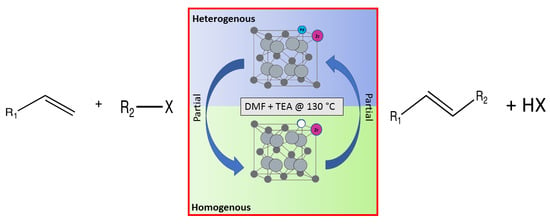
1. Introduction
The Heck reactions involve the formation of a new carbon–carbon double bond connecting an aryl halide with an olefin. This process is referred to as the arylation of olefins, or the vinylation of aryl halides. Typically Heck reactions—as with many cross-coupling reactions—are palladium-catalysed [
1]. Classically, homogenous catalyst systems are used in the industrial applications of Heck reactions [
2,
3,
4,
5,
6,
7]. Naturally, while homogenous systems have high turn-over frequencies (TOFs), their major disadvantage is the inability to be efficiently seperated from the final product. Incorporation of the catalyst in turn decreases the quality of the final product in terms of purity [
5,
6,
8,
9,
10,
11,
12,
13]. For pharmaceutical products, incorporation of the catalyst can be detrimental, but most notable is the economic consideration, since palladium is a precious metal and is thus expensive and offers limited recycling prospects as a homogenous catalyst [
5,
14,
15].
The most obvious approach to solving this problem is the employment of a heterogeneous system [
6]. However, while this approach may work for many systems, heterogeneous catalyst systems under Heck conditions generally leach [
6,
15,
16,
17,
18]. This is a process in which the active metal dissolves in solution and becomes homogenized into the reaction medium as with homogeneous systems [
1,
14,
15,
19,
20,
21]. Once more, the problems of the incorporation of precious metal and limited recyclability emerge. Thus, this paper attempts to develop a heterogeneous catalyst system, which is sufficiently active for Heck reactions, yet displays minimal or no leaching.
Although a great variety of materials can be applied, the drawbacks of low turn over numbers (TONs) and turn over frequencies (TOFs) which the amount of product a catalyst can turn over per mol of catalyst ( and TOFs being TONs per unit time) are of great concern. Although the stability of catalysts has been improved by exploring ligand-free alternatives, these methodologies tend to complicate the synthesis procedure by requiring additives. The contribution by Mpungose of introducing quaternary ammonium salts, has illustrated excellent results under milder conditions [
22]. Furthermore, the use of ammonium salts as phase transfer agents has been reported to also provide stability to the active metal [
14]. This then introduces another facet to the heterogeneous catalyst debate, of whether a truly ligand-free catalyst is possible, since the active metal will coordinate to substrates, which then act as ligands. This limits the scope of substrates that can be employed by virtue of their ability to “ligate” and “dissociate”.
Fortunately, immobilised homogenous systems, through covalent bonding, encapsulation and/or complexation, provide a more stable catalyst that can be recycled [
5]. Furthermore, supported metal catalysts, where the active metal is on a solid support, have increased the palette of materials that can be applied [
5]. These materials range from carbon supports, to zeolites, porous glass, clays, metal oxides, organic polymers, molecular sieves, and various combinations of the materials mentioned. These supports offer a myriad of advantages, such as stability (both thermal and to moisture), tuneable selectivity, and recyclability. These supports were introduced in the early 1970s, and this has left this field well studied [
8,
9,
10,
11,
12,
13,
19]. The low TONs and TOFs of these materials have been a major disadvantage. These stable catalysts allow an increase in the operating temperature and pressure range that can be used to compensate for the activity, however, TONs are still incomparable to those of homogenous systems. Nevertheless, Pd/C has been shown, in the work of Perosa in coupling reactions, to give TONs that are suitable for industrial applications [
23]. Noteworthy is the apparent consequence of complete exclusion of homogeneous systems from heterogeneous systems comes in the form of an impractical loss in TONs, as illustrated in
Table 1.
