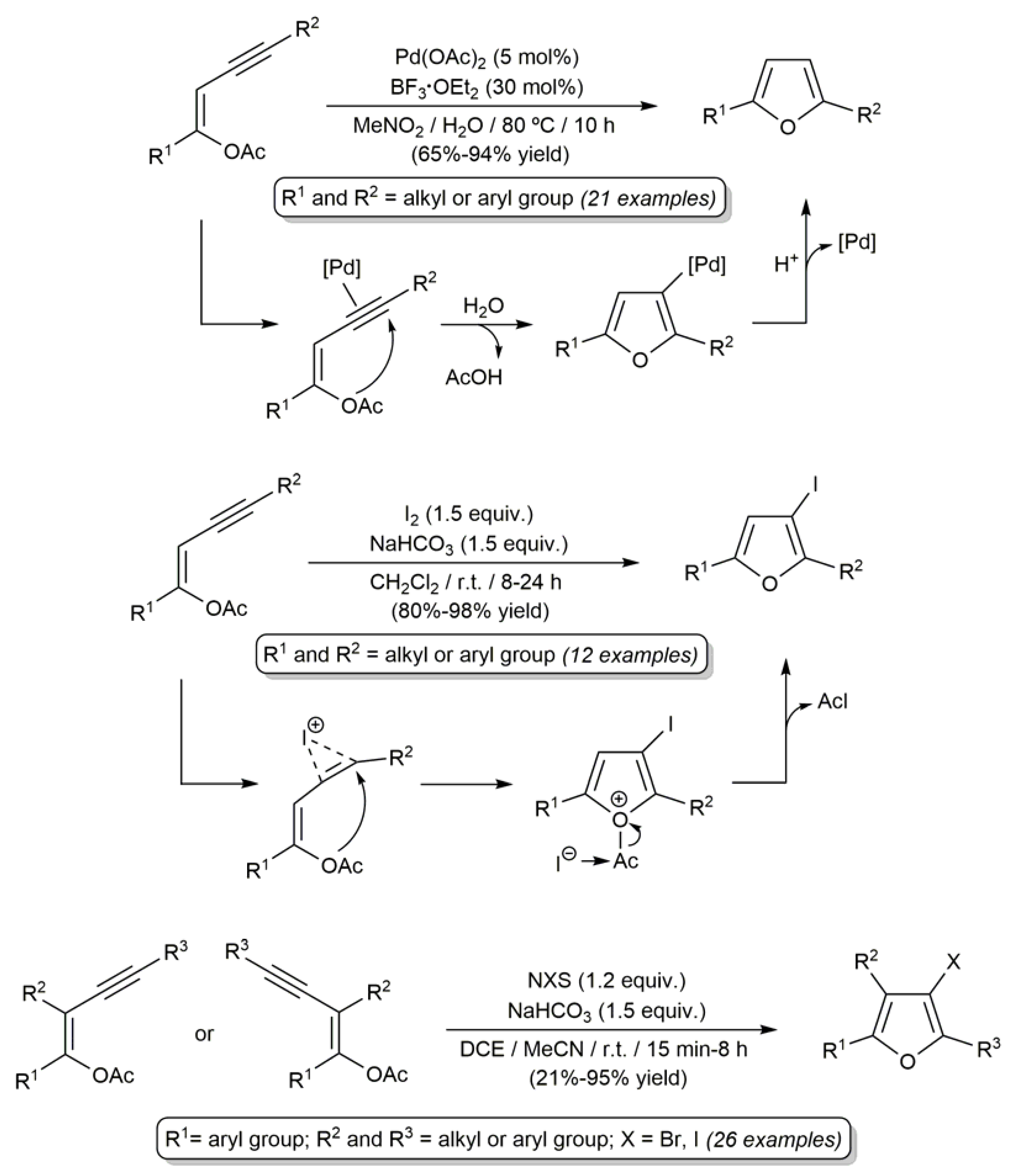
2.4. Pd-Catalyzed Synthesis of β-Haloenol Esters
3. Metal-Catalyzed Transformations of β-Haloenol Esters
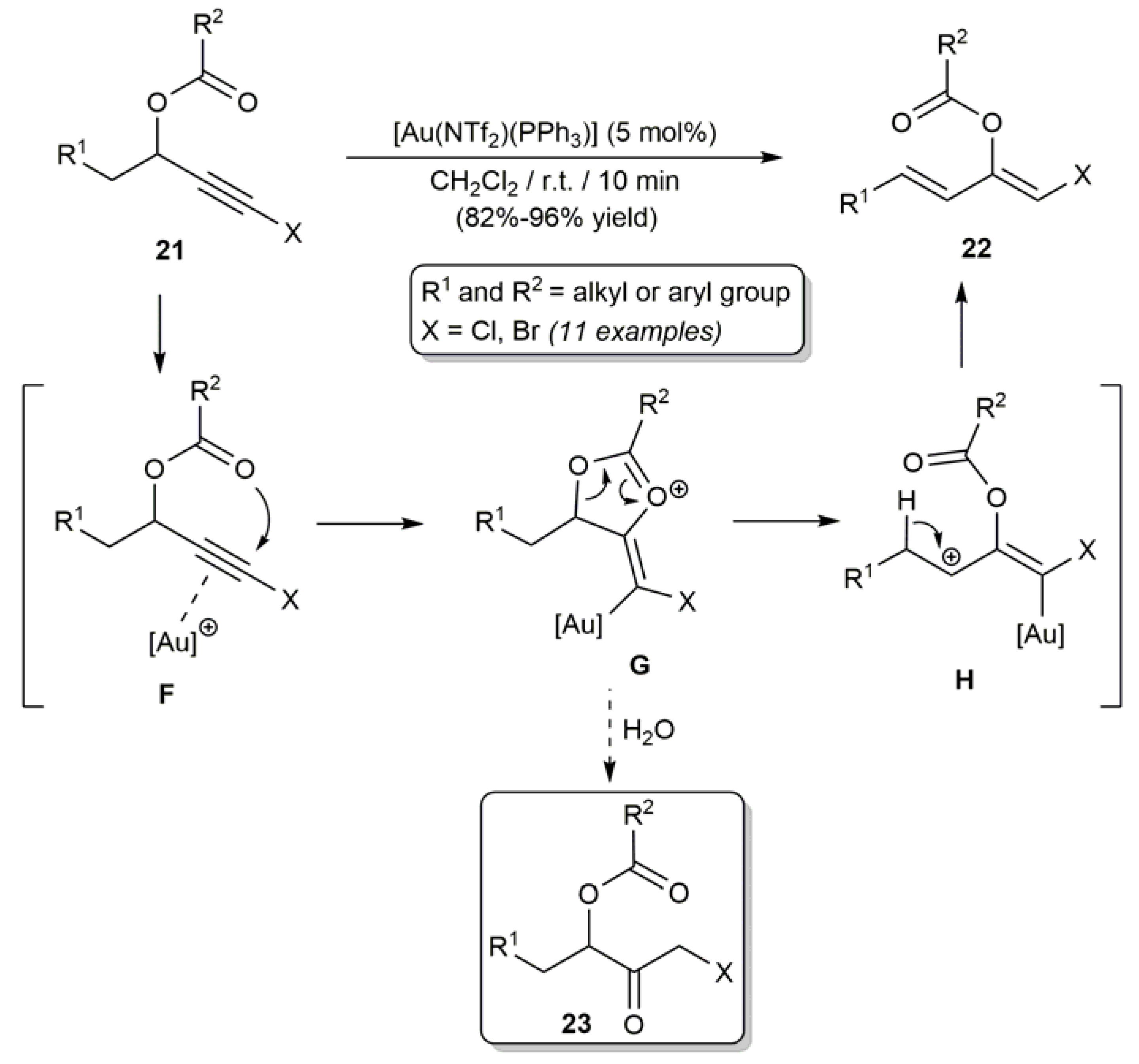
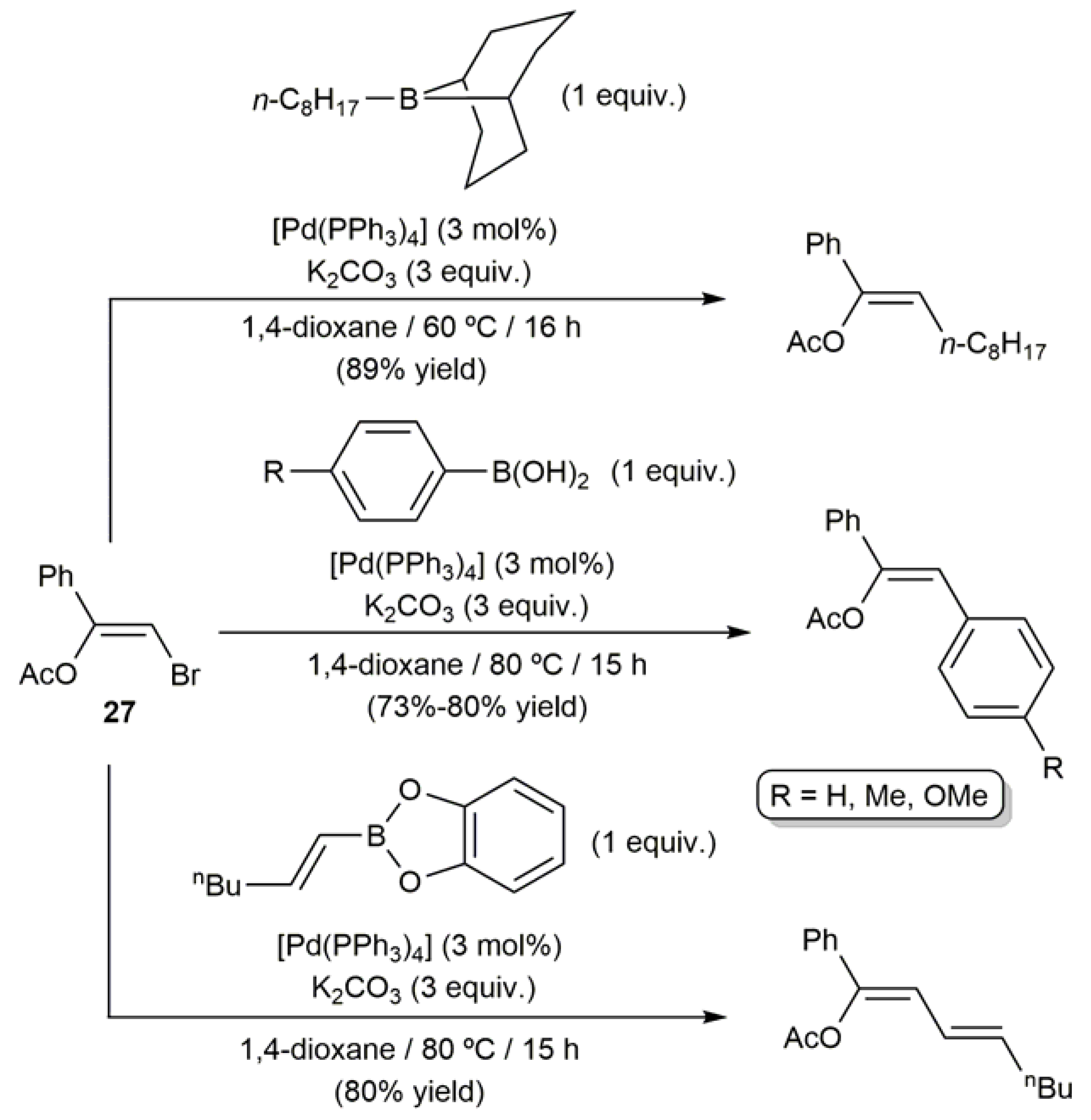
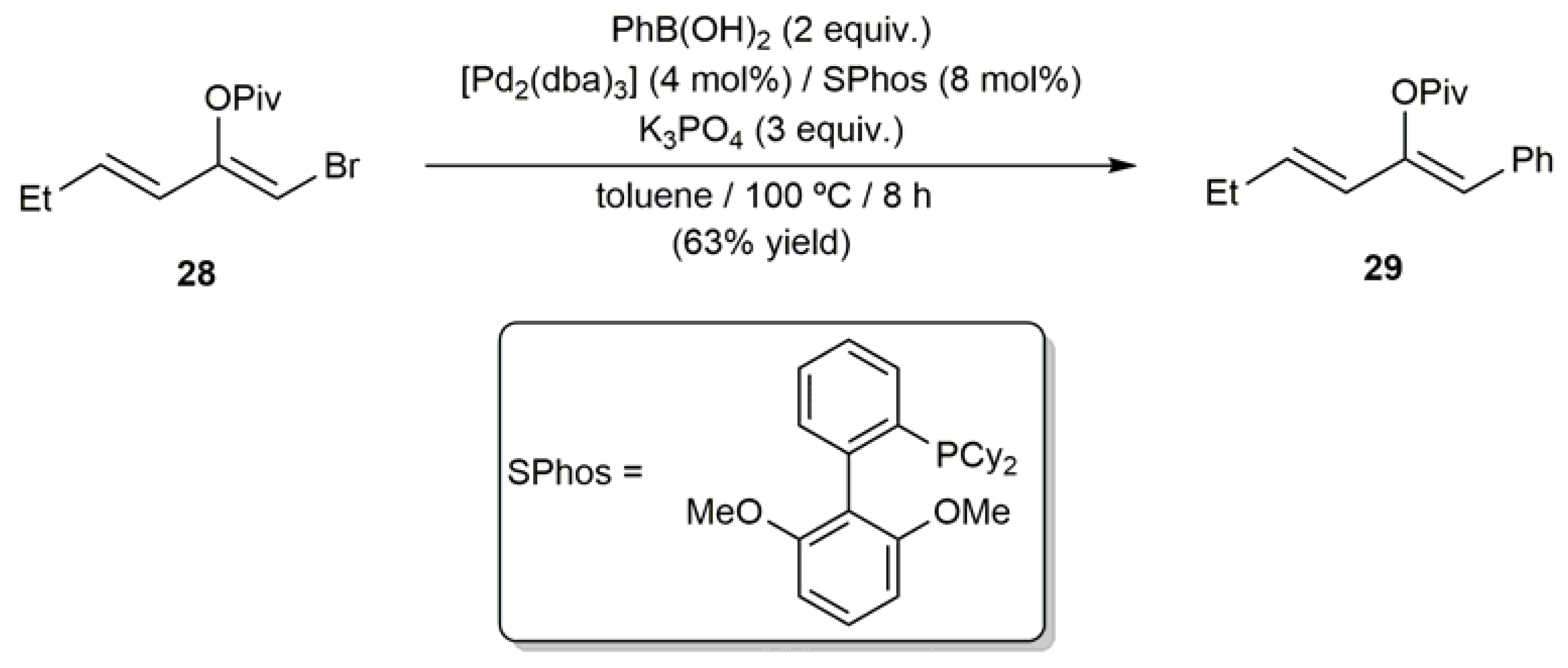
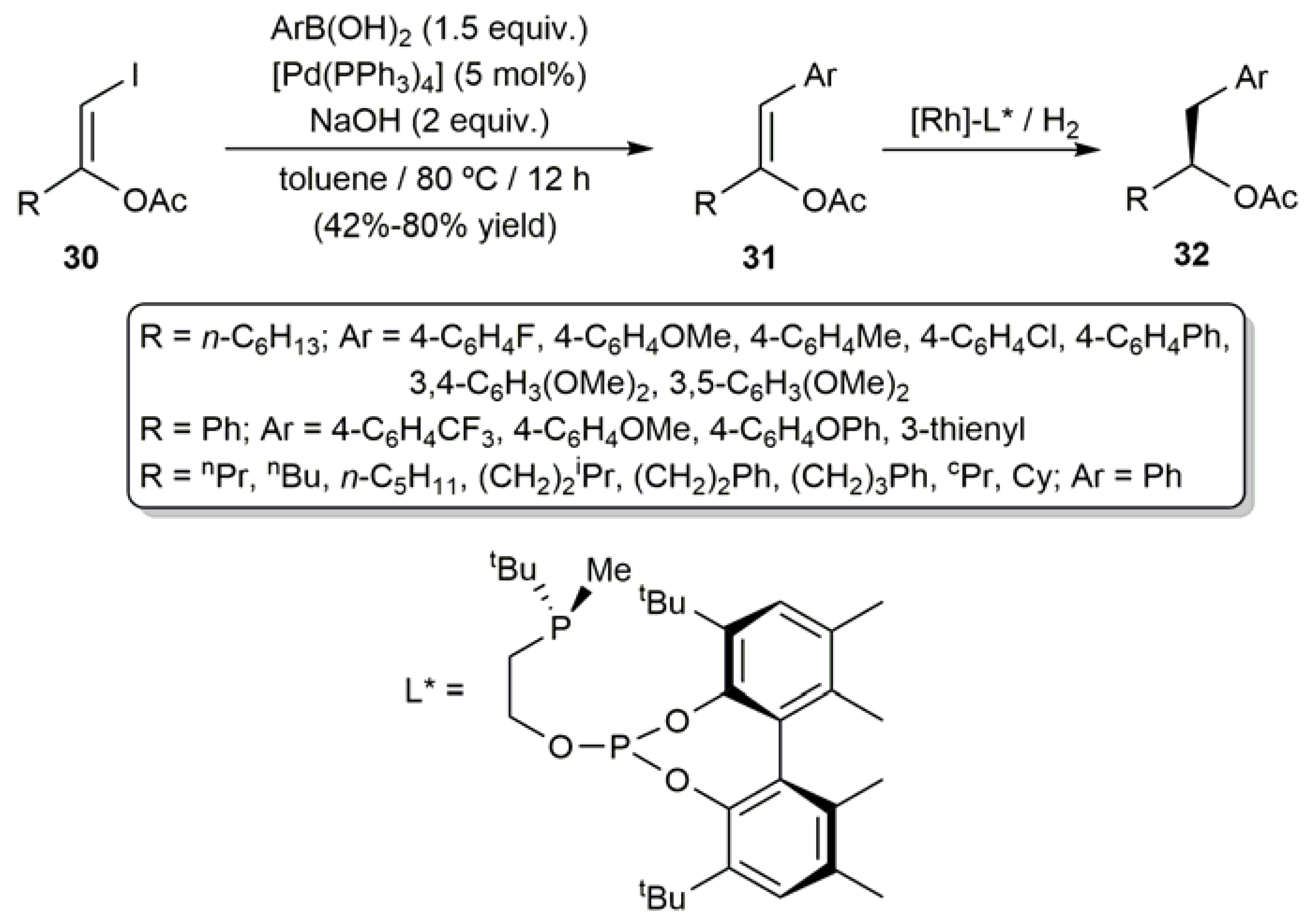
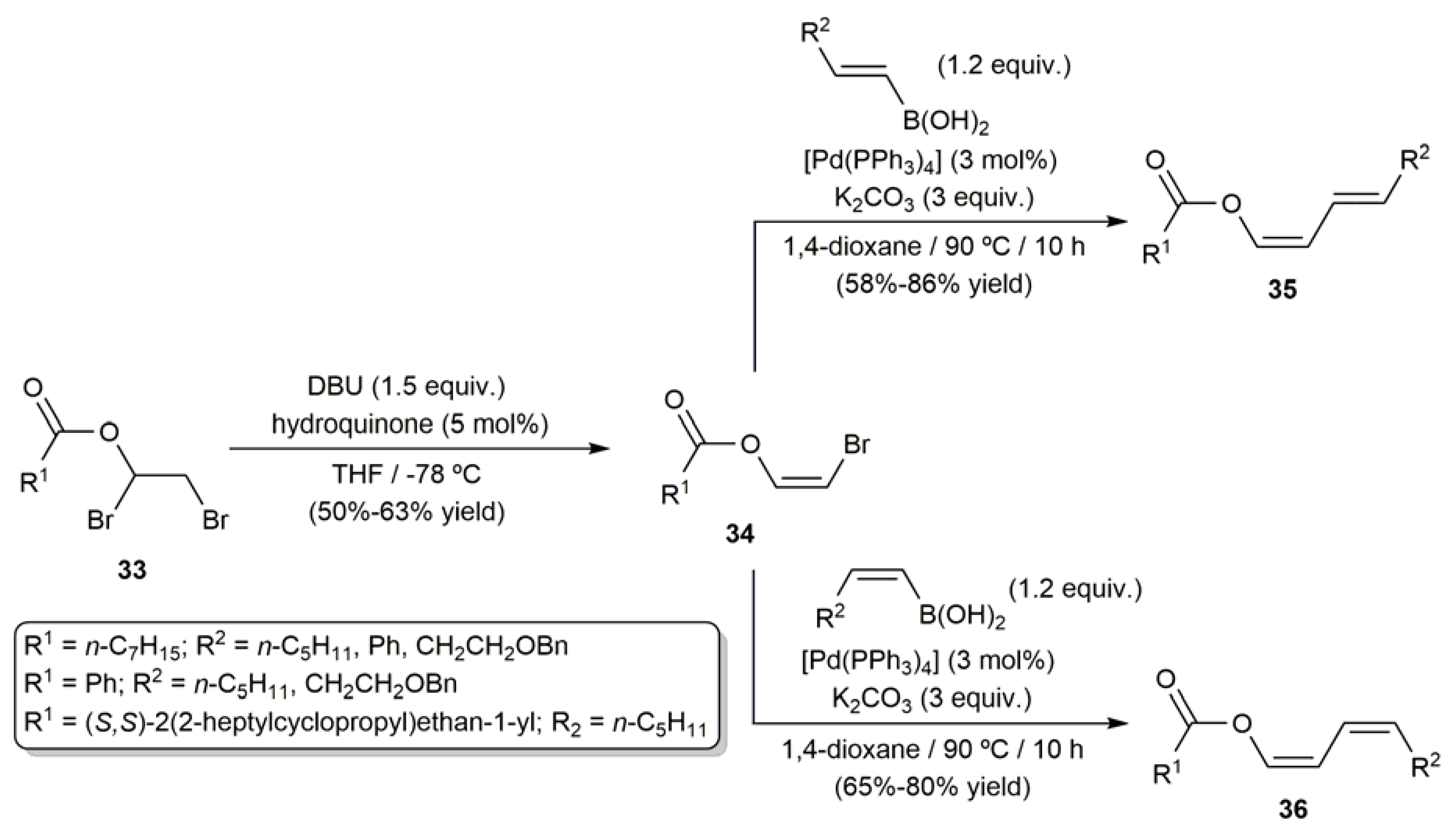
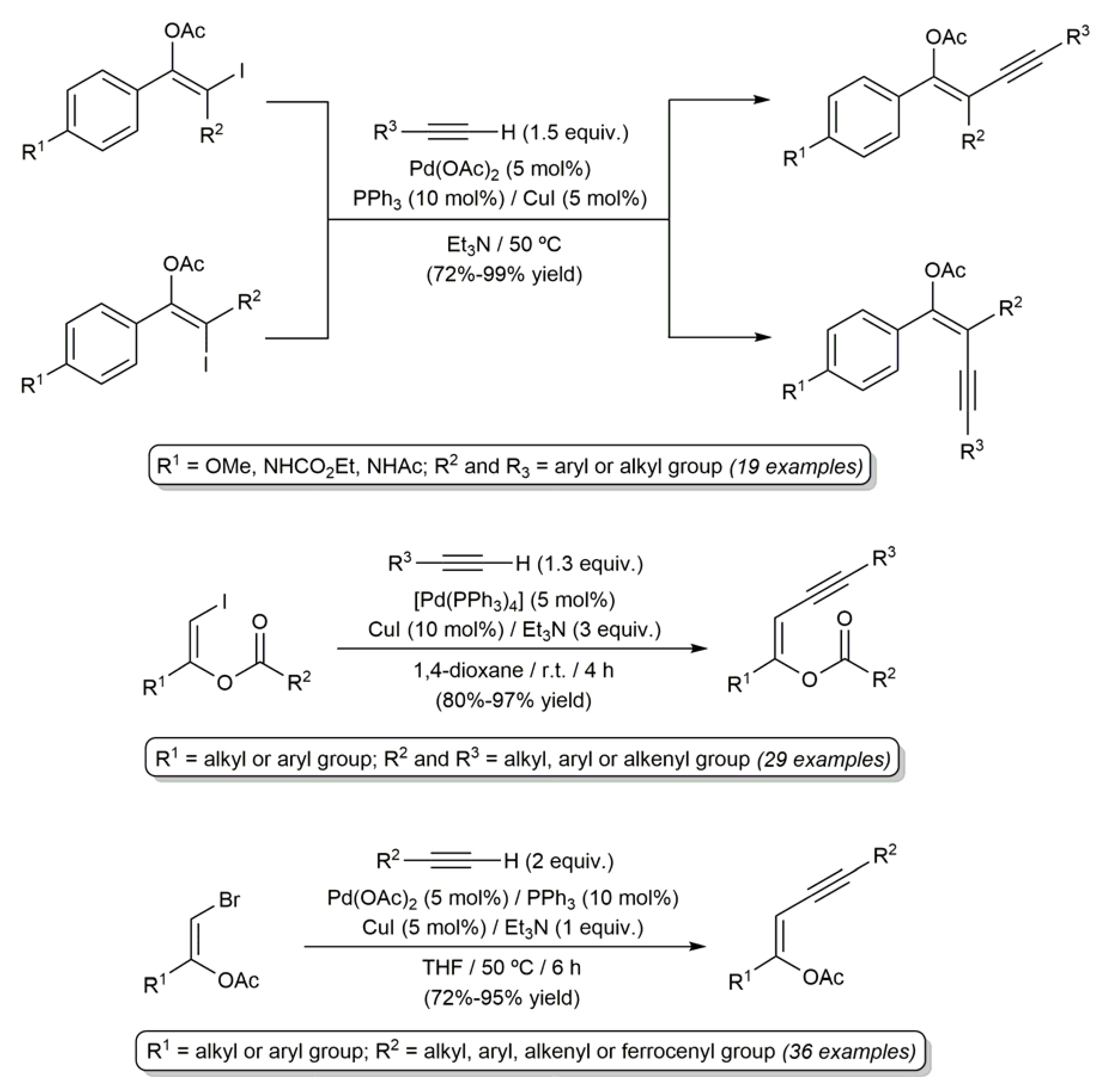
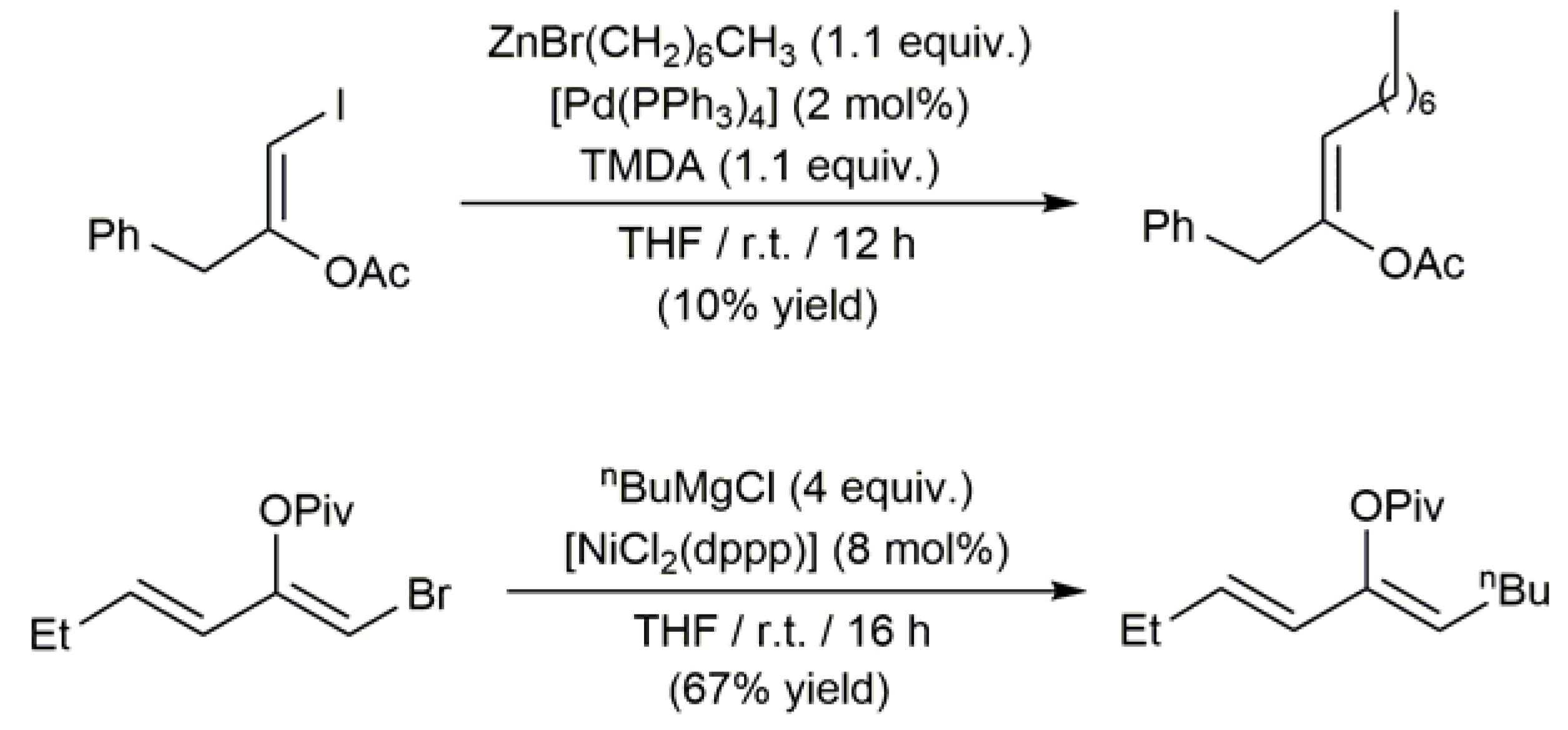
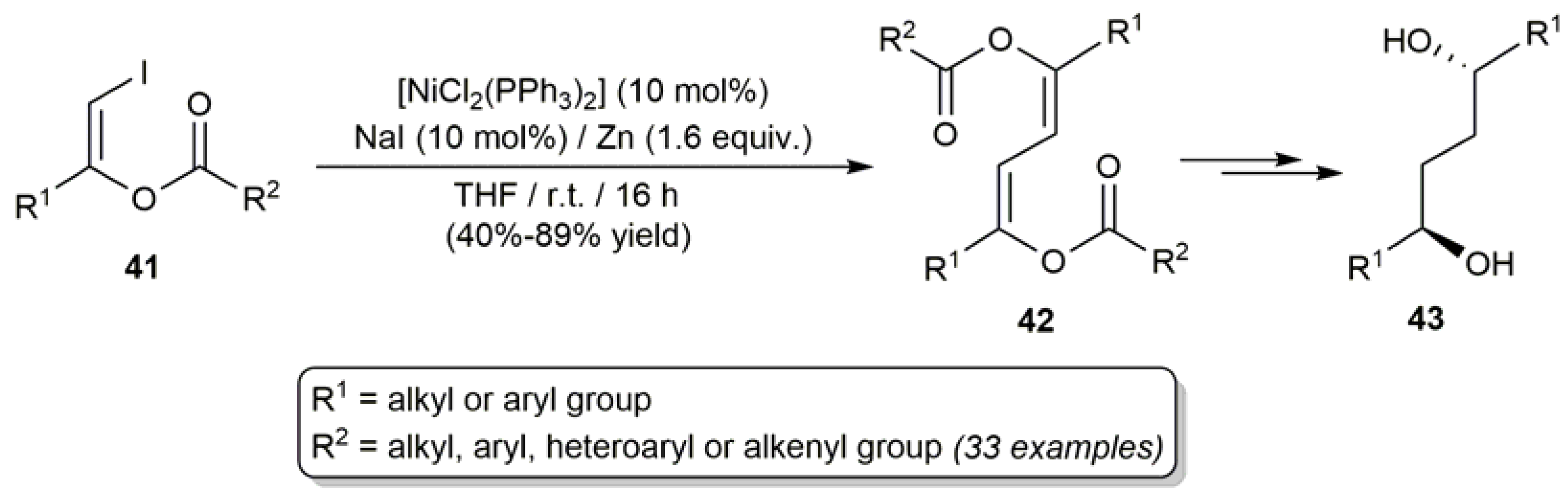
3.1. Acyclic β-Haloenol Esters
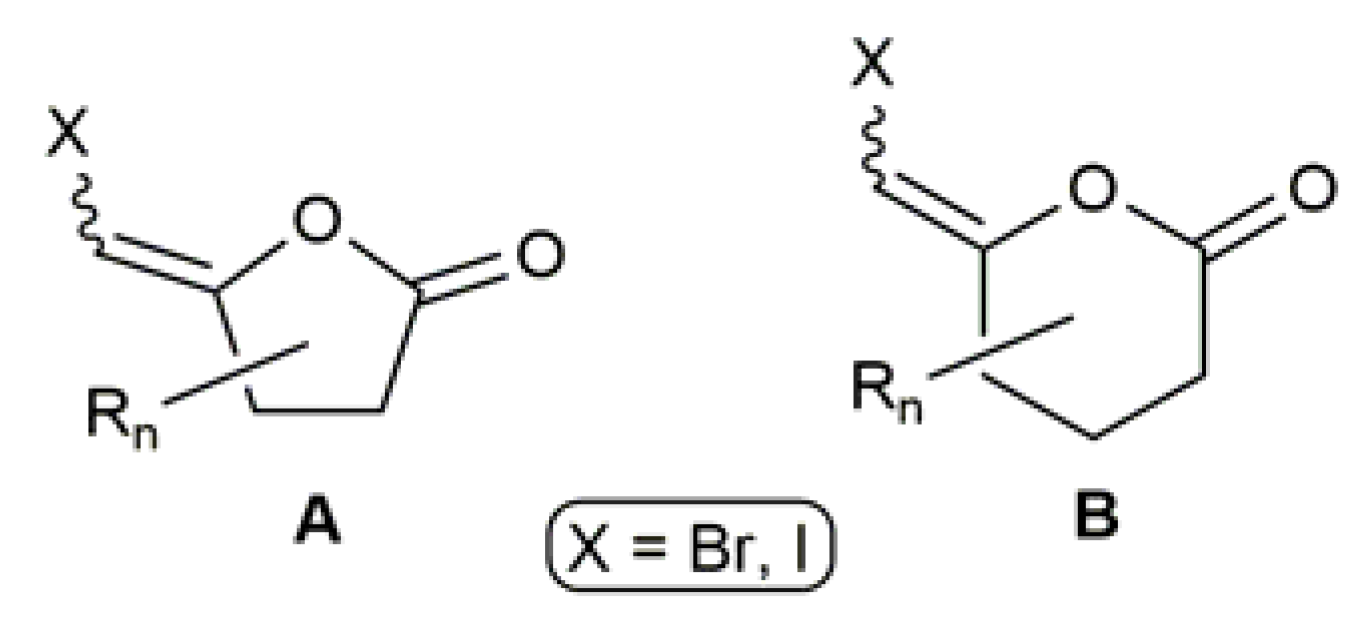
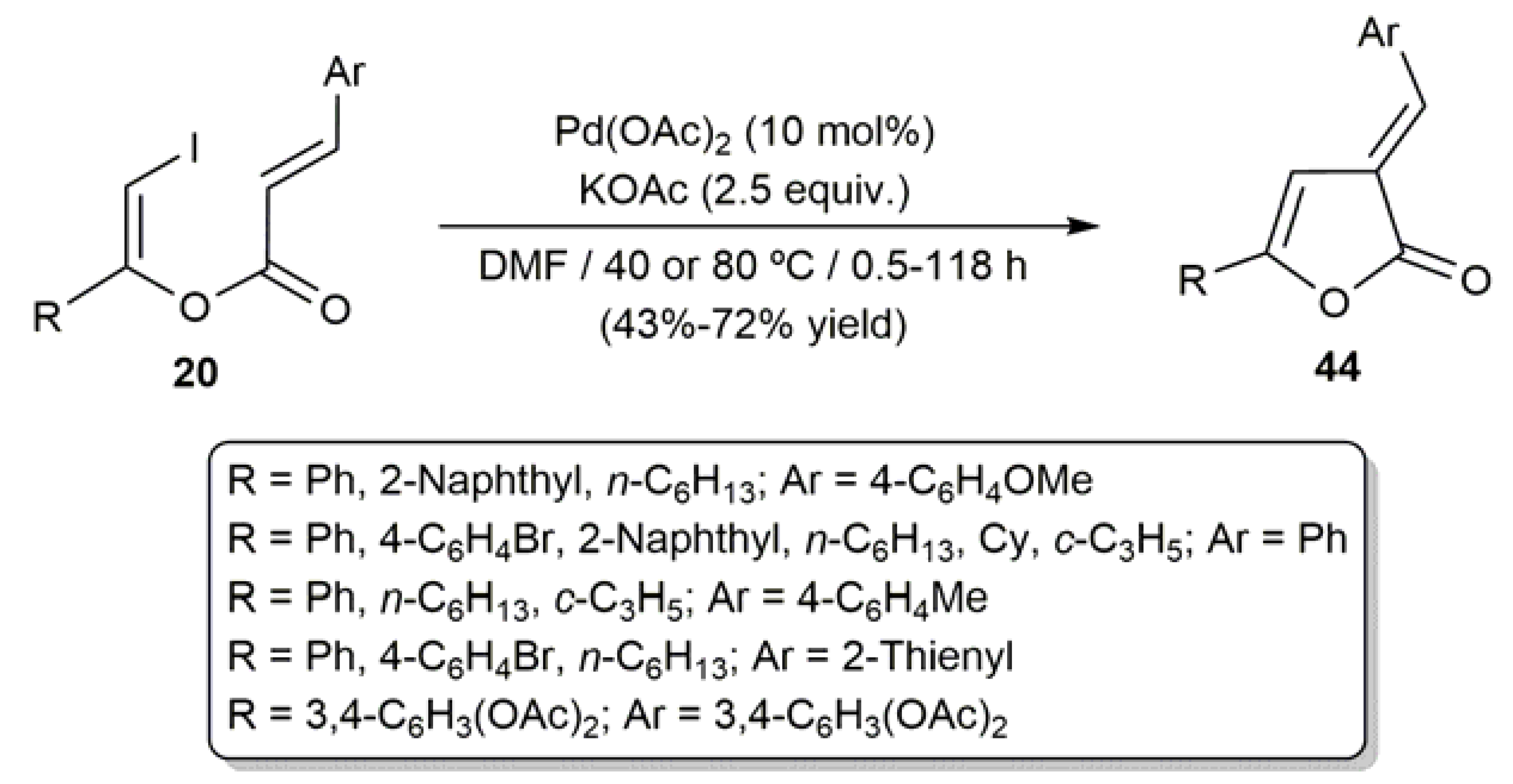
3.2. Cyclic β-Haloenol Esters
4. Conclusions
Funding
Conflicts of Interest
References
- De Meijer, A.; Diederich, F. (Eds.) Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Schlosser, M. (Ed.) Organometallics in Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Jeschke, J.; Gäbler, C.; Lang, H. Regioselective formation of enol esters from the ruthenium-catalyzed Markovnikov addition of carboxylic acids to alkynes. J. Org. Chem. 2016, 81, 476–484. [Google Scholar] [CrossRef]
- Francos, J.; Cadierno, V. Metal-catalyzed intra- and intermolecular addition of carboxylic acids to alkynes in aqueous media: A review. Catalysts 2017, 7, 328. [Google Scholar] [CrossRef]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. The intermolecular hydro-oxycarbonylation of internal alkynes: Current state of the art. Arkivoc 2018, 2, 17–39. [Google Scholar]
- Takai, K.; Kokumai, R.; Nobunaka, T. Reactions of coordinated geminal dichromium reagents with aldehydes: Stereoselective formation of (Z)-2-chloroalk-2-en-1-ols. Chem. Commun. 2001, 1128–1129. [Google Scholar] [CrossRef]
- Bejot, R.; Tisserand, S.; Reddy, L.M.; Barma, D.K.; Baati, R.; Falck, J.R.; Mioskowski, C. Stereoselective transformations of trihalomethylcarbinols induced by chromous chloride. Angew. Chem. Int. Ed. 2005, 44, 2008–2011. [Google Scholar] [CrossRef]
- Bejot, R.; Anjaiah, S.; Falck, J.R.; Mioskowski, C. α-Haloenol acetates: Versatile reactants for oxetan-2-one, azetidin-2-one and isoxazolidin-5-one synthesis. Eur. J. Org. Chem. 2007, 101–107. [Google Scholar] [CrossRef]
- Kashinath, D.; Mioskowski, C.; Falck, J.R.; Goli, M.; Meunier, S.; Baati, R.; Wagner, A. Highly stereoselective synthesis of (Z,E)-1-halo-1,3-dienol esters via rearrangement of Fischer chromium chloro-carbenes using microwave irradiation. Org. Biomol. Chem. 2009, 7, 1771–1774. [Google Scholar] [CrossRef]
- Joshi, G.C.; Chambers, W.D.; Warnhoff, E.W. Camphor enol and homoenol acetates. Tetrahedron Lett. 1967, 8, 3613–3617. [Google Scholar] [CrossRef]
