Strong Activity Enhancement of the Photocatalytic Degradation of an Azo Dye on Au/TiO2 Doped with FeOx



 , and
, and
Abstract
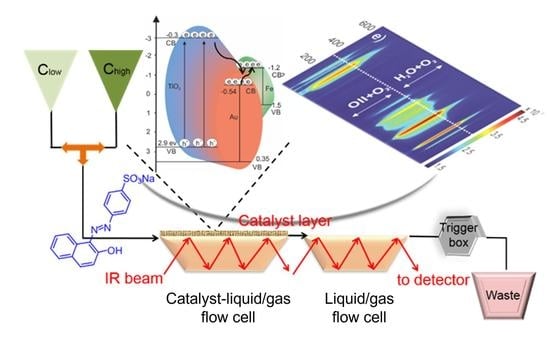
:
1. Introduction
2. Results
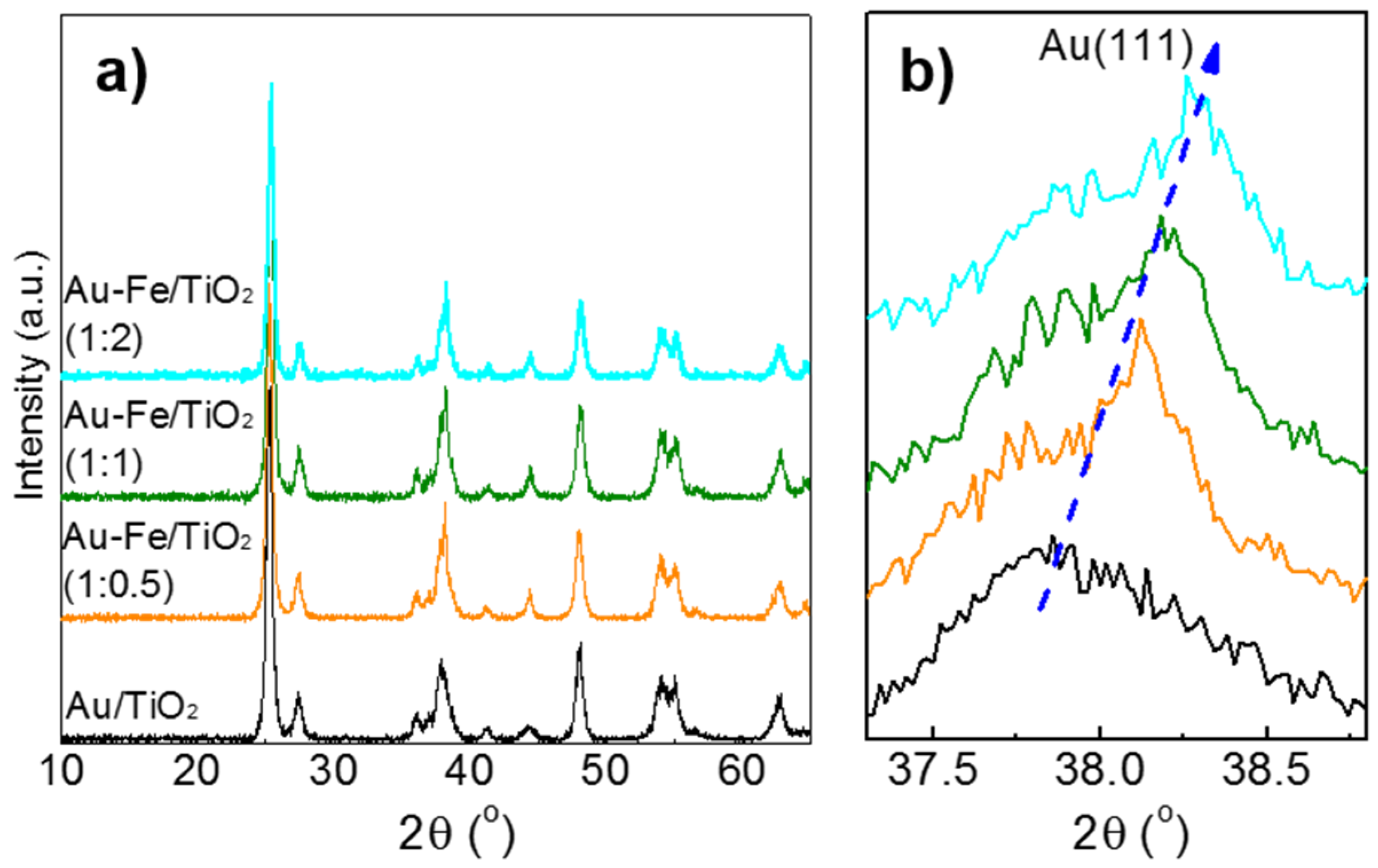
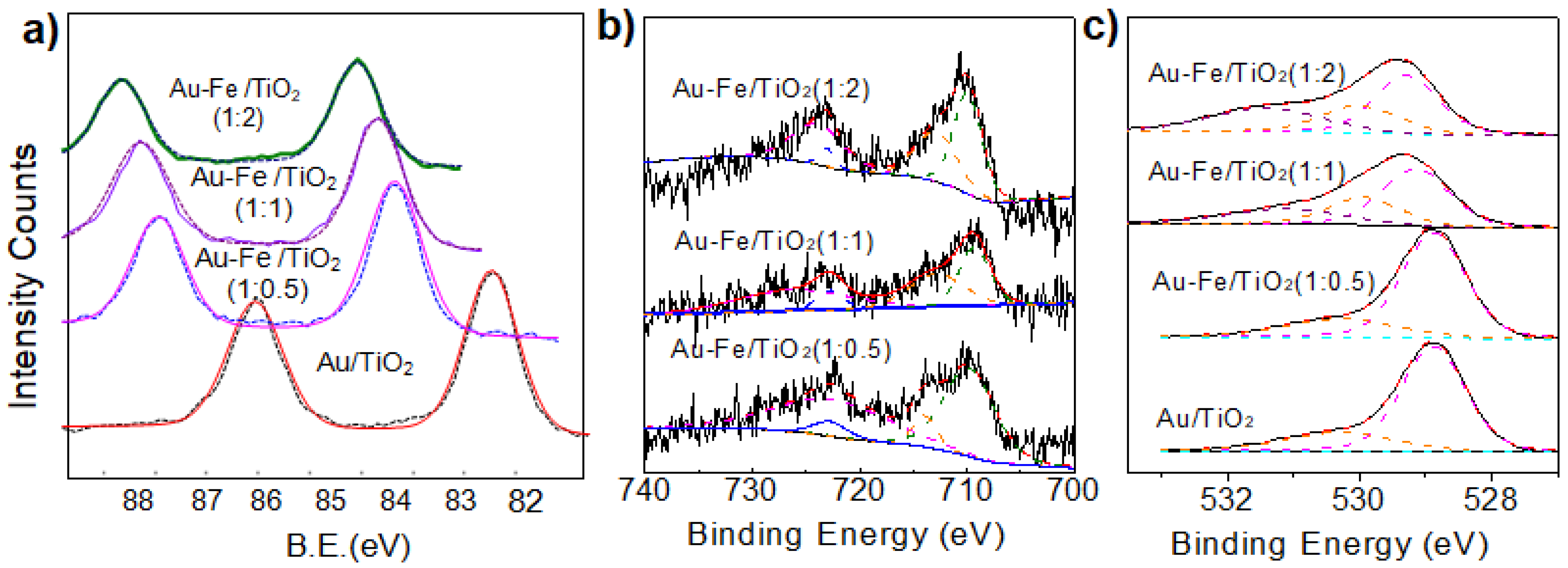
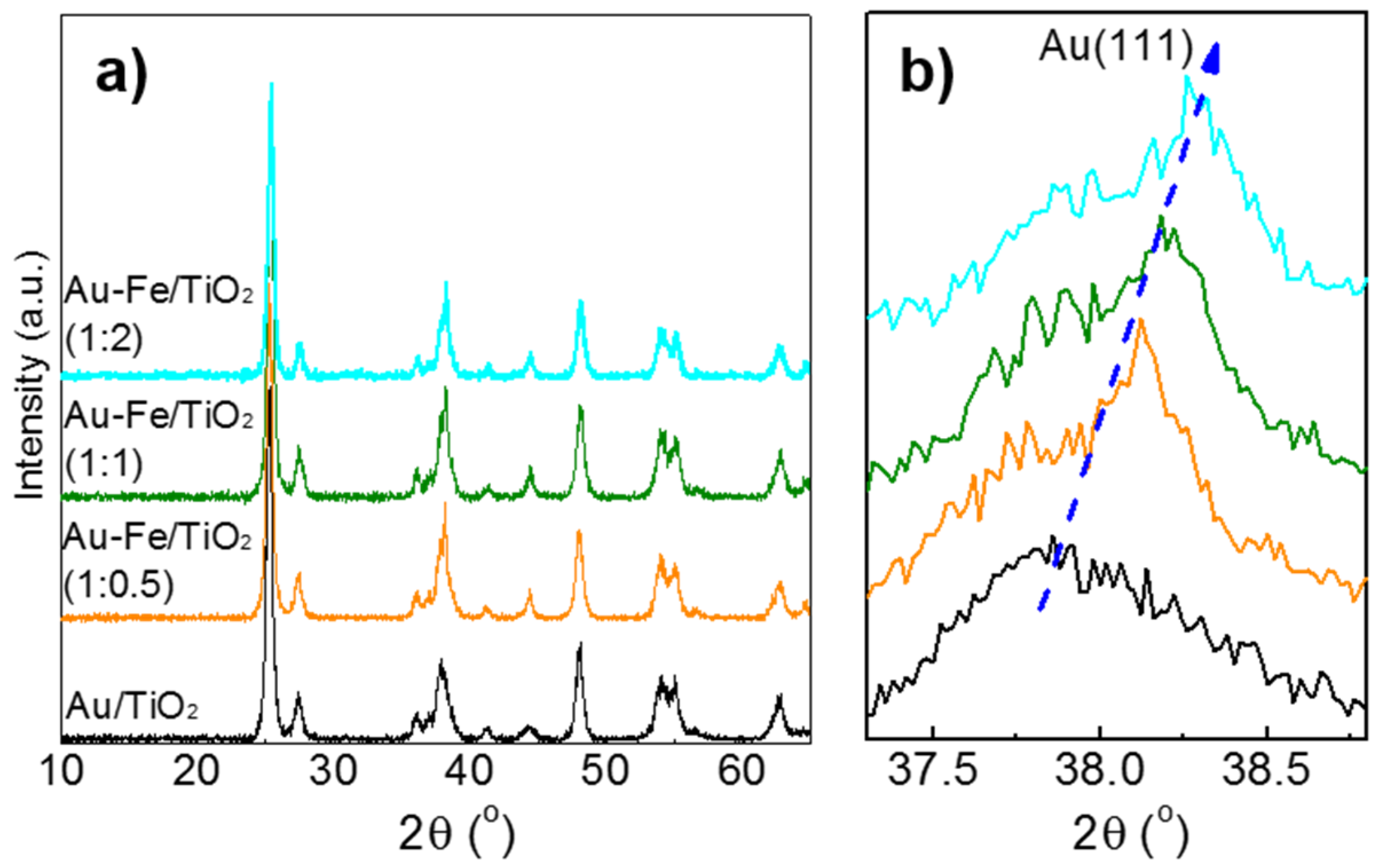
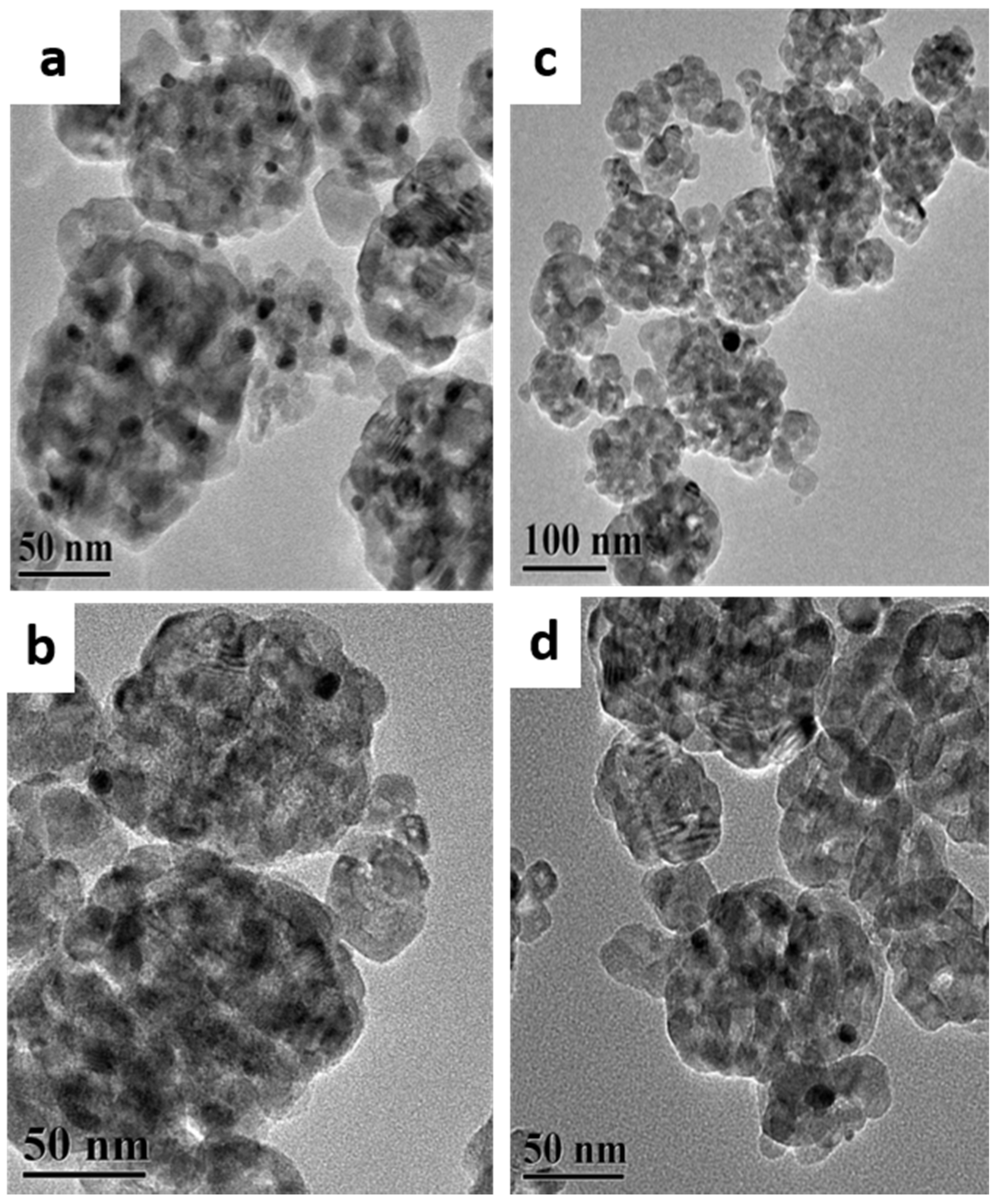
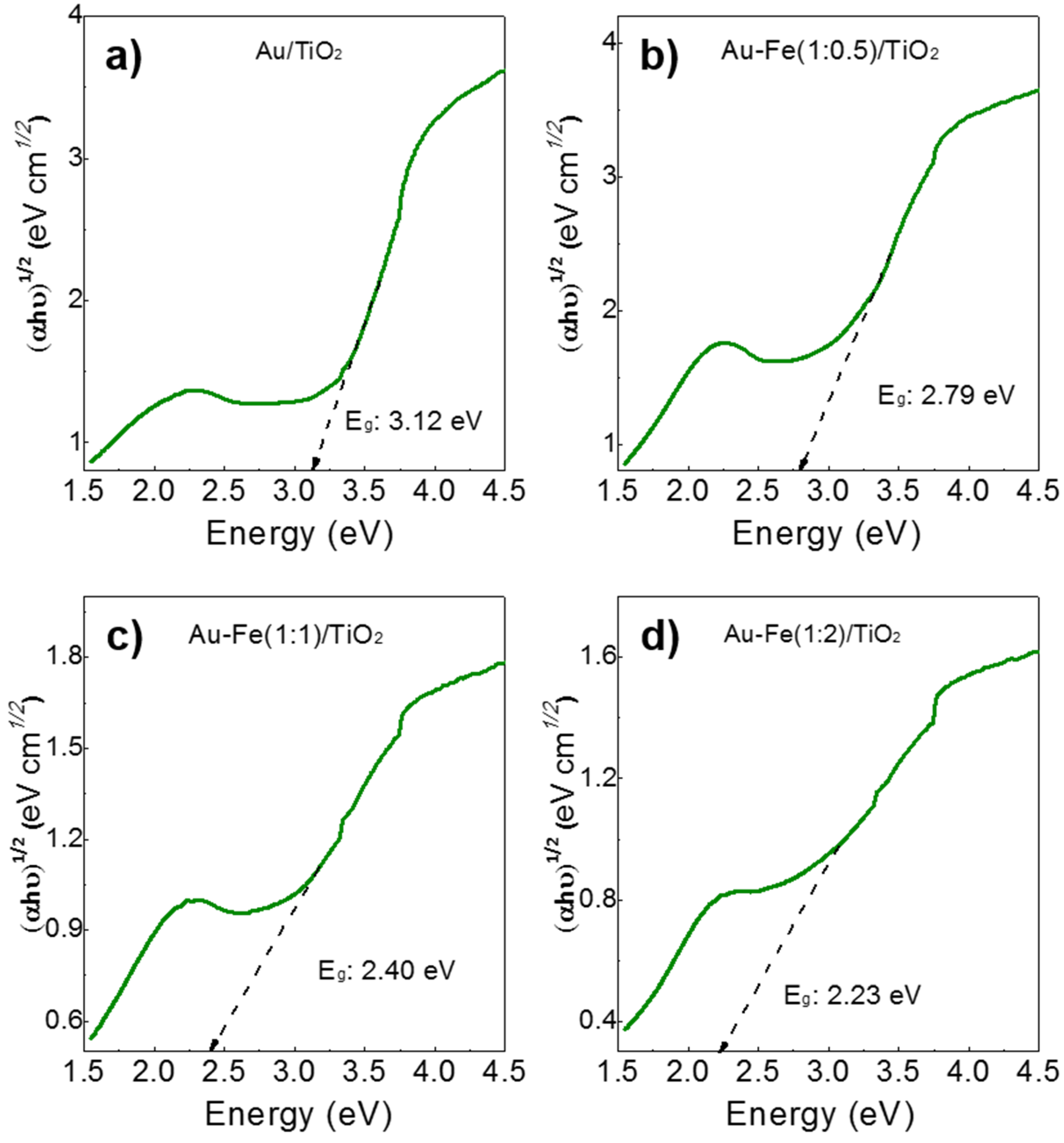
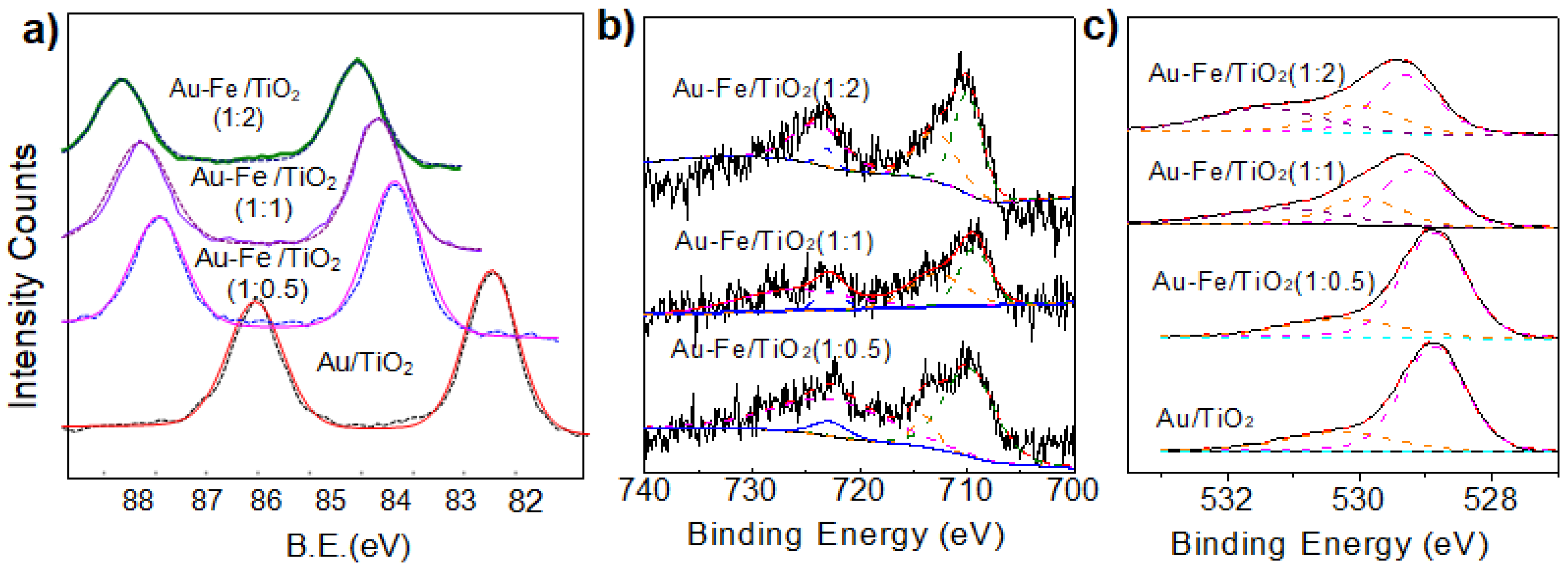
2.1. Structural Properties and Bandgap Energies of Catalysts
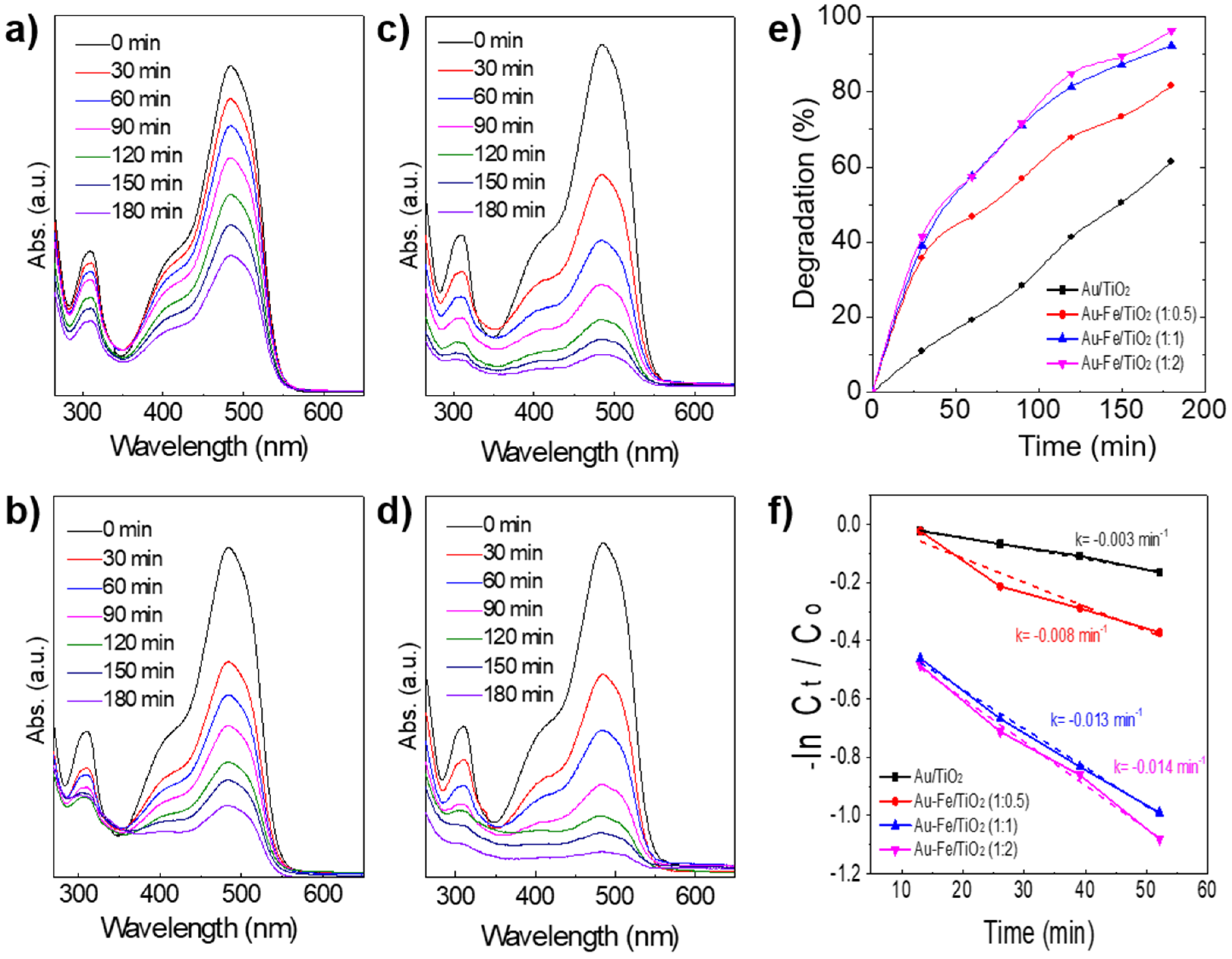
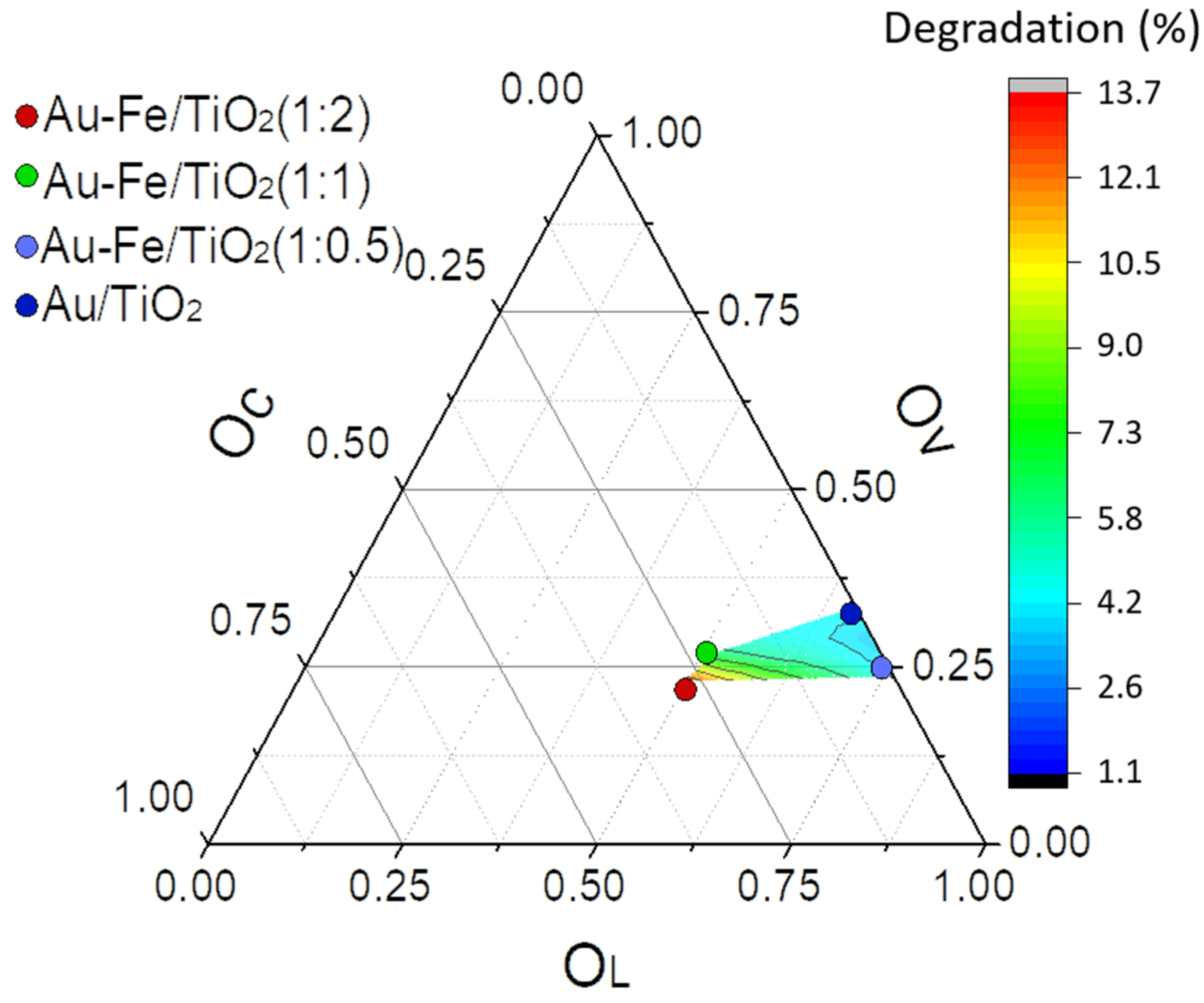
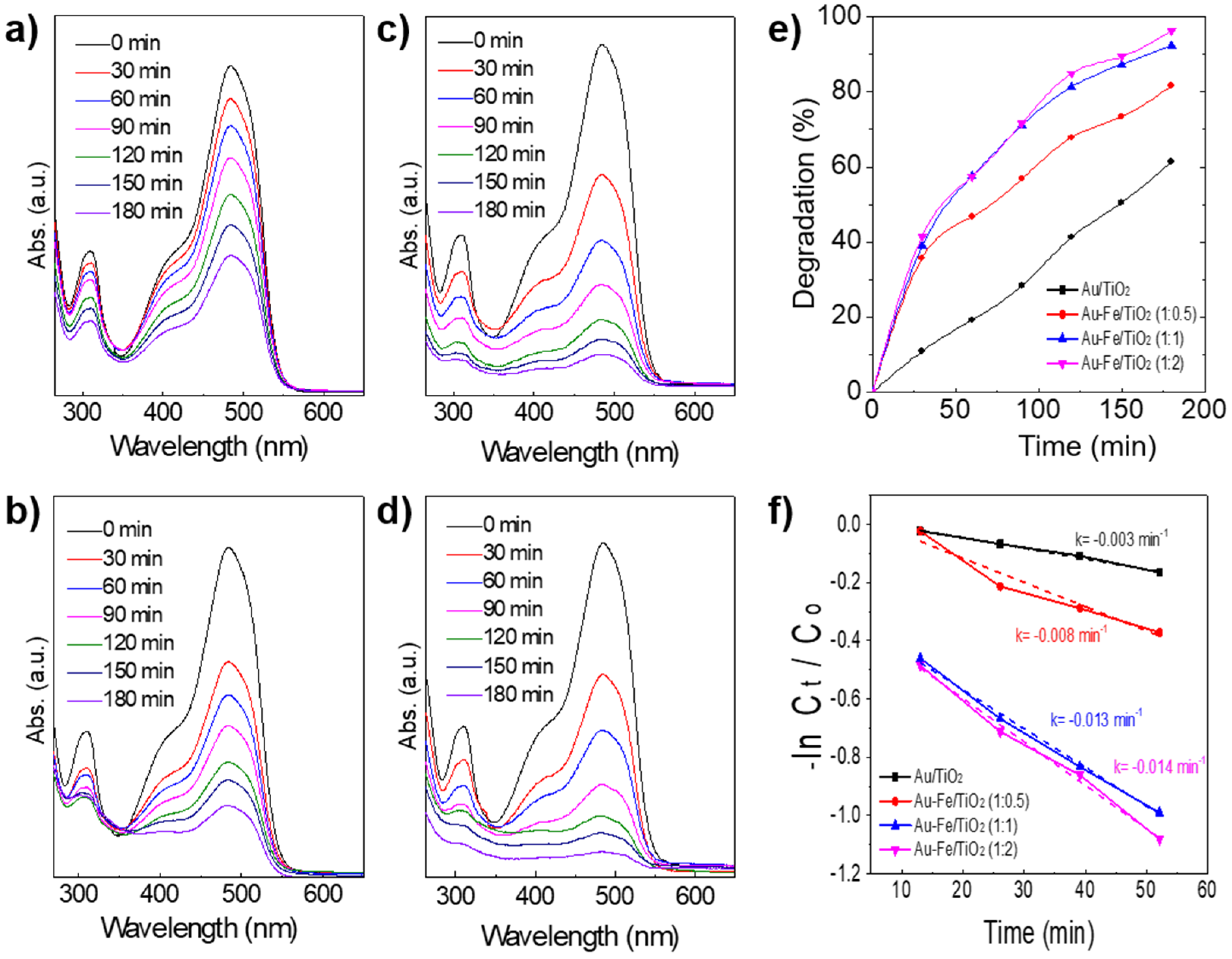
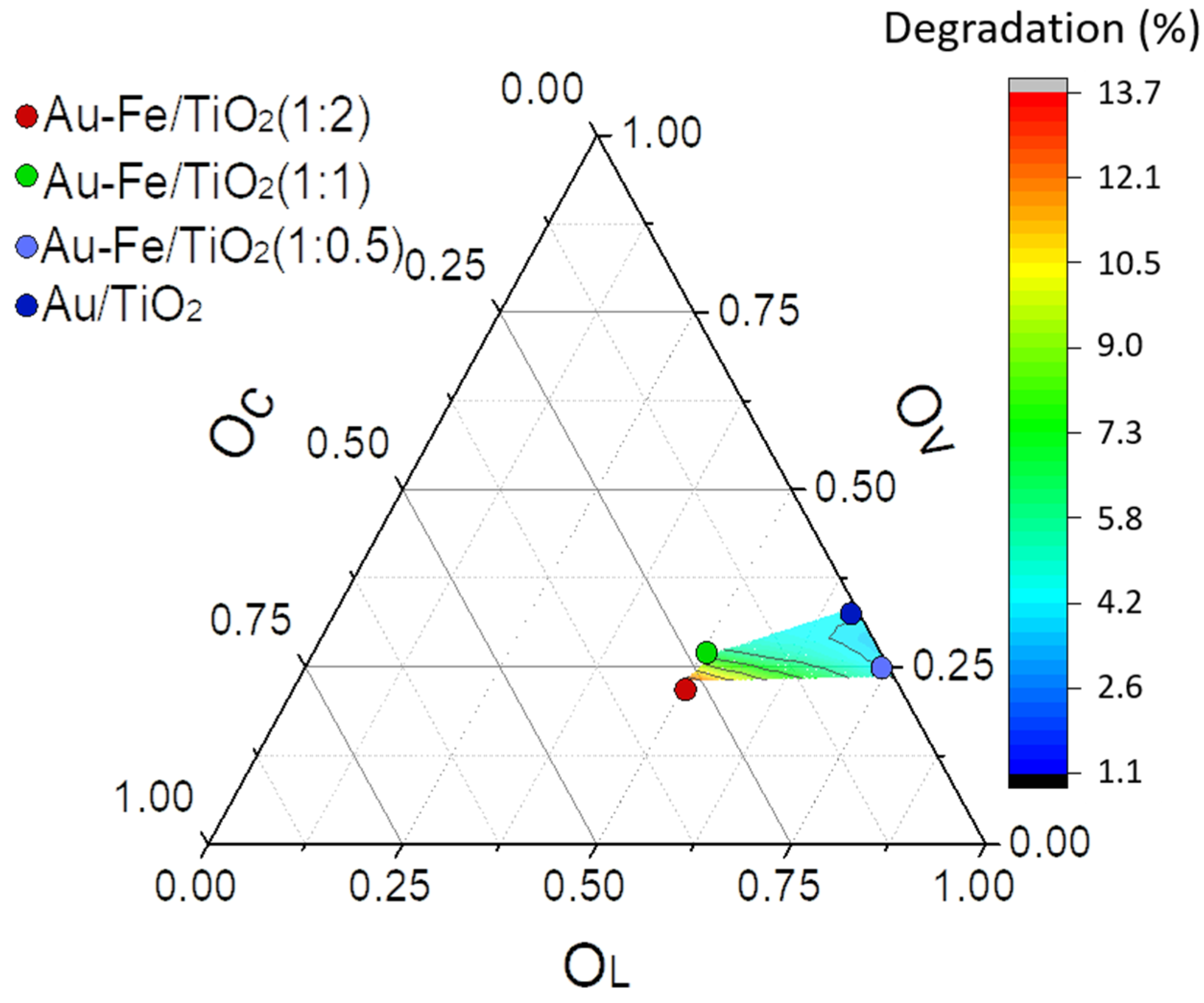
2.2. Catalytic Performance in Photo-Degradation of Orange II
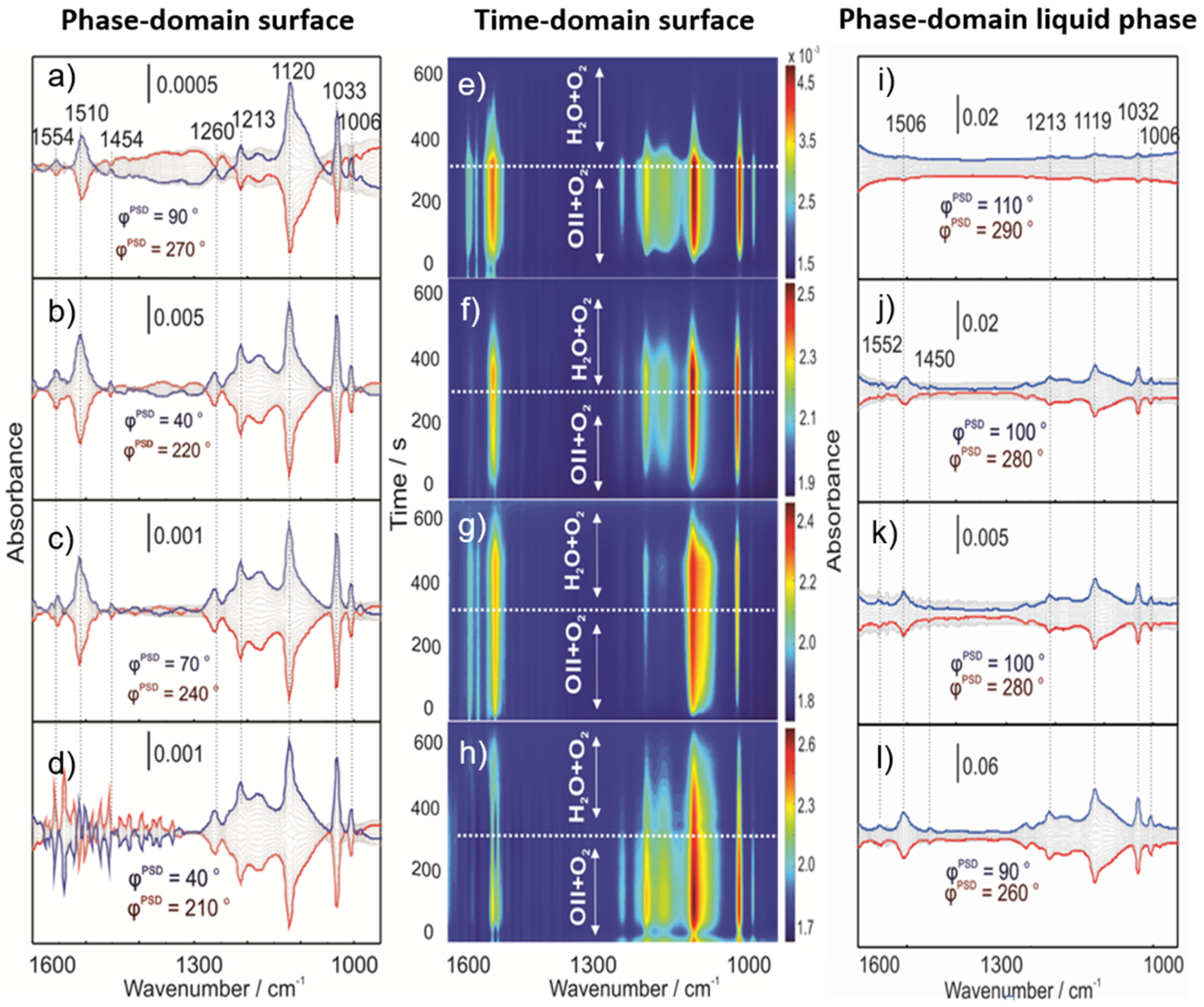
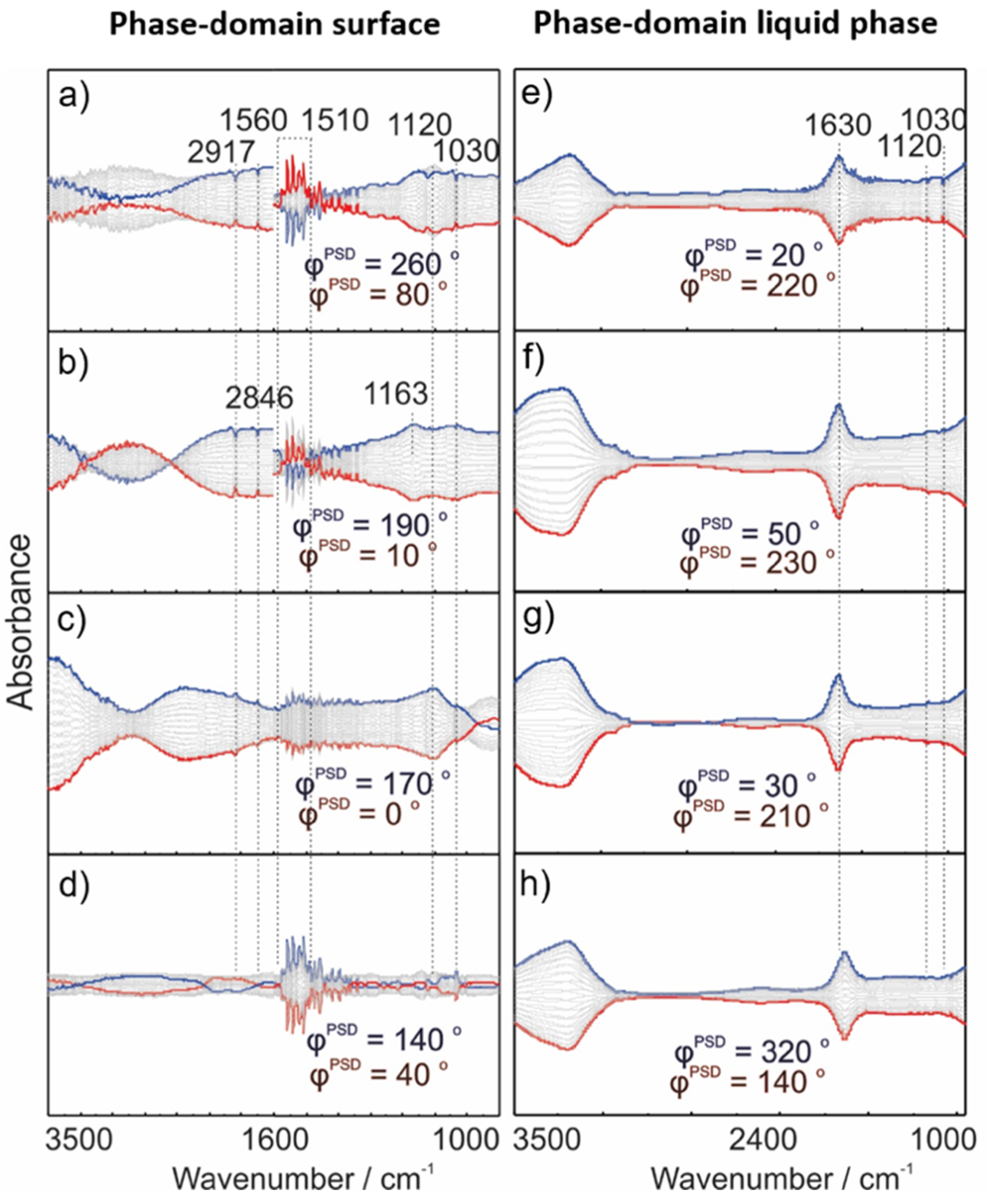
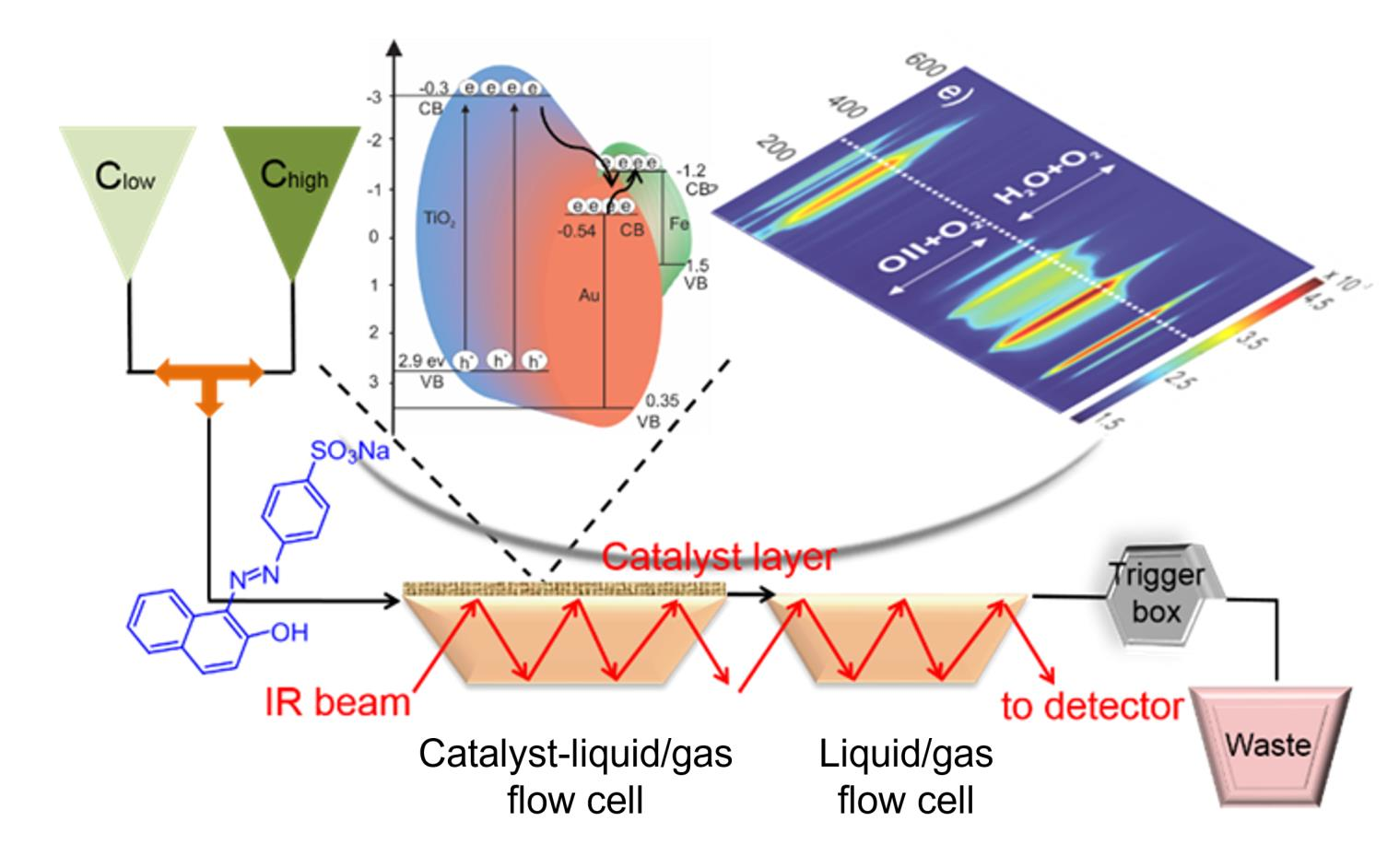
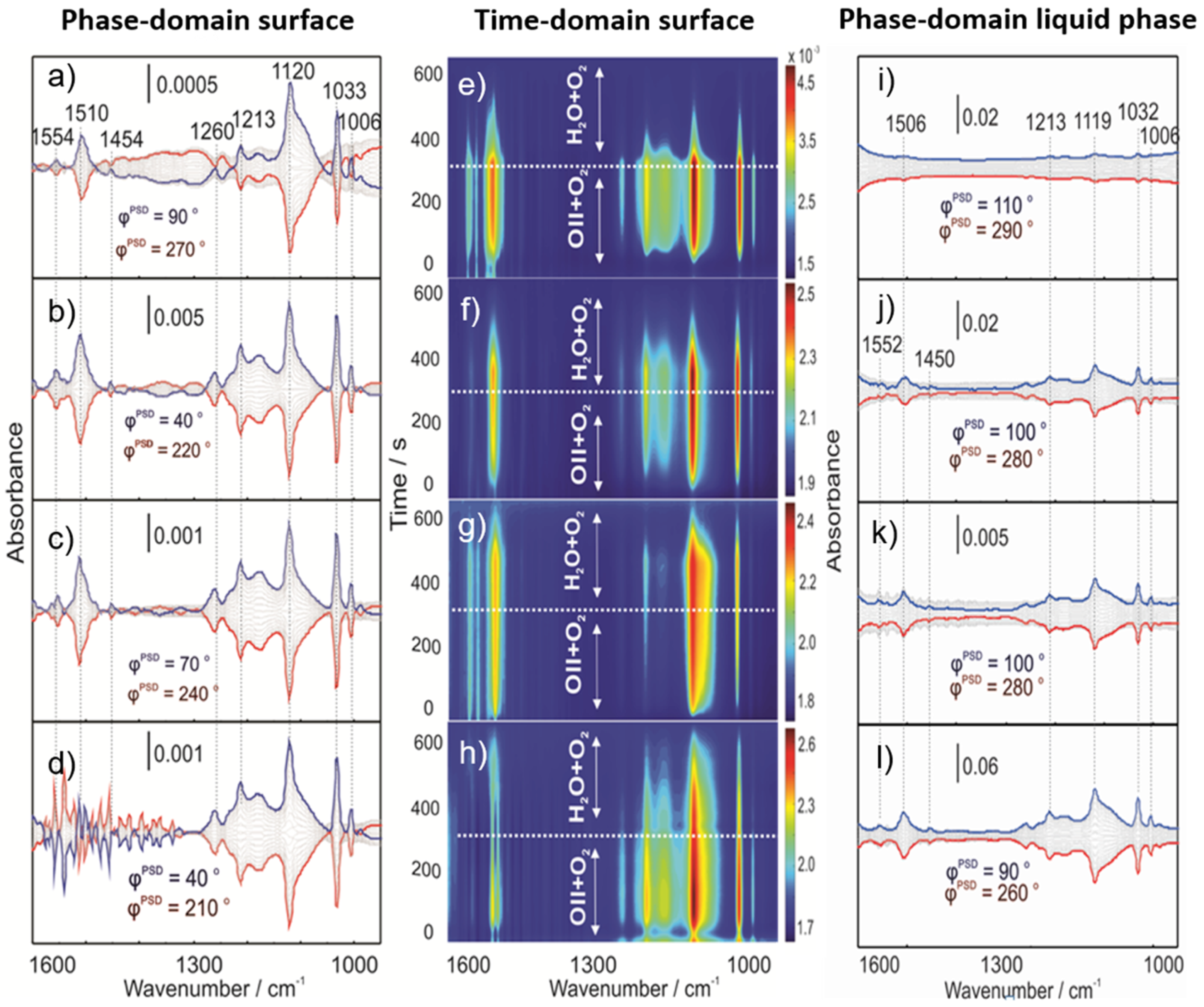
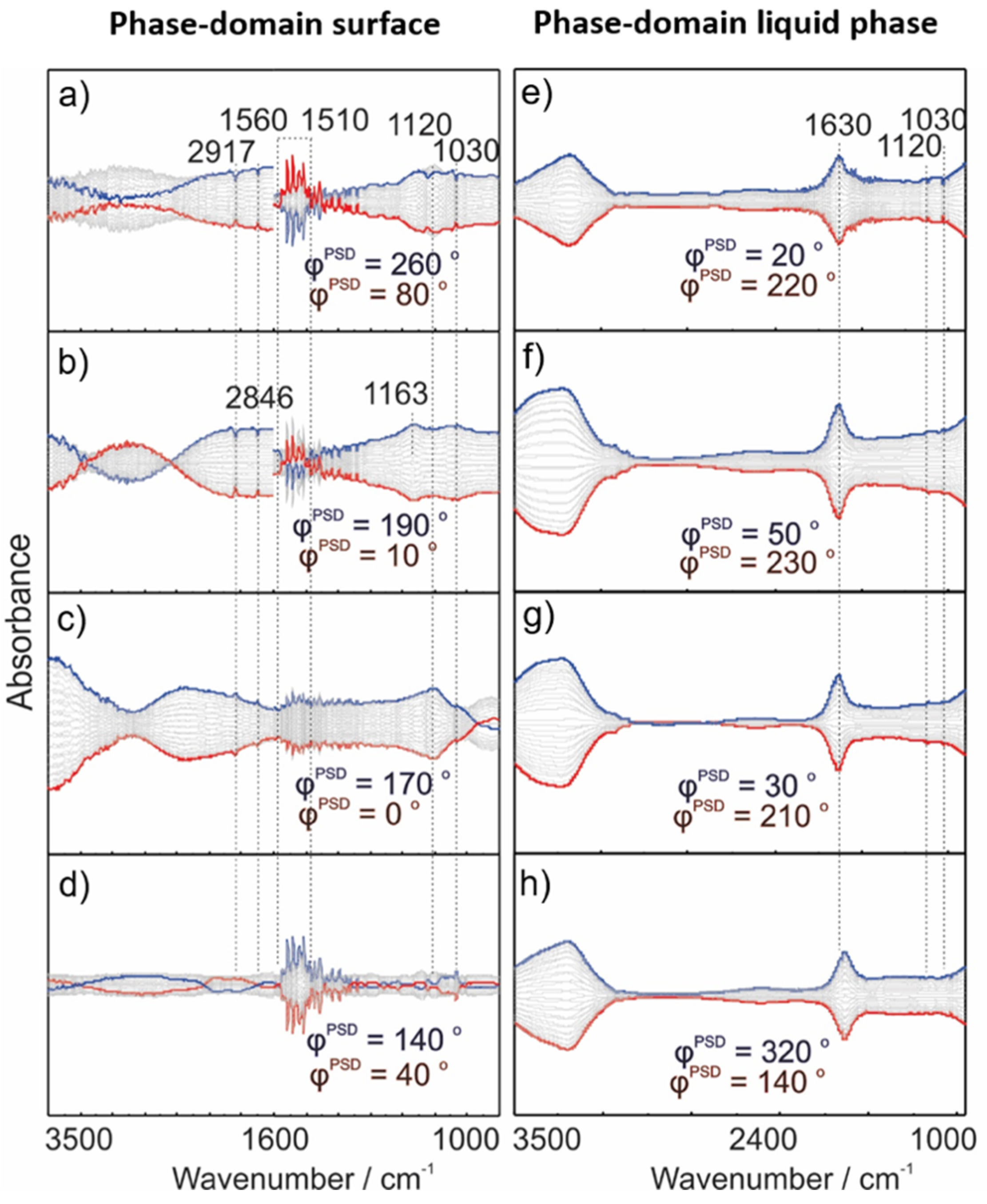
2.3. Operando MES-ATR-IR Spectroscopy
2.3.1. Concentration Modulation
2.3.2. Gas Modulation
3. Experimental
3.1. Chemicals and Materials
3.2. Preparation of Au/TiO2 and Au-FeOx/TiO2 Catalysts
3.3. Characterization
3.4. Catalytic Photodegradation
