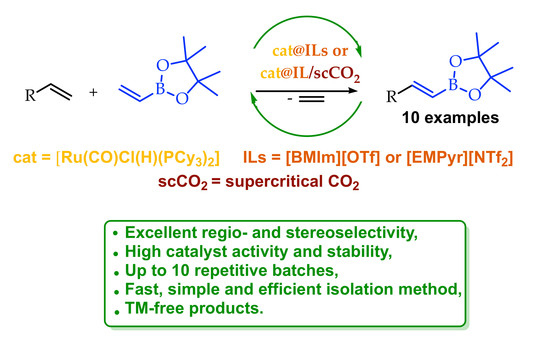
Ru-Catalyzed Repetitive Batch Borylative Coupling of Olefins in Ionic Liquids or Ionic Liquids/scCO2 Systems


Abstract
:
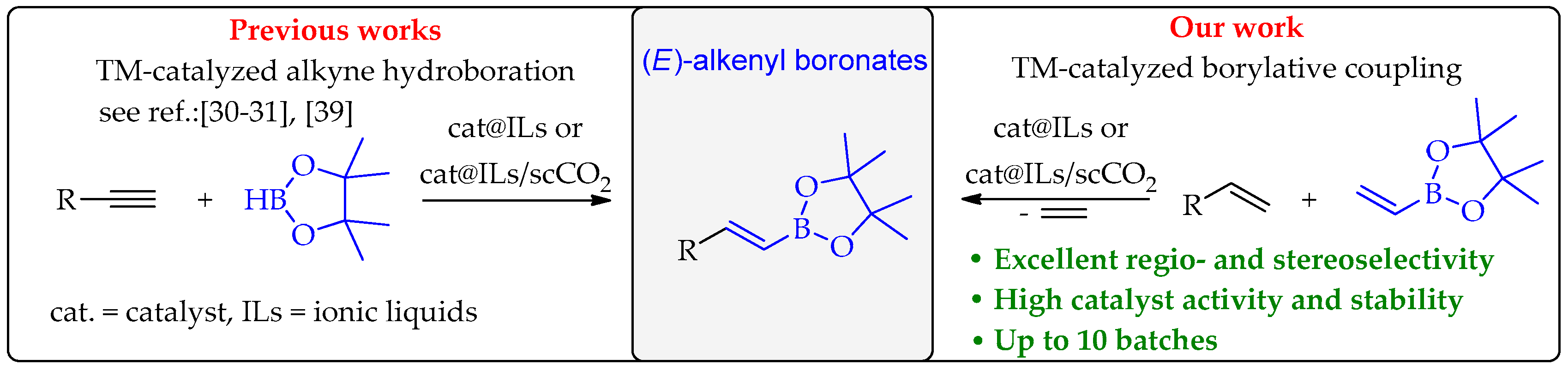
1. Introduction
2. Results and Discussion
2.1. Reaction and Extraction Conditions Screening
2.2. Scope of Substrates Investigations
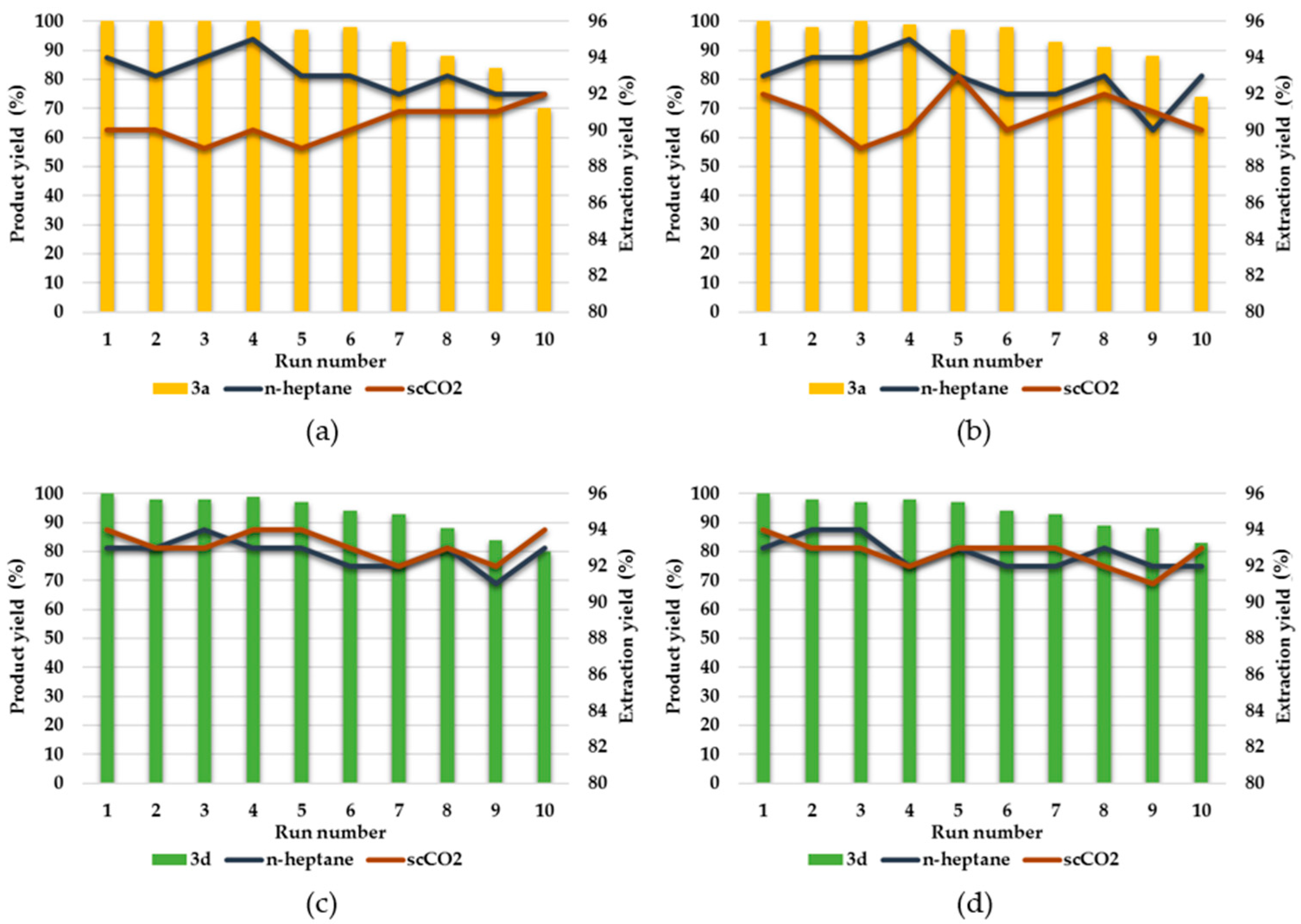
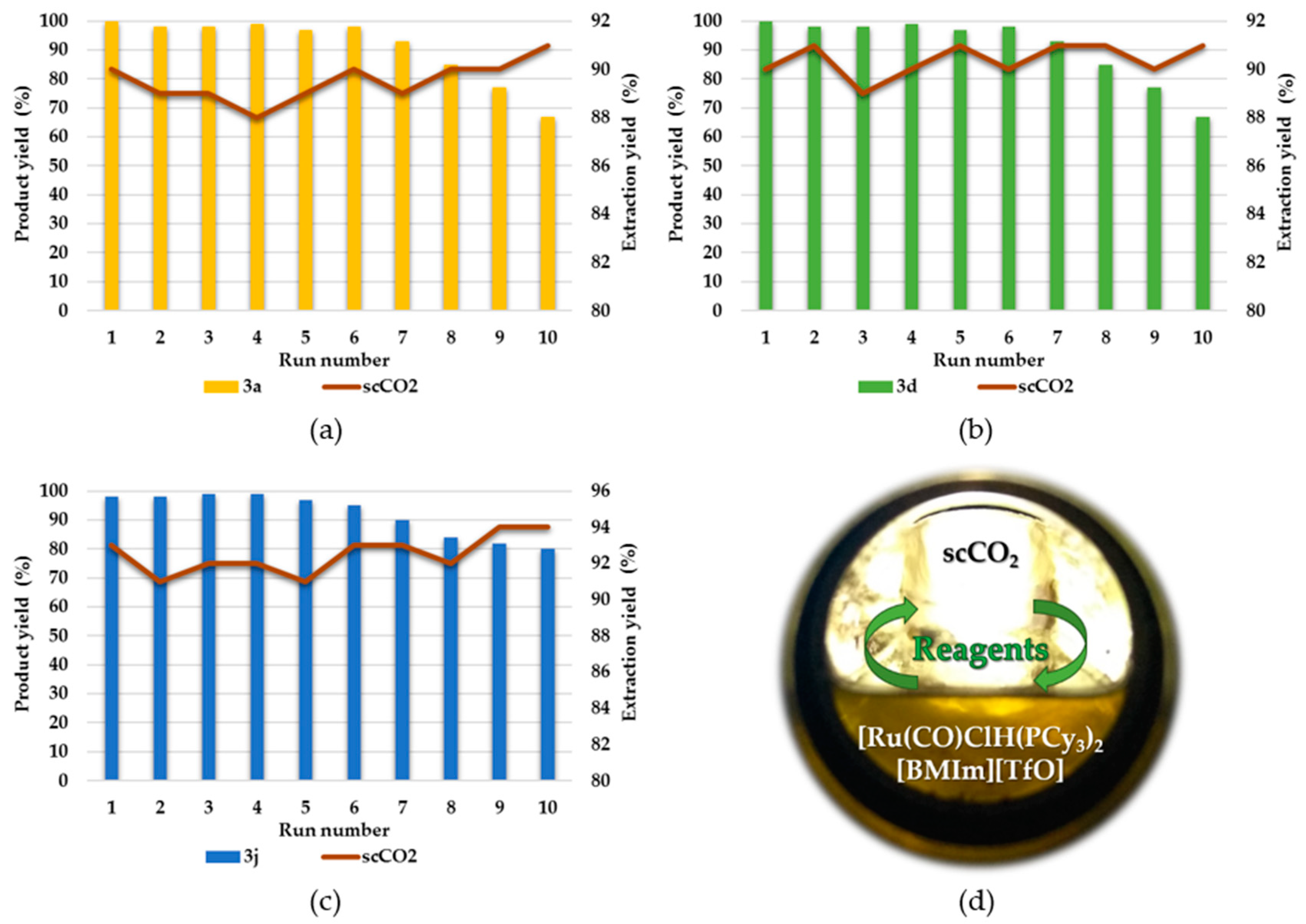
2.3. Repetitive Batch Borylative Coupling in the Monophasic Solvent System—[Ru(CO)Cl(H)(PCy3)2]@ILs
2.4. Repetitive Batch Borylative Coupling in the Biphasic Solvent System—[Ru(CO)Cl(H)(PCy3)2]@ILs/scCO2
3. Materials and Methods
3.1. Materials
3.2. General Procedures
3.2.1. Borylative Coupling in the [Ru(CO)Cl(H)(PCy3)2]@ILs System under Optimized Reaction/Extraction Conditions with Subsequent n-Heptane Extraction
3.2.2. Borylative Coupling in the [Ru(CO)Cl(H)(PCy3)2]@ILs/scCO2 System with Subsequent scCO2 Extraction
3.2.3. Repetitive Batch Borylative Coupling in [Ru(CO)Cl(H)(PCy3)2]@ILs with Subsequent Organic Solvent Extraction
3.2.4. Repetitive Batch Borylative Coupling in [Ru(CO)Cl(H)(PCy3)2]@ILs/scCO2 with Subsequent scCO2 Extraction
3.2.5. Suzuki Coupling of 3a with Bromobenzene
3.2.6. Iododeborylation of 3a with Molecular Iodine
3.3. Product Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhaduri, S.; Mukesh, D. Chemical industry and homogeneous catalysis. In Homogeneous Catalysis; Bhaduri, S., Mukesh, D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 1–21. [Google Scholar]
- Joshi, S.S.; Bhatnagar, A.; Ranade, V.V. Chapter 8—Catalysis for fine and specialty chemicals. In Industrial Catalytic Processes for Fine and Specialty Chemicals; Joshi, S.S., Ranade, V.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 317–392. [Google Scholar]
- Cole-Hamilton, D.J.; Tooze, R.P. Catalyst Separation, Recovery and Recycling: Chemistry and Process Design; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; Volume 30, pp. 1–247. [Google Scholar]