- Kowalski, C.J.; O’Dowd, M.L.; Burke, M.C.; Fields, K.W. α-Ketodianions. New reactive intermediates. J. Am. Chem. Soc. 1980, 102, 5411–5412. [Google Scholar] [CrossRef]
- Kowalski, C.J.; Haque, M.S.; Fields, K.W. Ester homologation via α-bromo α-keto dianion rearrangement. J. Am. Chem. Soc. 1985, 107, 1429–1430. [Google Scholar] [CrossRef]
- Kowalski, C.J.; Haque, M.S. Bromomethyl ketones and enolates: Alternative products from ester homologation reactions. J. Org. Chem. 1985, 50, 5140–5142. [Google Scholar] [CrossRef]
- Kowalski, C.J.; Reddy, R.E. Ester homologation revisited: A reliable, higher yielding and better understood procedure. J. Org. Chem. 1992, 57, 7194–7208. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; González, J.M.; Campos, P.J. A new electrophilic addition to acetylenes. Synthesis of 1,2-iodofunctionalized olefins. Tetrahedron Lett. 1986, 27, 3303–3306. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Synthesis of 2-functionalized 1,1-diiodo-1-alkenes. Generation and reactions of 1-iodo-1-lithio-1-alkenes and 1,1-dilithio-1-alkenes. J. Am. Chem. Soc. 1988, 110, 5567–5568. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Synthesis of 2-functionalized 1-chloro-1-iodo-1-alkenes from 1-chloro-1-alkynes and IPy2BF4. Tetrahedron Lett. 1990, 31, 2751–2754. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Electrophilic additions of positive iodine to alkynes through an iodonium mechanism. J. Org. Chem. 1990, 55, 3104–3106. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, M.A.; Campos, P.J. Stereoselective synthesis of 2-functionalized 1-bromo-1-iodo-1-alkenes by electrophilic iodination of 1-bromo-1-alkynes. Synthesis 1992, 270–272. [Google Scholar] [CrossRef]
- Pincock, J.A.; Yates, K. Kinetics and mechanism of electrophilic bromination of acetylenes. Can. J. Chem. 1970, 48, 3332–3348. [Google Scholar] [CrossRef]
- Ogata, Y.; Urasaki, I. The reaction of tolan with a mixture of iodine and peracetic acid. J. Org. Chem. 1971, 36, 2164–2168. [Google Scholar] [CrossRef]
- Yates, K.; Go, T.A. Vinyl cation intermediates in electrophilic additions to triple bonds. 2. Chlorination of alkylacetylenes. J. Org. Chem. 1980, 45, 2385–2391. [Google Scholar] [CrossRef]
- Heasley, G.E.; Codding, C.; Sheehy, J.; Gering, K.; Heasley, V.L.; Shellhamer, D.F.; Rempel, T. Chlorination of 1-hexyne and 3-hexyne in acetic acid and methanol. J. Org. Chem. 1985, 50, 1773–1776. [Google Scholar] [CrossRef]
- Hokamp, T.; Storm, A.T.; Yusubov, M.; Wirth, T. Iodine monoacetate for efficient oxyiodinations of alkenes and alkynes. Synlett 2018, 29, 415–418. [Google Scholar]