3.5. Operando ATR-IR Combined with Modulation-Excitation Spectroscopy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kamat, P.V. Photochemistry on nonreactive and reactive (semiconductor) surfaces. Chem. Rev. 1993, 93, 267–300. [Google Scholar] [CrossRef]
- Kaur, P.K.; Badru, R.; Singh, P.P.; Kaushal, S. Photodegradation of organic pollutants using heterojunctions: A review. J. Environ. Chem. Eng. 2020, 8, 103666. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M. Hierarchical photocatalysts. Chem. Soc. Rev. 2016, 45, 2603–2636. [Google Scholar] [CrossRef] [PubMed]
- Ben Mansour, H.; Boughzala, O.; Dridi, D.; Barillier, D.; Chekir-Ghedira, L.; Mosrati, R. Les colorants textiles sources de contamination de l’eau: CRIBLAGE de la toxicité et des méthodes de traitement. J. Water Sci. 2011, 24, 209–238. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.B.; Ng, L.Y.; Mohammad, A.W. A review of ZnO nanoparticles as solar photocatalysts: Synthesis, mechanisms and applications. Renew. Sust. Energ. Rev. 2018, 81, 536–551. [Google Scholar] [CrossRef]
- Vinodgopal, K.; Wynkoop, D.E.; Kamat, P.V. Environmental photochemistry on semiconductor surfaces: Photosensitized degradation of a textile azo dye, acid Orange 7, on TiO2 particles using visible light. Environ. Sci. Technol. 