Hence, a different approach to homogeneous catalysts, to give reusable heterogeneous catalysts, has been initiated [
19]. This different approach could combine the advantages of both homogeneous and heterogeneous catalysts. Such methods include a “ship in a bottle” catalyst concept. This notion sees the retention of a homogeneous catalyst system within zeolite pores [
8,
9,
10,
11,
12,
13]. The guest is retained in the zeolites because of sterical hindrance. Alternatively, impregnation of the active metal or compounds on a solid support can immobilise the active catalyst. Parallel to impregnation is the enhancement of physiosorption, via the choice of a porous material of suitable size for the compound to be deposited. However, these systems still suffer from leaching [
14,
19]. Additionally, the immobilisation of desired compounds by the formation of covalent bonds, which is known as grafting [
8,
9,
10,
11,
12,
13]. This can be done in the form of a linker, which increases the strength of the bond between the support and active compound or metal, theoretically minimsing leaching. The sol gel process can also b0e used, which involves the generation of a monomer with part of the active metal encapsulated.
Finally, the use of substituted materials or interstitials has been reported. The greatest feature of this method is the culmination of all the methodologies that have been discussed. The active metal is substituted at an atomic level in the crystal lattice of a host material. Theoretically, leaching should be reduced or eliminated. Furthermore, in the case of leaching, the active metal may leach and get redeposited into the pores of the host material, functioning as a supported material. Many reports claim a reversible “release-and-capture” process that occurs with such materials [
14,
34]. Therefore, great attention needs to be paid to the choice of host/support material. For instance, pores in zeolites are more tuneable than metal oxides, while metal oxides such as CeO
2 have an ability to move through phases, and are able to change their oxidation state to release electronic strain, and are thus suited to application in substituted materials [
8,
9,
10,
11,
12,
13,
35,
36,
37,
38]. The susceptibility of ceria to changed oxidation states allows for various metals to be substituted into the fluorite structure; however, this phenomena may indeed be the reason for leaching.
This work describes the substitution of zirconium and palladium atoms into the cerium host structure [
39,
40,
41], as illustrated in
Figure 1. The use of palladium as the active metal was appealing since most cross-coupling reactions are palladium-catalysed; additionally, the use of a basic metal oxide in conjunction with the active metal has been proven to be effective [
6].
Typically, ceria can tolerate no more than 20% of zirconium atoms; thereafter, mixed phases arise [
36,
42,
43,
44]. Therefore, this work aimed to minimise leaching of the active metal by incorporating a fractional amount of palladium into a zirconium-stabilised ceria host lattice. While the reaction mechanisms of heterogeneous systems have been widely speculated about, the true active species is still not known, and consequently this work models reactions after a Pd
2+ mechanism [
45,
46,
47,
48,
49].
2. Results
Following the synthesis of the material with the Pd
2+ ion encased in a ceria–zirconium solid solution, the metal loading was determined via ICP-OES, shown in
Table 2.
Imperative to this study was the incorporation of Pd in the cerium–zirconium lattice; therefore, a relatively high Pd loading was used to establish the affinity of Pd to the cerium–zirconium lattice. Surface Pd deposits from unsuccessful incorporation can easily be identified by P-XRD. Fortunately, where solid solution is concerned, preservation of the structural integrity of the cubic cerium lattice is only possible for dopants with below 20% of atoms before mixed phases arise [
36,
42,
43,
44]. In accordance with the Hume Rothery rules of substitution [
50], the percentage difference in the atomic radii of Pd
2+ and Ce
4+ is far smaller than that of Zr
4+, thus the addition of Pd
2+ is likely to introduce more defects in the host structure. A STEM Energy Dispersion X-ray spectroscopy (STEM-EDX) line scan was done on the Ce
0.89Zr
0.03Pd
0.08O
2-δ, as shown in
Figure 2.
This illustrates the abundance of elements in the sample across the selected area, marked by the yellow line, and the distribution of Ce, Pd and Zr. The line scan is characterised by a high Ce content throughout the area of interest, with sparse amounts of Zr. The Pd is uniformly dispersed with a constant intensity throughout the line scan. A line scan showing localised regions of metal distribution would be indicative of surface metal deposits. On a qualitative basis, the spectrum illustrates the abundance of the respective metals in ratios relatively consistent with those determined by ICP-OES.