- Jovtscheff, A.; Spassov, S.L. Heterolytische anlagerung an acetylen-verbindungen I: Stereochemie und kinetik der anlagerung im system diphenylacetylen-N-brom-succinimid-essigsäure-wasser. Monatsh. Chem. 1967, 98, 2272–2281. [Google Scholar] [CrossRef]
- Jovtscheff, A.; Spassov, S.L. Heterolytische anlagerungen an acetylen-verbindungen III: Über den mechanismus der umsetzung von acetylen-verbindungen mit N-bromsuccinimid in eisessig oder waβriger essigsäure. Monatsh. Chem. 1969, 100, 328–341. [Google Scholar] [CrossRef]
- Okamoto, N.; Miwa, Y.; Minami, H.; Takeda, K.; Yanada, R. Regio- and stereoselective multisubstituted enol ester synthesis. J. Org. Chem. 2011, 76, 9133–9138. [Google Scholar] [CrossRef] [PubMed]
- Merkushev, E.B.; Karpitskaya, L.G.; Novosel’tseva, G.I.; Raida, V.S. Conjugated iodoacetoxylation of triple bonds. Izv. Akad. Nauk Ser. Khim. 1978, 1153–1154. [Google Scholar] [CrossRef]
- Xia, X.-F.; Gu, Z.; Liu, W.; Wang, N.; Wang, H.; Xia, Y.; Gao, H.; Liu, X. Selective oxygenation of alkynes: A direct approach to diketones and vinyl acetate. Org. Biomol. Chem. 2014, 12, 9909–9913. [Google Scholar] [CrossRef]
- Priebbenow, D.L.; Gable, R.W.; Baell, J. Regio- and stereoselective iodoacyloxylations of alkynes. J. Org. Chem. 2015, 80, 4412–4418. [Google Scholar] [CrossRef]
- Crespo, L.T.C.; Nogueira, G.P.; de Mattos, M.C.S.; Esteves, P.M. Reaction of trihaloisocyanuric acids with alkynes: An efficient methodology for the preparation of β-haloenol acetates. Arkivoc 2018, 2, 205–214. [Google Scholar] [CrossRef]
- Chawla, R.; Singh, A.K.; Yadav, L.D.S. Catalyst- and metal-free rapid functionalizations of alkynes using TsNBr2. Synlett 2013, 24, 1558–1562. [Google Scholar]
- Daniels, S.B.; Cooney, E.; Sofia, M.J.; Chakravarty, P.K.; Katzenellenbogen, J.A. Haloenol lactones: Potent enzyme-activated irreversible inhibitors for α-chymotrypsin. J. Biol. Chem. 1983, 258, 15046–15053. [Google Scholar] [PubMed]
- Sofia, M.J.; Katzenellenbogen, J.A. Enol lactone inhibitors of serine proteases. The effect of regiochemistry on the inactivation behavior of phenyl-substituted (halomethylene)tetra- and -dihydrofuranones and (halomethylene)tetrahydropyranones toward α-chymotrypsin: Stable acyl enzyme intermediate. J. Med. Chem. 1986, 29, 230–238. [Google Scholar] [PubMed]
- Rai, R.; Katzenellenbogen, J.A. Guanidinophenyl-substituted enol lactones as selective, mechanism-based inhibitors of trypsin-like serine proteases. J. Med. Chem. 1992, 35, 4150–4159. [Google Scholar] [CrossRef]
- Wu, Z.; Minhas, G.S.; Wen, D.; Jiang, H.; Chen, K.; Zimniak, P.; Zheng, J. Design, synthesis, and structure-activity relationships of haloenol lactones: Site-directed and isozyme-selective glutathione S-transferase inhibitors. J. Med. Chem. 2004, 47, 3282–3294. [Google Scholar] [CrossRef]