- Centi, G.; Perathoner, S. Methods and tools of sustainable industrial chemistry: Catalysis. Sustain. Ind. Chem. 2009, 73–198. [Google Scholar] [CrossRef]
- Shende, V.S.; Saptal, V.B.; Bhanage, B.M. Recent advances utilized in the recycling of homogeneous catalysis. Chem. Rec. 2019, 19, 2022–2043. [Google Scholar] [CrossRef] [PubMed]
- Cole-Hamilton, D.J. Homogeneous catalysis—New approaches to catalyst separation, recovery, and recycling. Science 2003, 299, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and sustainable solvents in chemical processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Walkowiak, J.; Franciò, G.; Leitner, W. Supercritical fluids as advanced media for reaction and separation in homogeneous catalysis. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes; Wiley-VCH Verlag GmbH & Co. KGaA.: Weinheim, Germany, 2018; pp. 1221–1258. [Google Scholar]
- Bermúdez, M.-D.; Jiménez, A.-E.; Sanes, J.; Carrión, F.-J. Ionic liquids as advanced lubricant fluids. Molecules 2009, 14, 2888–2908. [Google Scholar] [CrossRef]
- Zhou, F.; Liang, Y.; Liu, W. Ionic liquid lubricants: Designed chemistry for engineering applications. Chem. Soc. Rev. 2009, 38, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bostrom, T. Application of ionic liquids in solar cells and batteries: A review. Curr. Org. Chem. 2015, 19, 556–566. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.S.Y.; MacFarlane, D.R. Ionic liquids in biomass processing. In Ionic Liquids; Springer: Berlin/Heidelberg, Germany, 2009; pp. 311–339. [Google Scholar]
- Tadesse, H.; Luque, R. Advances on biomass pretreatment using ionic liquids: An overview. Energy Environ. Sci. 2011, 4, 3913–3929. [Google Scholar] [CrossRef]
- Vekariya, R.L. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. [Google Scholar] [CrossRef]
- Welton, T. Ionic liquids in catalysis. Coord. Chem. Rev. 2004, 248, 2459–2477. [Google Scholar] [CrossRef]
- Pârvulescu, V.I.; Hardacre, C. Catalysis in ionic liquids. Chem. Rev. 2007, 107, 2615–2665. [Google Scholar] [CrossRef]