Figure 3 illustrates the X-ray diffractograms of (a) CeO
2, (b) Ce
0.97Zr
0.03O
2-δ, (c) Ce
0.89Zr
0.03Pd
0.08O
2-δ and (d) Pd/Ce
0.97Zr
0.03O
2-δ, with the reflective planes indexed. Each pattern shows the dominant face-centred cubic (fcc) pattern of ceria between the 2-theta values of 27° and 80°, consistent with the JCPDS card (81–0792) [
37,
51,
52] belonging to the Fm3m space group, and no additional peaks from tetragonal zirconia and PdO, implying the complete incorporation of both Zr and Pd into the fluorite native structure [
51,
52,
53,
54]. The exception is the diffractogram at (d), the material intended to have Pd surface deposits, which shows surface Pd-O as a shoulder at 2-theta = 31° [
51]. Therefore, surface deposits of palladium show an additional phase manifesting as a shoulder in the 200 reflection plane of cerium oxide.
Additional data from the diffractograms, namely d-spacings, crystallites sizes and average lattice strain values calculated from the Williamson–Hall plot, are listed in
Table 3. The addition of Zr into the cerium lattice leads to no change in the overall strain observed or the strain averages. This is because Zr is of a similar size, valency and electronegativity as the Ce and thus does not appear as a “foreign” atom in the host structure. Palladium has a lower solubility due to the difference in size and electronegativity. However, metals with higher valencies can dissolve metals with lower valency, as is the case here with cerium and palladium [
55]. This then translates to lower electrostatic repulsion between Ce and Pd, which thus decreases strain.
The larger crystallite size of the cerium–zirconium solid solution resulted from the presence of the oxygen vacancies that formed, which cause the interplanar distances to decrease [
54]. However, when palladium is incorporated into the ceria–zirconia solid solution through solution combustion, instead of depositing onto the surface, the palladium resides in the vacancies of the solid solution. As a result of filling the vacancies with palladium atoms, ionic repulsion is decreased, making the overall crystallite size smaller than that of the ceria–zirconia solid solution [
43,
44,
53]. This phenomena was also observed by Gulyaev [
38], where the addition of palladium resulted in strain relief given the low valency of palladium, which decreased electrostatic repulsion in the host lattice [
54]. Furthermore, Gulyvaev attributed the increase in d-spacings to the increase in defects, which were caused by the addition of the Pd ions in the ceria–zirconia solid solution in an attempt to relieve strain in the lattice [
38]. Thus, an increase in the interplanar spacings was observed, as in this work, and with the incorporation of the smaller Pd atom it is observed that the crystallite size decreases, while the edge lengths of the fcc lattice increase.
The representative TEM image from the particle size analysis of Ce
0.89Zr
0.03Pd
0.08O
2-δ is shown in
Figure 4a, showing spherical particles with distinctive grain boundaries and an average grain size of 7.73 nm, with a low standard deviation of ±0.69 nm, indicating the uniform size of the material [
38,
52,
56]. The inter-planar spacing values of 3.0 Å, 2.6 Å and 2.0 Å, measured from the HRTEM images in
Figure 4a, were correspondingly assigned to the (111), (200) and (220) planes of the fluorite structure of cerium oxide, indicating substitution [
38,
52].
The contrast in the image is uniform, without dense regions that would indicate palladium nanoparticles on the surface of the cerium–zirconium solid solution. The surface morphology illustrated by the SEM micrograph also shows spheres of variable sizes, suggesting sintering of the material, and thus the surface area of the material is expected to be low. Furthermore, the material also shows hollow regions that indicate large pore diameters. The Scanning Electron Microscope–Dark Field (STEM-DF) image (
Figure 4c) shows a material of uniform brightness, while bright localised regions would indicate surface metal deposits. The geometry of the diffraction pattern of
Figure 4d relates to the [011] zone axis [
36,
57]. The electron diffraction pattern, from previous reports of the fcc structure, is characterised by electron diffraction rings with interplanar spacings corresponding to ca. 3.1 Å, related to the 111 reflection plane, 2.7 Å related to the 200 reflection plane, and 1.9 Å related to the 220 reflection plane [
36,
57]. With the presence of other phases, such as the tetragonal phases of PdO and ZrO
2, additional reflections would be observed. Reports of cerium–zirconium solid-solutions describe a single cubic phase for substitutions below 20%, before monoclinic and tetragonal phases become apparent [
42,
43,
53]. Thus, the fcc structure of cerium oxide is confirmed.