- Mock, J.N.; Taliaferro, J.P.; Lu, X.; Patel, S.K.; Cummings, B.S.; Long, T.E. Haloenol pyranones and morpholinones as antineoplastic agents. Bioorg. Med. Chem. Lett. 2012, 22, 4854–4858. [Google Scholar] [CrossRef]
- Albrecht, L.; Albrecht, A.; Janecki, T. α-Alkylidene-γ- and δ-lactones and lactams. In Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity; Janecki, T., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 147–192. [Google Scholar]
- Ranganathan, S.; Muraleedharan, K.M.; Vaish, N.K.; Jayaraman, N. Halo- and selenolactonisation: The two major strategies for cyclofunctionalisation. Tetrahedron 2004, 60, 5273–5308. [Google Scholar] [CrossRef]
- Halder, J.; Das, D.; Nanda, S. A distinctive transformation based diversity oriented synthesis of small ring carbocycles and heterocycles from biocatalytically derived enantiopure α-substituted-β-hydroxyesters. Org. Biomol. Chem. 2018, 16, 2549–2575. [Google Scholar] [CrossRef]
- Wilking, M.; Mück-Lichtenfeld, C.; Daniliuc, C.G.; Hennecke, U. Enantioselective, desymmetrizing bromolactonization of alkynes. J. Am. Chem. Soc. 2013, 135, 8133–8136. [Google Scholar] [CrossRef]
- Wilking, M.; Daniliuc, C.G.; Hennecke, U. Monomeric cinchona alkaloid-based catalysts for highly enantioselective bromolactonisation of alkynes. Chem. Eur. J. 2016, 22, 18601–18607. [Google Scholar] [CrossRef]
- Fricke, C.; Wilking, M.; Daniliuc, C.G.; Hennecke, U. An enantioselective iodolactonization/cross-coupling protocol for the synthesis of highly substituted enol lactones. Eur. J. Org. Chem. 2018, 3158–3166. [Google Scholar] [CrossRef]
- Wu, W.; Jiang, H. Haloalkynes: A powerful and versatile building block in organic synthesis. Acc. Chem. Res. 2014, 47, 2483–2504. [Google Scholar] [CrossRef] [PubMed]
- Petko, D.; Koh, S.; Tam, W. Transition metal-catalyzed reactions of alkynyl halides. Curr. Org. Synth. 2019, 16, 546–582. [Google Scholar] [CrossRef] [PubMed]
- Cadierno, V. Metal-catalyzed hydrofunctionalization reactions of haloalkynes. Eur. J. Inorg. Chem. 2020, 886–898. [Google Scholar] [CrossRef]
- Uemura, S.; Tara, H.; Okano, M.; Ichikawa, K. Acetoxythallation of acetylenes and the proto- and halogenodethallation of the products. Bull. Chem. Soc. Jpn. 1974, 47, 2663–2671. [Google Scholar] [CrossRef]
- Amos, R.A.; Katzenellenbogen, J.A. An efficient synthesis of γ-methylene-γ-butyrolactone (α’-angelicalactone). Application to the synthesis of deoxyobtusilactone and deoxyisoobtusilactone. J. Org. Chem. 1978, 43, 560–564. [Google Scholar] [CrossRef]
- Krafft, G.A.; Katzenellenbogen, J.A. Synthesis of halo enol lactones. Mechanism-based inactivators of serine proteases. J. Am. Chem. Soc. 1981, 103, 5459–5466. [Google Scholar] [CrossRef]