- Blanchard, L.A.; Brennecke, J.F. Recovery of organic products from ionic liquids using supercritical carbon dioxide. Ind. Eng. Chem. Res. 2001, 40, 287–292. [Google Scholar] [CrossRef]
- Jutz, F.; Andanson, J.-M.; Baiker, A. Ionic liquids and dense carbon dioxide: A beneficial biphasic system for catalysis. Chem. Rev. 2011, 111, 322–353. [Google Scholar] [CrossRef] [PubMed]
- Jessop, P.G.; Stanley, R.R.; Brown, R.A.; Eckert, C.A.; Liotta, C.L.; Ngo, T.T.; Pollet, P. Neoteric solvents for asymmetric hydrogenation: Supercritical fluids, ionic liquids, and expanded ionic liquids. Green Chem. 2003, 5, 123–128. [Google Scholar] [CrossRef]
- Geier, D.; Schmitz, P.; Walkowiak, J.; Leitner, W.; Franciò, G. Continuous flow asymmetric hydrogenation with supported ionic liquid phase catalysts using modified co2 as the mobile phase: From model substrate to an active pharmaceutical ingredient. ACS Catal. 2018, 8, 3297–3303. [Google Scholar] [CrossRef]
- Muzart, J. Ionic liquids as solvents for catalyzed oxidations of organic compounds. Adv. Synth. Catal. 2006, 348, 275–295. [Google Scholar] [CrossRef]
- Rieger, B.; Plikhta, A.; Castillo-Molina, D.A. Ionic liquids in transition metal-catalyzed hydroformylation reactions. In Ionic Liquids (ILs) in Organometallic Catalysis; Dupont, J., Kollár, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 95–144. [Google Scholar]
- Li, J.; Yang, S.; Wu, W.; Jiang, H. Recent advances in Pd-catalyzed cross-coupling reaction in ionic liquids. Eur. J. Org. Chem. 2018, 2018, 1284–1306. [Google Scholar] [CrossRef]
- Maciejewski, H.; Szubert, K.; Marciniec, B.; Pernak, J. Hydrosilylation of functionalised olefins catalysed by rhodium siloxide complexes in ionic liquids. Green Chem. 2009, 11, 1045–1051. [Google Scholar] [CrossRef]
- Maciejewski, H.; Szubert, K.; Marciniec, B. New approach to synthesis of functionalised silsesquioxanes via hydrosilylation. Catal. Commun. 2012, 24, 1–4. [Google Scholar] [CrossRef]
- Maciejewski, H.; Szubert, K.; Fiedorow, R.; Giszter, R.; Niemczak, M.; Pernak, J.; Klimas, W. Diallyldimethylammonium and trimethylvinylammonium ionic liquids—Synthesis and application to catalysis. Appl. Catal. A 2013, 451, 168–175. [Google Scholar] [CrossRef]