1996, 30, 1660–1666. [Google Scholar] [CrossRef]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Konstantinou, I.K.; Albanis, T.A. TiO2-assisted photocatalytic degradation of azo dyes in aqueous solution: Kinetic and mechanistic investigations—A review. Appl. Catal. B 2004, 49, 1–14. [Google Scholar] [CrossRef]
- Shan, A.Y.; Ghazi, T.I.M.; Rashid, S.A. Immobilisation of titanium dioxide onto supporting materials in heterogeneous photocatalysis: A review. Appl. Catal. A Gen. 2010, 389, 1–8. [Google Scholar] [CrossRef]
- Horikoshi, S.; Serpone, N. Can the photocatalyst TiO2 be incorporated into a wastewater treatment method? Background and prospects. Catal. Today 2020, 340, 334–346. [Google Scholar] [CrossRef]
- Fernandez, J.; Kiwi, J.; Baeza, J.; Freer, J.; Lizama, C.; Mansilla, H.D. Orange II photocatalysis on immobilised TiO2 effect of the pH and H2O2. Appl. Catal. B 2004, 48, 205–211. [Google Scholar] [CrossRef]
- Li, G.; Qu, J.; Zhang, X.; Liu, H.; Liu, H. Electrochemically assisted photocatalytic degradation of Orange II: Influence of initial pH values. J. Mol. Catal. A Chem. 2006, 259, 238–244. [Google Scholar] [CrossRef]
- Liu, S.; Wang, S.; Sun, X.; Li, W.; Ni, Y.; Wang, W.; Wang, M.; Yao, S. Mechanism and kinetics studies on the degradation of acid Orange 7. Spectrosc. Spect. Anal. 2005, 25, 776–779. [Google Scholar]