- Spencer, R.W.; Tam, F.T.; Thomas, E.; Robinson, V.J.; Krantz, A. Ynenol lactones: Synthesis and investigation of reactions relevant to their inactivation of serine proteases. J. Am. Chem. Soc. 1986, 108, 5589–5597. [Google Scholar] [CrossRef]
- Barluenga, J.; Martínez-Gallo, J.M.; Nájera, C.; Yus, M. Stereoselective bifunctionalization of alkynes by means of the mercury(II) salt-iodine combination. J. Chem. Soc. Perkin Trans. 1 1987, 1017–1019. [Google Scholar] [CrossRef]
- Dai, W.; Katzenellenbogen, J.A. Stereoselective Z- and E-bromo enol lactonization of alkynois acids. J. Org. Chem. 1991, 56, 6893–6896. [Google Scholar] [CrossRef]
- Chen, Z.; Li, J.; Jiang, H.; Zhu, S.; Li, Y.; Qi, C. Silver-catalyzed difunctionalization of terminal alkynes: Highly regio- and stereoselective synthesis of (Z)-β-haloenol acetates. Org. Lett. 2010, 12, 3262–3265. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Toullec, P.Y.; Antoniotti, S.; Brancour, C.; Genêt, J.-P.; Michelet, V. Room temperature Au(I)-catalyzed exo-selective cycloisomerization of acetylenic acids: An entry to functionalized γ-lactones. J. Am. Chem. Soc. 2006, 128, 3112–3113. [Google Scholar] [CrossRef] [PubMed]
- Harkat, H.; Weibel, J.-M.; Pale, P. A mild access to γ- and δ-alkylidene lactones through gold catalysis. Tetrahedron Lett. 2006, 47, 6273–6276. [Google Scholar] [CrossRef]
- Harkat, H.; Dembelé, A.Y.; Weibel, J.-M.; Blanc, A.; Pale, P. Cyclization of alkynoic acids with gold catalysts: A surprising dichotomy between AuI and AuIII. Tetrahedron 2009, 65, 1871–1879. [Google Scholar] [CrossRef]
- Gasperini, D.; Maggi, L.; Dupuy, S.; Veenboer, R.M.P.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P. Gold(I)-catalysed cyclisation of alkynoic acids: Towards an efficient and eco-friendly synthesis of γ-,δ- and ε-lactones. Adv. Synth. Catal. 2016, 358, 3857–3862. [Google Scholar] [CrossRef]
- González-Liste, P.J.; León, F.; Arribas, I.; Rubio, M.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Highly stereoselective synthesis and hydrogenation of (Z)-1-alkyl-2-arylvinyl acetate: A wide scope procedure for the preparation of chiral homobenzylic esters. ACS Catal. 2016, 6, 3056–3060. [Google Scholar] [CrossRef]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. Gold-catalyzed regio- and stereoselective addition of carboxylic acids to iodoalkynes: Access to (Z)-β-iodoenol esters and 1,4-disubstituted (Z)-enynyl esters. J. Org. Chem. 2017, 82, 1507–1516. [Google Scholar] [CrossRef]