- Rogalski, S.; Żak, P.; Miętkiewski, M.; Dutkiewicz, M.; Fiedorow, R.; Maciejewski, H.; Pietraszuk, C.; Śmiglak, M.; Schubert, T.J. Efficient synthesis of E-1, 2-bis (silyl) ethenes via ruthenium-catalyzed homocoupling of vinylsilanes carried out in ionic liquids. Appl. Catal. A 2012, 445, 261–268. [Google Scholar] [CrossRef]
- Szyling, J.; Walkowiak, J.; Sokolnicki, T.; Franczyk, A.; Stefanowska, K.; Klarek, M. PEG-mediated recyclable borylative coupling of vinyl boronates with olefins. J. Catal. 2019, 376, 219–227. [Google Scholar] [CrossRef]
- Szyling, J.; Franczyk, A.; Stefanowska, K.; Maciejewski, H.; Walkowiak, J.d. Recyclable hydroboration of alkynes using RuH@ IL and RuH@ IL/scCO2 catalytic systems. ACS Sustain. Chem. Eng. 2018, 6, 10980–10988. [Google Scholar] [CrossRef]
- Szyling, J.; Franczyk, A.; Stefanowska, K.; Klarek, M.; Maciejewski, H.; Walkowiak, J. An effective catalytic hydroboration of alkynes in supercritical CO2 under repetitive batch mode. ChemCatChem 2018, 10, 531–539. [Google Scholar] [CrossRef]
- Szyling, J.; Franczyk, A.; Stefanowska, K.; Walkowiak, J. A recyclable Ru (CO) Cl (H)(PPh3) 3/PEG catalytic system for regio-and stereoselective hydroboration of terminal and internal alkynes. Adv. Synth. Catal. 2018, 360, 2966–2974. [Google Scholar] [CrossRef]
- Stefanowska, K.; Franczyk, A.; Szyling, J.; Salamon, K.; Marciniec, B.; Walkowiak, J. An effective hydrosilylation of alkynes in supercritical CO2–A green approach to alkenyl silanes. J. Catal. 2017, 356, 206–213. [Google Scholar] [CrossRef]
- Szyling, J.; Franczyk, A.; Pawluć, P.; Marciniec, B.; Walkowiak, J. A stereoselective synthesis of (E)-or (Z)-β-arylvinyl halides via a borylative coupling/halodeborylation protocol. Org. Biomol. Chem. 2017, 15, 3207–3215. [Google Scholar] [CrossRef] [Green Version]
- Szyling, J.; Walkowiak, J. Effective one-pot synthesis of (E)-poly (vinyl arylenes) via trans-borylation/Suzuki coupling protocol. Green Proc. Synth. 2017, 6, 301–310. [Google Scholar] [CrossRef]