- Lucarelli, L.; Nadtochenko, V.; Kiwi, J. Environmental photochemistry: Quantitative adsorption and ftir studies during the TiO2-photocatalyzed degradation of orange II. Langmuir 2000, 16, 1102–1108. [Google Scholar] [CrossRef]
- Rauf, M.A.; Meetani, M.A.; Hisaindee, S. An overview on the photocatalytic degradation of azo dyes in the presence of TiO2 doped with selective transition metals. Desalination 2011, 276, 13–27. [Google Scholar] [CrossRef]
- Stylidi, M.; Kondarides, D.I.; Verykios, X.E. Pathways of solar light-induced photocatalytic degradation of azo dyes in aqueous TiO2 suspensions. Appl. Catal. B 2003, 40, 271–286. [Google Scholar] [CrossRef]
- Stylidi, M.; Kondarides, D.I.; Verykios, X.E. Visible light-induced photocatalytic degradation of acid Orange 7 in aqueous TiO2 suspensions. Appl. Catal. B 2004, 47, 189–201. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Yu, H.-Q.; Li, Q.-R. Radiolytic degradation of Acid Orange 7: A mechanistic study. Chemosphere 2005, 61, 1003–1011. [Google Scholar] [CrossRef]
- Zhao, H.-Z.; Sun, Y.; Xu, L.-N.; Ni, J.-R. Removal of acid Orange 7 in simulated wastewater using a three-dimensional electrode reactor: Removal mechanisms and dye degradation pathway. Chemosphere 2010, 78, 46–51. [Google Scholar] [CrossRef]