The optimum reaction conditions were determined and listed in
Table 4, with screening solvents, bases and temperatures, using the reaction between iodobenzene and methylacrylate as the standard reaction. All conversions exceeded 99%.
The optimum temperature for this system was found to be 130 °C. Although complete conversion is possible at room temperature, the higher the temperature, the shorter the reaction times required [
5,
58]. Thus, the reaction time at 25 °C was 24 h, at 80 °C the reaction time was halved to 12 h, while a 1 h reaction time was observed at 130 °C, when DMF was used as a solvent and TEA was used as a base. With respect to solvents, polar aprotic solvents have been reported as the ideal solvents for Heck reactions [
5], hence the 1 h reaction time observed for DMF. In contrast, the polar aprotic DMSO showed longer reaction times compared to DMF [
5]. This is because, in addition to DMF being aprotic, this solvent also has a coordinating ability and acts a ligand which stabilises the catalyst–base intermediate, which is formed in the pre-activation step and influences the migratory insertion pathway [
59,
60]. Typically, the challenge with the use of protic solvents is their labile nature, due to their acidic protons. The protons make the reaction media acidic, resulting in the decomposition of the organopalladium complex formed by oxidative addition [
60], hence the necessity of adding a base. Ethylene glycol and water, on the other hand, have poor miscibility with the substrates, resulting in a biphasic system, leading to the partitioning of the substrates between the two phases, making sampling dependent on the partitioning coefficient of the cross-coupled product between the two solvent mediums. Additionally, the use of water resulted in a longer reaction time of 22 h. With respect to the use of non-polar solvents, these solvents do not coordinate to the catalyst, which is important for the stability of intermediates that is usually provided by the ligands in organometallic catalysts, as well as regioselectivity. Thus, while toluene gives a shorter reaction time, non-polar solvents favour insertion at the β carbon, decreasing selectivity to the trans product [
61]. Finally, with respect to the base selection, triethylamine gave a reaction time of 1 h. Although the strong base NaOH was effective, because alkenes are stable under basic conditions, esters are susceptible to hydrolysis in the presence of a strong base. Once hydrolysis occurs on the ester “segment” of an acrylate to form a carboxylic acid, this new substrate will have a different reactivity to cross-coupling reactions, hence the long reaction times when NaOH was used. Oxidative cross-coupling is typically observed when NaOH is used [
62].
Stoichiometric amounts of tetraammonium salts are additives that have been reported to improve reaction times when used in conjunction with an inorganic base [
22]. However, the use of tetrabutylammonium bromide or tetrabutylammonium hydroxide as a base rather than an additive resulted in reaction times of 24 h and 28 h, respectively. Substantially longer reaction times (6 h) compared to triethylamine (1 h) were found when using the inorganic base K
2CO
3 [
63].
Catalyst loading was then investigated to determine the minimum loading required to achieve >99% conversion in 1 h. Employing the optimum reaction conditions, based on the base, solvent and temperature investigations, various loadings of catalyst were then explored. The base used was triethylamine with DMF as a solvent, at 130 °C.
Figure 5 shows the reaction profile of the various catalyst loadings studied at 10-min reaction intervals.
The catalyst was found to be very active at loadings as low as 0.05 mol% of Pd (>99% in 75 min), while a loading of 0.01 mol% of Pd was less effective, since these reactions required 12 h to reach completion. The general activity of the catalysts with Pd loadings between 0.05 and 1 mol% is characterised by a sharp gradient in the first 10 minutes, and then the rate of reaction slows down with the depletion of the substrate. While the 0.01 mol% Pd-loaded catalyst displays an induction period, likely due to the transformation of the catalyst to the “true” catalyst [
64,
65], at higher loadings this induction period is not observed, likely due to the rate of reaction. Therefore, testing for the Heck activity of the catalyst was limited to the use of iodobenzene with various substituents to control electron density, and the conditions were set at 0.05 mol% Pd in DMF and 1.5 mol equivalence triethylamine at 130 °C.
Several electron-donating or electron-withdrawing aryl iodides afforded cross-coupling products with excellent yields. Of interest is the activity of the catalyst in the presence of substrates of varying electronics and sterical hindrance. To establish the optimal conditions with respect to the electronic contributions of coupling partners, substituents on the iodobenzene were changed based on their electronic strength, while keeping the olefin constant. Thereafter, olefins were varied based on their functional groups.