- Denmark, S.E.; Tymonko, S.A. Sequential cross-coupling of 1, 4-bissilylbutadienes: Synthesis of unsymmetrical 1, 4-disubstituted 1, 3-butadienes. J. Am. Chem. Soc. 2005, 127, 8004–8005. [Google Scholar] [CrossRef]
- Carreras, J.; Caballero, A.; Pérez, P.J. Alkenyl boronates: Synthesis and applications. Chem. Asian J. 2019, 14, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Pawluć, P.; Franczyk, A.; Walkowiak, J.; Hreczycho, G.; Kubicki, M.; Marciniec, B. (E)-9-(2-Iodovinyl)-9H-carbazole: A new coupling reagent for the synthesis of π-conjugated carbazoles. Org. Lett. 2011, 13, 1976–1979. [Google Scholar] [CrossRef]
- Aubin, S.; Le Floch, F.; Carrié, D.; Guegan, J.P.; Vaultier, M. Transition-metal-catalyzed hydrosilylation and hydroboration of terminal alkynes in ionic liquids. In Ionic Liquids; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2002; Volume 818, pp. 334–346. [Google Scholar]
- Marciniec, B.; Jankowska, M.; Pietraszuk, C. New catalytic route to functionalized vinylboronates. Chem. Commun. 2005, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, B.; Walkowiak, J. Ruthenium (II) complex catalyzed O-borylation of alcohols with vinylboronates. Synlett 2009, 2009, 2433–2436. [Google Scholar] [CrossRef]
- Marciniec, B.; Walkowiak, J. New catalytic route to borasiloxanes. Chem. Commun. 2008, 2695–2697. [Google Scholar] [CrossRef] [PubMed]
- Walkowiak, J.; Marciniec, B. A new catalytic method for the synthesis of boroxanes. Tetrahedron Lett. 2010, 51, 6177–6180. [Google Scholar] [CrossRef]
- Blanchard, L.A.; Gu, Z.; Brennecke, J.F. High-pressure phase behavior of ionic liquid/CO2 systems. J. Phys. Chem. B 2001, 105, 2437–2444. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.S.; Lee, D.W.; Chen, Y. Hydrovinylation and [2 + 2] cycloaddition reactions of alkynes and alkenes catalyzed by a well-defined cationic ruthenium—Alkylidene complex. Organometallics 1999, 18, 2043–2045. [Google Scholar] [CrossRef]