- Mercyrani, B.; Hernandez-Maya, R.; Solis-Lopez, M.; Th-Th, C.; Velumani, S. Photocatalytic degradation of Orange G using TiO2/Fe3O4 nanocomposites. J. Mater. Sci. Mater. Electron. 2018, 29, 15436–15444. [Google Scholar]
- May-Lozano, M.; Mendoza-Escamilla, V.; Garcia, E.R.; Medina, R.L.; Rivadeneyra-Romero, G.; Martinez-Delgadillo, S.A. Sonophotocatalytic degradation of Orange II dye using low cost photocatalyst. J. Clean. Prod. 2017, 148, 836–844. [Google Scholar] [CrossRef]
- Bansal, J.; Hafiz, A.K.; Sharma, S.N. Photoreduction of dye with noble metal gold permeated with metal oxide titania. J. Nanosci. Nanotechnol. 2020, 20, 3896–3901. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, X. Photocatalytic properties of gold/gold ion-modified titanium dioxide for wastewater treatment. Appl. Catal. A Gen. 2002, 228, 15–27. [Google Scholar] [CrossRef]
- Subramanian, V.; Wolf, E.E.; Kamat, P.V. Catalysis with TiO2/gold nanocomposites. effect of metal particle size on the Fermi level equilibration. J. Am. Chem. Soc. 2004, 126, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yue, L.; Liu, S.; Huang, B.; Zhang, X. Hydrothermal preparation and photocatalytic activity of mesoporous Au–TiO2 nanocomposite microspheres. J. Colloid Interface Sci. 2009, 334, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Adekoya, D.O.; Tahir, M.; Amin, N.A.S. G-C3C4/(Cu/TiO2) nanocomposite for enhanced photoreduction of CO2 to CH3OH and HCOOH under uv/visible light. J. CO2 Util. 2017, 18, 261–274. [Google Scholar] [CrossRef]
- Dutta, I.; Munns, C.B.; Dutta, G. An X-ray diffraction (xrd) study of vapor deposited gold thin films on aluminum nitride (aln) substrates. Thin Solid Films 1997, 304, 229–238. [Google Scholar] [CrossRef]
- Kosyachenko, L.A. Sollar cells thin-film technology. IntechOpen 2011. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Rasul, M.; Martens, W.N.; Brown, R.; Hashib, M. Heterogeneous photocatalytic degradation of phenols in wastewater: A review on current status and developments. Desalination 2010, 261, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Nasralla, N.; Yeganeh, M.; Astuti, Y.; Piticharoenphun, S.; Shahtahmasebi, N.; Kompany, A.; Karimipour, M.; Mendis, B.; Poolton, N.; Šiller, L. Structural and spectroscopic study of Fe-doped TiO2 nanoparticles prepared by sol–gel method. Sci. Iran. 2013, 20, 1018–1022. [Google Scholar]
- Han, X.-G.; He, H.-Z.; Kuang, Q.; Zhou, X.; Zhang, X.-H.; Xu, T.; Xie, Z.-X.; Zheng, L.-S. Controlling morphologies and tuning the related properties of nano/microstructured ZnO crystallites. J. Phys. Chem. C 2008, 113, 584–589. [Google Scholar] [CrossRef]