Table 5 lists the TOFs obtained when aryl iodides of various electronics were reacted with methylacrylate. Establishing coupling pairs based on electron density, coupling reactions can then be predicted based on substrate type for this catalyst. Although regioselectivity in these reactions was controlled by the electron-withdrawing substrates, stereoselectivity is reversible and results from the torsal strain experienced by the product; therefore, a minor cis configuration product is observed. Product yield was calculated based on the
1H-NMR of isolated yields, observing the ratio of the
cis and
trans peaks in the mixture.
Entries 1–5 in
Table 5 show the reaction of methyl acrylate, with substituted iodobenzenes with varying substituents in the para position, with methyl acrylate, which is moderately electron-withdrawing and has generally been used as a model reaction because of its high reactivity.
Table 5 lists the yields, TONs and turn over frequencies TOFs at the >99% conversion of iodobenzene, exclusively for the trans product yield.
The addition of an electron-donating substituents to the aryl halide results in polarisation of the C-X bond, making the C-X bond more reactive and thus susceptible to oxidative addition [
61,
66,
67], hence the trend in the reaction time observed from entries 1–5. From entries 2 and 3, the substituent begins to withdraw electrons more strongly, relative to entry 1 (H). As a result, bond-polarisation decreases and the C-X bond is less reactive, resulting in longer reaction times. Iodobenzene with the nitro group in the para position (entry 5) is strongly deactivated because of the high electron-withdrawing potential of the NO
2 competing with the I group for electrons, which thus strengthens the C-I bond and decreases the reactivity. This explains the longer reaction times observed for the substrates with more electron-withdrawing groups.
However, the lower trans yields observed for iodobenzene with electron-withdrawing groups is due to their susceptibility to undergo homocoupling, where the aryl halide preferentially couples with itself [
61,
68,
69]. Therefore, the reaction shows a fast depletion of the iodobenzene, but without giving the desired product. Consequently, as a result of the low concentration of the aryl halide in solution, free olefin decomposes over longer reaction times. Most notable is the reaction shown in entry 5, with the nitro substituent para to the halide, with the reaction time exceeding 24 h but yielding no product. This was expected because, while NH
2, OH and OCH
3 withdraw electrons, they also donate electrons by resonance. Conversely, the NO
2 is strongly deactivating. However, when the position of the nitro substituent was changed from para to meta, reaction times were observed to decrease.
Table 6 shows the change in reaction and trans product selectivity under the stated reaction conditions and catalyst loading.
The reaction times observed show a significant decrease, but without significant change in trans yield. This suggests the electron-donating potential of the m-nitro-iodobenzene group, resulting in a decrease in reaction time by a factor of two compared to the more withdrawing p-nitro-iodobenzene group. Therefore, the positions of the substituents of the aryl halide are responsible for activating or deactivating the C-I bond for oxidative addition. The m-nitro-group contributes to electron density by resonance, resulting in a more activated substrate.
It is, however, premature to assign priority to the electronics of the halide as the rate-determining step, since the influence of the olefin is still to be investigated, and migratory insertion is also an important step in the mechanism. For this investigation,
Table 7 lists the substrates tested under the specified reaction conditions, where the electron density’s contribution to the alkene is investigated, since that is where coupling occurs.
Table 7, entries 1–4 are arranged from least reactive electron pairs to most reactive. This catalyst system shows high trans yields and short reaction times for electron-deficient olefins [
69]. This is clearly seen in the reaction with the neutral styrene, where both reaction time and product selectivity are compromised. Secondary coupling due to the tendency of styrene to polymerise is typically found with stilbene (the styrene and iodobenzene coupling product), and this manifests in an apparent decrease in product yield. Initially, the desired product forms, and it then cross-couples further with styrene in a competing reaction. Entries 2 and 3 are for the reactions of methyl acrylate and acrylamide, which are electron-deficient; the trend seems to be that the more electron-deficient the molecule, the more susceptible it is to migratory insertion. Additionally, the preference for the trans isomer increases with the more electron-deficient substrates. Furthermore, the absence of branched products could be attributed to the steric hindrance on the alpha carbon, as well polarisation on the beta carbon due to the electron-withdrawing potential of the substituent on the alkene.