- León, F.; González-Liste, P.J.; García-Garrido, S.E.; Arribas, I.; Rubio, M.; Cadierno, V.; Pizzano, A. Broad scope synthesis of esters precursors of nonfunctionalized chiral alcohols based on the asymmetric hydrogenation of α,β-dialkyl-, α,β-diaryl-, and α-alkyl-β-aryl-vinyl esters. J. Org. Chem. 2017, 82, 5852–5867. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Toste, F.D. (Eds.) Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Muthusamy, G.; Pansare, S.V. Stereoselective synthesis of E-3-(arylmethylidene)-5-(alkyl/aryl)-2(3H)-furanones by sequential hydroacyloxylation-Mizoroki-Heck reactions of iodoalkynes. Org. Biomol. Chem. 2018, 16, 7971–7983. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, B.; Zhang, L. The use of Br/Cl to promote regioselective gold-catalyzed rearrangement of propargylic carboxylates: An efficient synthesis of (1Z, 3E)-1-bromo/cloro-2-carboxy-1,3-dienes. Chem. Commun. 2010, 46, 9179–9181. [Google Scholar] [CrossRef][Green Version]
- Jiang, J.; Liu, Y.; Hou, C.; Li, Y.; Luan, Z.; Zhao, C.; Ke, Z. Rationalization of the selectivity between 1,3- and 1,2-migration: A DFT study on gold(I)-catalyzed propargylic ester rearrangement. Org. Biomol. Chem. 2016, 14, 3558–3563. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Nayak, S.; Prabagar, B.; Sahoo, A.K. Regioselective hydration of terminal halo-substituted propargyl carboxylates by gold catalyst: Synthesis of α-acyloxy α’-halo ketones. J. Org. Chem. 2014, 79, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, Y.; Zhao, X. Theoretical insight into the Au(I)-catalyzed hydration of halo-substituted propargyl acetate: Dynamic water-assisted mechanism. RSC Adv. 2016, 6, 89836–89846. [Google Scholar]
- Chen, Z.-W.; Ye, D.-N.; Ye, M.; Zhou, Z.-G.; Li, S.-H.; Liu, L.-X. AgF/TFA-promoted highly efficient synthesis of α-haloketones from haloalkynes. Tetrahedron Lett. 2014, 55, 1373–1375. [Google Scholar] [CrossRef]
- Zeng, M.; Huang, R.-X.; Li, W.-Y.; Liu, X.-W.; He, F.-L.; Zhang, Y.-Y.; Xiao, F. In(OTf)3/acid co-catalyzed hydration of 1-haloalkynes to α-halomethyl ketones. Tetrahedron 2016, 72, 3818–3822. [Google Scholar] [CrossRef]
- Zou, H.; He, W.; Dong, Q.; Wang, R.; Yi, N.; Jiang, J.; Pen, D.; He, W. First catalyzed hydration of haloalkynes by a recyclable catalytic system. Eur. J. Org. Chem. 2016, 116–121. [Google Scholar] [CrossRef]
- Chen, X.; Chen, D.; Lu, Z.; Kong, L.; Zhu, G. Palladium-catalyzed coupling of haloalkynes with allyl acetate: A regio- and stereoselective synthesis of (Z)-β-haloenol acetates. J. Org. Chem. 2011, 76, 6338–6343. [Google Scholar] [CrossRef]