- Valencia, S.; Marin, J.M.; Restrepo, G. Study of the bandgap of synthesized titanium dioxide nanoparticules using the sol-gel method and a hydrothermal treatment. Open Mater. Sci. J. 2009, 4, 9–14. [Google Scholar] [CrossRef]
- Zheng, J.; Jiang, Q.; Lian, J. Synthesis and optical properties of flower-like ZnO nanorods by thermal evaporation method. Appl. Surf. Sci. 2011, 257, 5083–5087. [Google Scholar] [CrossRef]
- Bauer, C.; Jacques, P.; Kalt, A. Investigation of the interaction between a sulfonated azo dye (AO7) and a TiO2 surface. Chem. Phys. Lett. 1999, 307, 397–406. [Google Scholar] [CrossRef]
- Wang, X.; Maeda, N.; Baiker, A. Synergistic effects of Au and FeOx nanocomposites in catalytic NO reduction with CO. ACS Catal. 2016, 6, 7898–7906. [Google Scholar] [CrossRef]
- Andanson, J.M.; Baiker, A. Exploring catalytic solid/liquid interfaces by in situ attenuated total reflection infrared spectroscopy. Chem. Soc. Rev. 2010, 39, 4571–4584. [Google Scholar] [CrossRef] [Green Version]
- Buergi, T.; Baiker, A. Attenuated total reflection infrared spectroscopy of solid catalysts functioning in the presence of liquid-phase reactants. In Advances in Catalysis; Gates, B.C., Knozinger, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 50, pp. 227–283. [Google Scholar]