- Ahmad, N.; Levison, J.J.; Robinson, S.; Uttley, M.; Wonchoba, E.; Parshall, G. Complexes of ruthenium, osmium, rhodium, and iridium containing hydride carbonyl, or nitrosyl ligands. Inorg. Synth. 1974, 15, 45–64. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ionic Liquid | Conversion of 1 [%] a | Yield of 3a [%] b |
|---|---|---|---|
| 1 | [EMPyr][OTf] | 95 | 95 |
| 2 | [EMPyr][NTf2] | 100 | 100 |
| 3 | [BMPyr][OTf] | 91 | 91 |
| 4 | [BMPyr][NTf2] | 93 | 93 |
| 5 | [BMIm][NTf2] | 97 | 97 |
| 6 | [BMIm][OTf] | 100 | 100 |
| 7 | [BMIm][PF6] | 88 | 88 |
| 8 | [BMIm][BF4] | 54 | 54 |
| 9 | [BMIm]Cl | trace | n.d |
| Entry | Ionic Liquid | Temperature [°C] | Time [h] | 1:2a [mol] | Catalyst Loading [mol%] | Conversion of 1 [%] a | Yield of 3a [%] b |
|---|---|---|---|---|---|---|---|
| 1 | [EMPyr][NTf2] | 120 | 1:4 | 3 | 100 | 100 | |
| 2 | [EMPyr][NTf2] | 110 | 24 | 1:4 | 3 | 100 | 100 |
| 3 | [EMPyr][NTf2] | 100 | 24 | 1:4 | 3 | 96 | 96 |
| 4 | [EMPyr][NTf2] | 80 | 24 | 1:4 | 3 | 88 | 88 |
| 5 | [EMPyr][NTf2] | 110 | 18 | 1:4 | 3 | 100 | 100 |
| 6 | [EMPyr][NTf2] | 110 | 6 | 1:4 | 3 | 100 | 100 |
| 7 | [EMPyr][NTf2] | 110 | 3 | 1:4 | 3 | 94 | 94 |
| 8 | [EMPyr][NTf2] | 110 | 6 | 1:3 | 3 | 100 | 100 |
| 9 | [EMPyr][NTf2] | 110 | 6 | 1:2 | 3 | 100 | 100 |
| 10 | [EMPyr][NTf2] | 110 | 6 | 1:2 | 2 | 100 | 100 |
| 11 | [EMPyr][NTf2] | 110 | 6 | 1:1.5 | 2 | 100 | 100 |
| 12 | [EMPyr][NTf2] | 110 | 6 | 1:1.5 | 1 | 100 | 100 |
| 13 | [EMPyr][NTf2] | 110 | 6 | 1:1.5 | 0.5 | 93 | 93 |
| 14 | [EMPyr][NTf2] | 110 | 6 | 1:1 | 1 | 100 | 83 c |
| 15 d | [EMPyr][NTf2] | 110 | 6 | 1:1.5 | 1 | 46 | 37 e |
| 16 | [BMIm][OTf] | 110 | 6 | 1:1.5 | 1 | 100 | 100 |
| 17 | [BMIm][OTf] | 80 | 6 | 1:1.5 | 1 | 53 | 53 |
| 18 | [BMIm][OTf] | 110 | 3 | 1: 1.5 | 1 | 91 | 91 |
| 19 | [BMIm][OTf] | 110 | 6 | 1:1 | 1 | 100 | 88 c |
| 20 d | [BMIm][OTf] | 110 | 6 | 1: 1.5 | 1 | 41 | 33 e |
| 21 | [BMIm][OTf] | 110 | 6 | 1: 1.5 | 0.5 | 89 | 89 |
| Entry | Solvent | [EMPyr][NTf2] | [BMIm][OTf] |
|---|---|---|---|
| 1 a | n-Hexane | − | − |
| 2 a | n-Heptane | − | − |