To investigate the effect of sterical hindrance on the double bond,
Table 8 lists the acrylates that were used for this investigation with iodobenzene. Acrylates consist of an alkene segment, where cross-coupling occurs, and an ester segment. Therefore, two positions can be investigated, namely, the region around the alkene segment on the terminal carbon, as well as the ester segment of the molecule. However, cross-coupling is only concerned with the alkene segment. Considering the classical Pd
2+ Heck mechanism approach, which assumes the migratory insertion of the terminal carbons, it then follows that the addition of a methyl group on the alkene segment should limit the progress of the reaction more so than the addition of a methyl group on the ester segment of the acrylate [
60]. Furthermore, since cross-coupling is largely concerned with the alkene segment, because that is the centre of cross-coupling, steric hindrance at the ester segment should be negligible. Reaction times, as expected, increase significantly with the addition of a methyl group around the alkene segment.
Table 8 summarises the results of the steric investigations.
Entry 1 shows a short reaction time with a good yield where the double bond is not hindered. Surprisingly, for entry 2, where the carbon labelled 2 is hindered, reaction times increase significantly. However, reaction times greater than 34 h are observed when the terminal carbon is hindered, resulting in an olefin that is reluctant to cross-couple. This is because the double bond is completely hindered, while in entry 2 the isobutyl methacrylate still has the terminal carbon available for insertion. Therefore, steric hindrance is only a factor if the region around the double bond is substituted [
60]. Additionally, the reactions described in
Table 6 are attributed to the electron density endowed upon the double bond. To gain further understanding of the influence of electron density, both olefin and aryliodobenzenes were varied.
Table 9 outlines the reaction parameters and substrates used. This was to establish the substrate scope. As it stands, electron-rich iodobenzenes with electron-poor olefins seem ideal for this catalyst system. For these reactions, only the TOFs and TONs were considered, as shown in
Table 9, which gives a summary of the influence of olefins and halides on the extent of the reaction and linear trans product selectivity, reported as TONs and TOFs.
It is important to note the various stages at which each substrates is involved in the reaction, in order to better understand the relationships observed. Aryl halides are involved in the oxidative addition step that is governed by the reactivity of the C-X bond [
59,
70,
71]. Thus, an electron-withdrawing group should decrease the reactivity of the C-X bond, increasing the rate of the oxidation step, since the C-X bond does not get polarised, as is the case for the electron-donating substrates. Considering the reactions of methylacrylate with the various substituents, the reactions reach completion in 1 h, with the exception of the reaction with the strongly withdrawing acetophenone, which completed in 2 h. The TONs, however, are lowest for the electron-donating iodoaniline, and highest for the neutral iodobenzene. Substituents with electron-withdrawing groups in this case showed the lowest TOFs, as oxidative addition occurs much slower, as seen in
Table 5, because the substituents are meta-directing. Additionally, yields for the desired product are low because of homocoupling typical of electron-withdrawing substrates.
On the other hand, olefins are involved in the reaction cycle at the migratory insertion step. However, this work focuses on the trans product yield, which requires regioselectivity control, and which is determined by the choice of olefin, hence the use of electron-deficient substrates [
59]. Stereoselectivity is an irreversible kinetic phenomenon, and is determined in the syn elimination step. Pre-isomerisation may occur as soon as the olefin is introduced into the reaction media; secondly, it may occur once it is coordinated to the Pd-H centre, and finally, isomerisation may also occur when the cross-coupled product has formed [
66]. However, stereoselectivity management is beyond the scope of this work. Noteworthy was the low trans yield observed for the reactions with styrene as the olefin [
58]. The reaction resulted in a myriad of products, including the desired trans isomer, as well as homocoupling products characterised by an increase in the product peak given the absence of depletion of the aryly halide. Furthermore, considering the reactions of unsubstituted iodobenzene with the olefins of variable electron densities, reactions with a high, strongly electron-deficient acrylonitrile gave the best TOFs [
72]. However, selectivity was low due to the size of the substituent. Methyl acrylate is more bulky, and thus the cis isomer is not favoured [
73]. Most notable was the absence of the reaction between acrylonitrile and iodophenol; despite the substrates satisfying all the parameters discussed in this work, no reaction was observed. In this case, the iodide is cleaved faster than the rate at which the migratory insertion step can insert the acrylonitrile. After 1 h reaction time all the idodophenol is cleaved, forming phenols in solution that cannot react any further, and the desired reaction is not accomplished.