- Espinosa-Jalapa, N.A.; Ke, D.; Nebra, N.; Le Goanvic, L.; Mallet-Ladeira, S.; Monot, J.; Martin-Vaca, B.; Bourissou, D. Enhanced catalytic performance of indenediide palladium pincer complexes for cycloisomerization: Efficient synthesis of alkylidene lactams. ACS Catal. 2014, 4, 3605–3611. [Google Scholar] [CrossRef]
- Monot, J.; Brunel, P.; Kefalidis, C.E.; Espinosa-Jalapa, N.A.; Maron, L.; Martin-Vaca, B.; Bourissou, D. A case study of proton shuttling in palladium catalysis. Chem. Sci. 2016, 7, 2179–2187. [Google Scholar] [CrossRef]
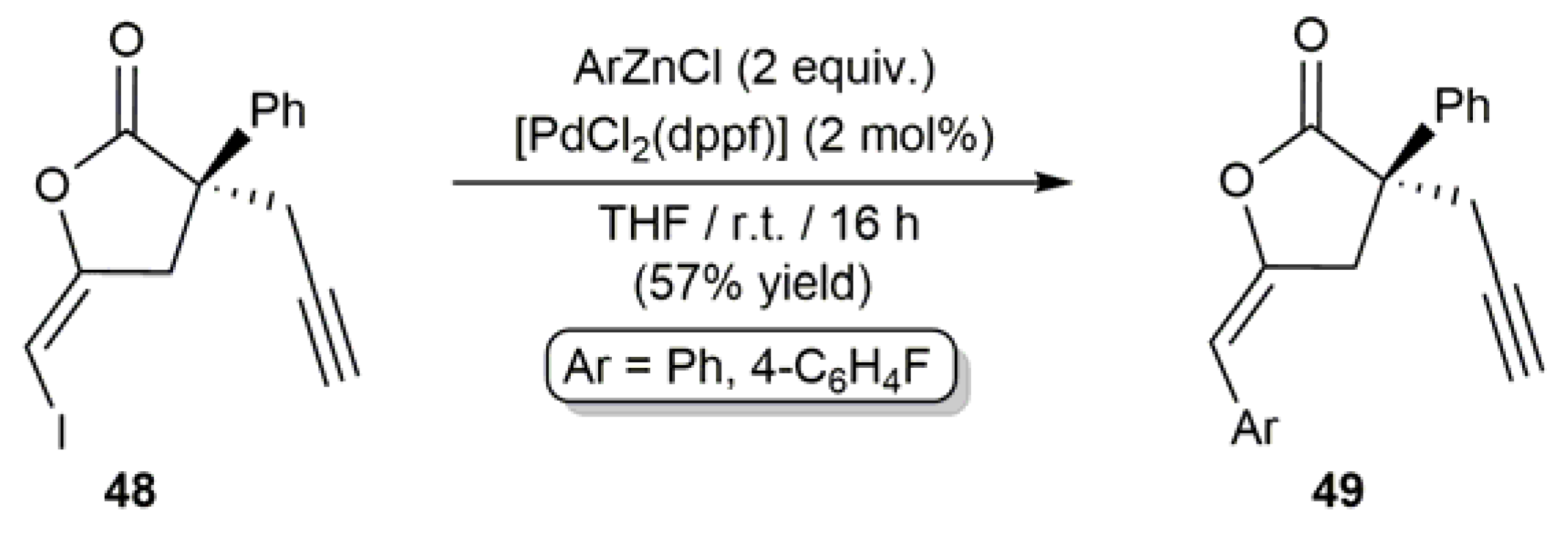
- Abe, S.; Miyaura, N.; Suzuki, A. The palladium-catalyzed cross-coupling reaction of enol acetates with 1-alkenyl-, aryl-, or alkylboron compounds; A facile synthesis of ketones and their enol acetates. Bull. Chem. Soc. Jpn. 1992, 65, 2863–2865. [Google Scholar] [CrossRef]
- He, R.; Deng, M.-Z. A novel method for the construction of (Z,E)- or (Z,Z)-conjugated alkadienyl carboxylates. Org. Lett. 2002, 4, 2759–2762. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, G.; Jiang, H.; Huang, H.; Pan, X. Synthesis of 2,5-disubstituted 3-iodofurans via palladium-catalyzed coupling and iodocyclization of terminal alkynes. J. Org. Chem. 2011, 76, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Yanada, R. Multisubstituted furan formation from (Z)- or (E)-enynyl acetates: Tandem reactions accelerated by electron-donating groups on aromatic rings. J. Org. Chem. 2012, 77, 3944–3951. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Chen, H.-C.; Ye, D.-N.; Hu, Q.-S. (Z)-1,4-Diphenylbut-1-en-3-ynyl acetate. Acta Cryst. 2012, E68, o2847. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-W.; Luo, M.-T.; Wen, Y.-L.; Ye, M.; Zhou, Z.-G.; Liu, L.-X. A highly efficient synthesis of 2,5-disubstituted furans from enyne acetates catalyzed by Lewis acid and palladium. Synlett 2014, 25, 2341–2344. [Google Scholar] [CrossRef]
- Lee, H.; Yi, Y.; Jun, C.-H. Copper(II)-promoted, one-pot conversion of 1-alkynes with anhydrides or primary amines to the respective 2,5-disubstituted furans or pyrroles under microwave irradiation conditions. Adv. Synth. Catal. 2015, 357, 3485–3490. [Google Scholar] [CrossRef]
- Okamoto, N.; Sueda, T.; Yanada, R. Tandem reaction of enynyl acetate: Precursor of allenyl ketones. Chem. Pharm. Bull. 2016, 64, 941–946. [Google Scholar] [CrossRef]
- Stefani, H.A.; Guarezemini, A.S.; Cella, R. Homocoupling reactions of alkynes, alkenes and alkyl compounds. Tetrahedron 2010, 66, 7871–7918. [Google Scholar] [CrossRef]
- Takagi, K.; Hayama, N.; Sasaki, K. Ni(0)-trialkylphosphine complexes. Efficient homo-coupling catalyst for aryl, alkenyl, and heteroaromatic halides. Bull. Chem. Soc. Jpn. 1984, 57, 1887–1890. [Google Scholar] [CrossRef]