| 3 a | Toluene | + | + |
| 4 b | Dichloromethane | + | + |
| 5 a | Tetrahydrofuran | + | + |
| 6 c | Supercritical CO2 | − | − |
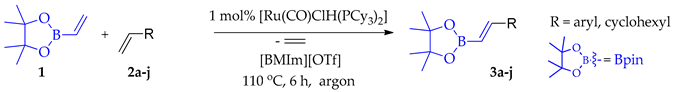
| Entry | Olefin | 2 | Product | 3 | Conversion of 1 [%] a | Extraction Yield [%] b | Isolation Yield [%] c |
|---|---|---|---|---|---|---|---|
| 1 | 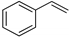 | a |  | a | 100 | 94 d 90 e | 81 |
| 2 | 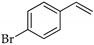 | b | 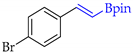 | b | 99 | 91 d 88 e | 77 |
| 3 | 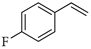 | c | 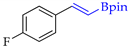 | c | 99 | 90 d 91 e | 78 |
| 4 |  | d | 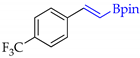 | d | 100 | 93 d 94 e | 83 |
| 5 | 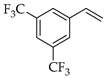 | e |  | e | 99 | 90 d 92 e | 81 |
| 6 |  | f | 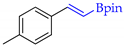 | f | 100 | 93 d 91 e | 79 |
| 7 | 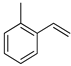 | g | 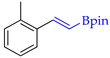 | g | 98 | 84 d 81 e | 69 |
| 8 | 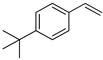 | h | 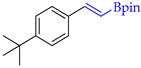 | h | 93 | 81 d 77 e | 73 |
| 9 |  | i | 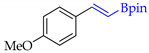 | i | 89 | 74 d 75 e | 71 |
| 10 |  | j |  | j | 100 | 95 d 96 e | 87 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szyling, J.; Sokolnicki, T.; Franczyk, A.; Walkowiak, J. Ru-Catalyzed Repetitive Batch Borylative Coupling of Olefins in Ionic Liquids or Ionic Liquids/scCO2 Systems. Catalysts 2020, 10, 762. https://doi.org/10.3390/catal10070762
Szyling J, Sokolnicki T, Franczyk A, Walkowiak J. Ru-Catalyzed Repetitive Batch Borylative Coupling of Olefins in Ionic Liquids or Ionic Liquids/scCO2 Systems. Catalysts. 2020; 10(7):762. https://doi.org/10.3390/catal10070762
Chicago/Turabian StyleSzyling, Jakub, Tomasz Sokolnicki, Adrian Franczyk, and Jędrzej Walkowiak. 2020. "Ru-Catalyzed Repetitive Batch Borylative Coupling of Olefins in Ionic Liquids or Ionic Liquids/scCO2 Systems" Catalysts 10, no. 7: 762. https://doi.org/10.3390/catal10070762