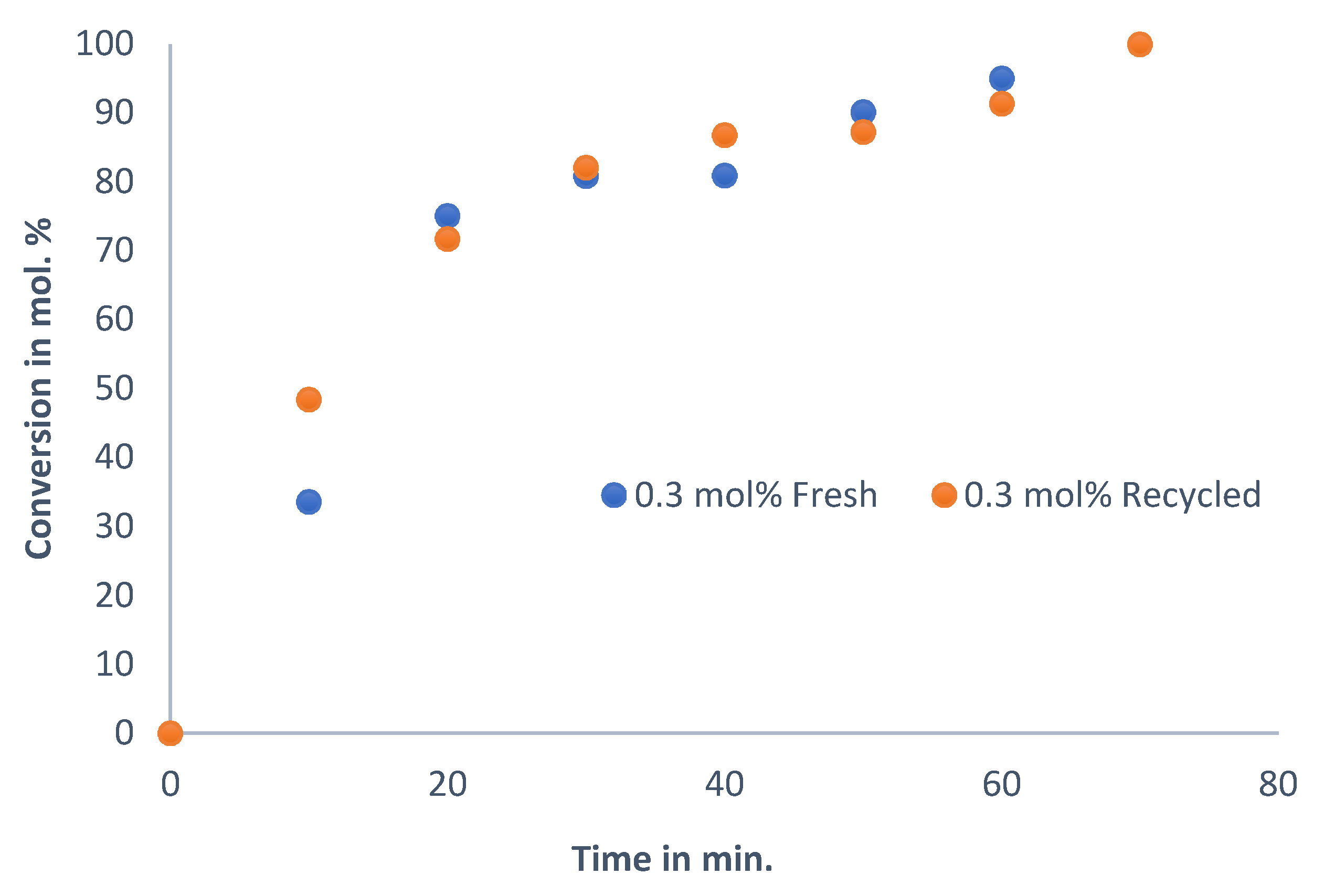
To investigate the extent to which the catalyst can be reused, recyclability tests were carried out. In this work, the catalyst was recycled three times, meaning that it was used in four consecutive reactions.