- Takagi, K.; Mimura, H.; Inokawa, S. The in situ-generated nickel(0)-catalyzed homo-coupling of alkenyl halides with zinc powder. A specific outcome in stereochemistry. Bull. Chem. Soc. Jpn. 1984, 57, 3517–3522. [Google Scholar] [CrossRef]
- Sasaki, K.; Nakao, K.; Kobayashi, Y.; Sakai, M.; Uchino, N.; Sakakibara, Y.; Takagi, K. Ni(0)-triphenylphosphine complex-catalyzed homo-coupling of 1-alkenyl halides with zinc powder. Bull. Chem. Soc. Jpn. 1993, 66, 2446–2448. [Google Scholar] [CrossRef]
- Francos, J.; Cadierno, V. Nickel-catalyzed homocoupling of (Z)-β-iodoenol esters: Stereoselective access to (Z,Z)-buta-1,3-diene-1,4-diyl diesters. Synthesis 2019, 51, 3117–3126. [Google Scholar] [CrossRef]
- León, F.; Francos, J.; López-Serrano, J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Double asymmetric hydrogenation of conjugated dienes: A self-breeding chirality route to C2 symmetric 1,4-diols. Chem. Commun. 2019, 55, 786–789. [Google Scholar] [CrossRef] [PubMed]
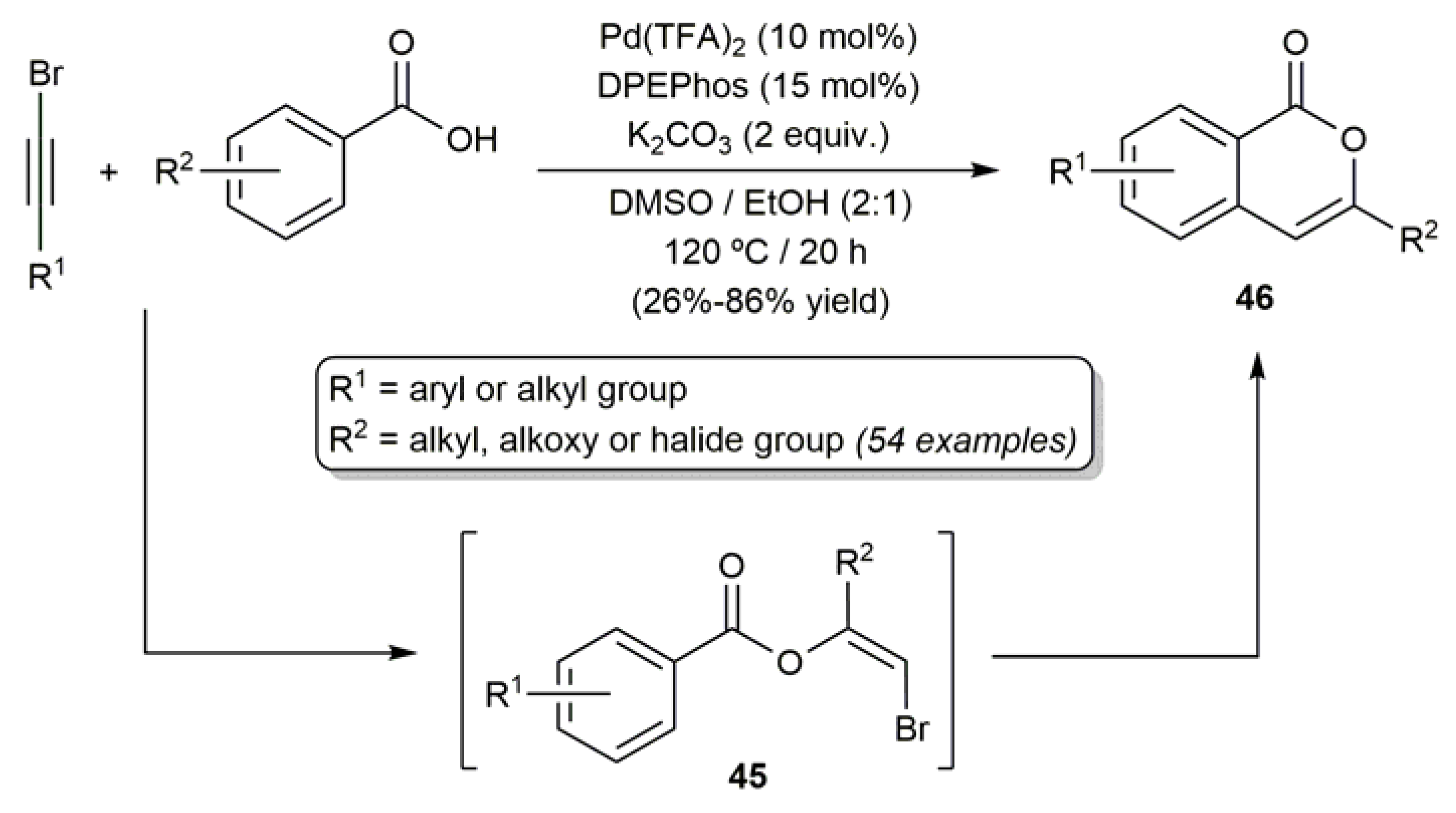
- Jiang, G.; Li, J.; Zhu, C.; Wu, W.; Jiang, H. Palladium-catalyzed sequential nucleophilic addition/oxidative annulation of bromoalkynes with benzoic acids to construct functionalized isocoumarins. Org. Lett. 2017, 19, 4440–4443. [Google Scholar] [CrossRef]
- Chen, Z.; Ye, D.; Xu, G.; Ye, M.; Liu, L. Highly efficient synthesis of 2,5-disubstituted pyrazines from (Z)-β-haloenol acetates. Org. Biomol. Chem. 2013, 11, 6699–6702. [Google Scholar] [CrossRef]
- Tam, F.T.; Spencer, R.W.; Thomas, E.M.; Copp, L.J.; Krantz, A. Novel suicide inhibitors of serine proteinases. Inactivation of human leukocyte elastase by ynenol lactones. J. Am. Chem. Soc. 1984, 106, 6849–6851. [Google Scholar] [CrossRef]
- Zupan, L.A.; Weiss, R.H.; Hazen, S.L.; Parnas, B.L.; Aston, K.W.; Lennon, P.J.; Getman, D.P.; Gross, R.W. Structural determinants of haloenol lactone-mediated suicide inhibition of canine myocardial calcium-independent phospholipase A2. J. Med. Chem. 1993, 36, 95–100. [Google Scholar] [CrossRef]
































© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cadierno, V. Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters. Catalysts 2020, 10, 399. https://doi.org/10.3390/catal10040399
Cadierno V. Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters. Catalysts. 2020; 10(4):399. https://doi.org/10.3390/catal10040399
Chicago/Turabian StyleCadierno, Victorio. 2020. "Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters" Catalysts 10, no. 4: 399. https://doi.org/10.3390/catal10040399
APA StyleCadierno, V. (2020). Metal-Catalyzed Synthesis and Transformations of β-Haloenol Esters. Catalysts, 10(4), 399. https://doi.org/10.3390/catal10040399