Figure 6 shows the result using the optimum reaction condition when DMF is used as a solvent, with trimethylamine as the base and with a 0.3 mol% catalyst loading at 130 °C.
The reaction profile of the fresh catalyst is the same as that of the recycled catalyst, even after four reaction cycles, characterised by a steep gradient for the first 20 min, and then, as the reaction reaches completion, the reaction slows down, as a function of a decrease in reactant concentration. The deviation between the recycled catalysts and the fresh catalyst is small, and the catalyst maintains its activity even after the fourth reaction cycle. Therefore, the active species is still present in appreciable amounts, and any leaching that may occur is insignificant. In order to assess the quantity and activity of the leached Pd, leaching tests were initiated. The quantities of Pd that remained in solution at the end of the reaction are presented in
Table 10. This shows the quantitative amounts of palladium that remained, as determined by ICP-OES analysis of the solutions, after each recycle, when the solid catalyst is centrifuged to separate the solid catalyst from the reaction medium.
The results show consistently low amounts of Pd leaching into the solution after every cycle. Various reports have cited a “release-and-capture” mechanism, where the active metal is released from its heterogeneous precursor into solution and can either perform catalysis as a Pd
0 nanoparticle, or is abstracted as a soluble Pd species during oxidative addition. This is often referred to as leached Pd, which often remains in solution after the solid catalyst is removed. Some reports noted that the amount of active metal leached is a function of the iodobenzene present in the solution [
16,
34,
74,
75,
76], and once the reaction reaches completion and all iodobenzene is consumed, the active species gets deposited on the surface of the solid support or host material. In this case, a decrease of Pd in solution should be observed.
Figure 7 shows the reaction profiles of the Hot Filtrate test and the Hg poisoning test, and the catalyst profile after quantitatively recycling the catalyst three times. Generally, for a catalyst to be deemed heterogeneous, the reaction has to cease with the removal of the solid catalyst. Hot filtration then monitors the reaction medium, from activation to completion, without the presence of the catalyst. Thus, the catalyst is allowed to react past its induction period, which was observed in
Figure 5 i.e., after 10 minutes for a 0.3 mol% loading, and then the catalyst is removed and the reaction medium monitored. If the removal of the solid catalyst stops the reaction, then the catalyst is completely heterogeneous. Additionally, Hg selectively poisons any Pd
0 species in the reaction medium when the solid catalyst is removed after it has been activated. This helps determine the Pd species that is leached. If the addition of Hg stops the reaction, then the active species are Pd
0 [
5,
16,
34,
65,
74,
75,
77].
Once the solid catalyst was removed after activation, the Hot Filtrate reaction still underwent an induction period [
65,
76,
78] lasting 30 min, and thereafter conversion took place peaking at 70%. The induction periods arise from the conversion of the catalyst material into the true catalyst prior to catalysis; thus the synthesised material acts as a host or pre-catalyst to improve the stability of the catalyst [
16,
64,
65]. It is noteworthy that the leaching tests were performed after 40% conversion; however, an induction period was still needed, meaning that the leached species is not immediately active, requiring further activation into a molecular Pd species rather than being capable of surface chemistry, as in the case of leached nanoparticles. However, since the reaction continues after the solid material has been removed, a homogeneous component to the reaction can be inferred. The Hg0-poisoned filtrate exhibits the same general reaction profile as that in the Hot Filtrate reaction, with an equal induction period and gradual conversion. However, conversion for the Hg0-poisoned filtrate is lower than in the Hot Filtrate reaction, with a 50% conversion after 70 min. When the Hot Filtrate and the Hg0-poisoned reactions are allowed to reach completion, as shown in
Figure 8, it is clear that these profiles match the reaction profiles of the fresh catalysts and recycled catalysts (
Figure 5 and
Figure 6), only taking much longer to reach completion, that is, 14 h and 18 h.
This result implies a rapid heterogeneous reaction from the solid catalyst, and a slower homogeneous reaction from the leached species, contributing to the overall reactivity of the catalyst.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











