- Meemken, F.; Müller, P.; Hungerbühler, K.; Baiker, A. Simultaneous probing of bulk liquid phase and catalytic gas-liquid-solid interface under working conditions using attenuated total reflection infrared spectroscopy. Rev. Sci. Instrum. 2014, 85, 084101. [Google Scholar] [CrossRef]
- Baurecht, D.; Fringeli, U.P. Quantitative modulated excitation Fourier transform infrared spectroscopy. Rev. Sci. Instrum. 2001, 72, 3782–3792. [Google Scholar] [CrossRef] [Green Version]
- Bürgi, T.; Baiker, A. In situ infrared spectroscopy of catalytic solid−liquid interfaces using phase-sensitive detection: Enantioselective hydrogenation of a pyrone over Pd/TiO2. J. Phys. Chem. B 2002, 106, 10649–10658. [Google Scholar] [CrossRef]
- Urakawa, A.; Bürgi, T.; Baiker, A. Sensitivity enhancement and dynamic behavior analysis by modulation excitation spectroscopy: Principle and application in heterogeneous catalysis. Chem. Eng. Sci. 2008, 63, 4902–4909. [Google Scholar] [CrossRef]
- Müller, P.; Hermans, L. Applications of modulation excitation spectroscopy in heterogeneous catalysis. Ind. Eng. Chem. Res. 2017, 56, 1123–1136. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
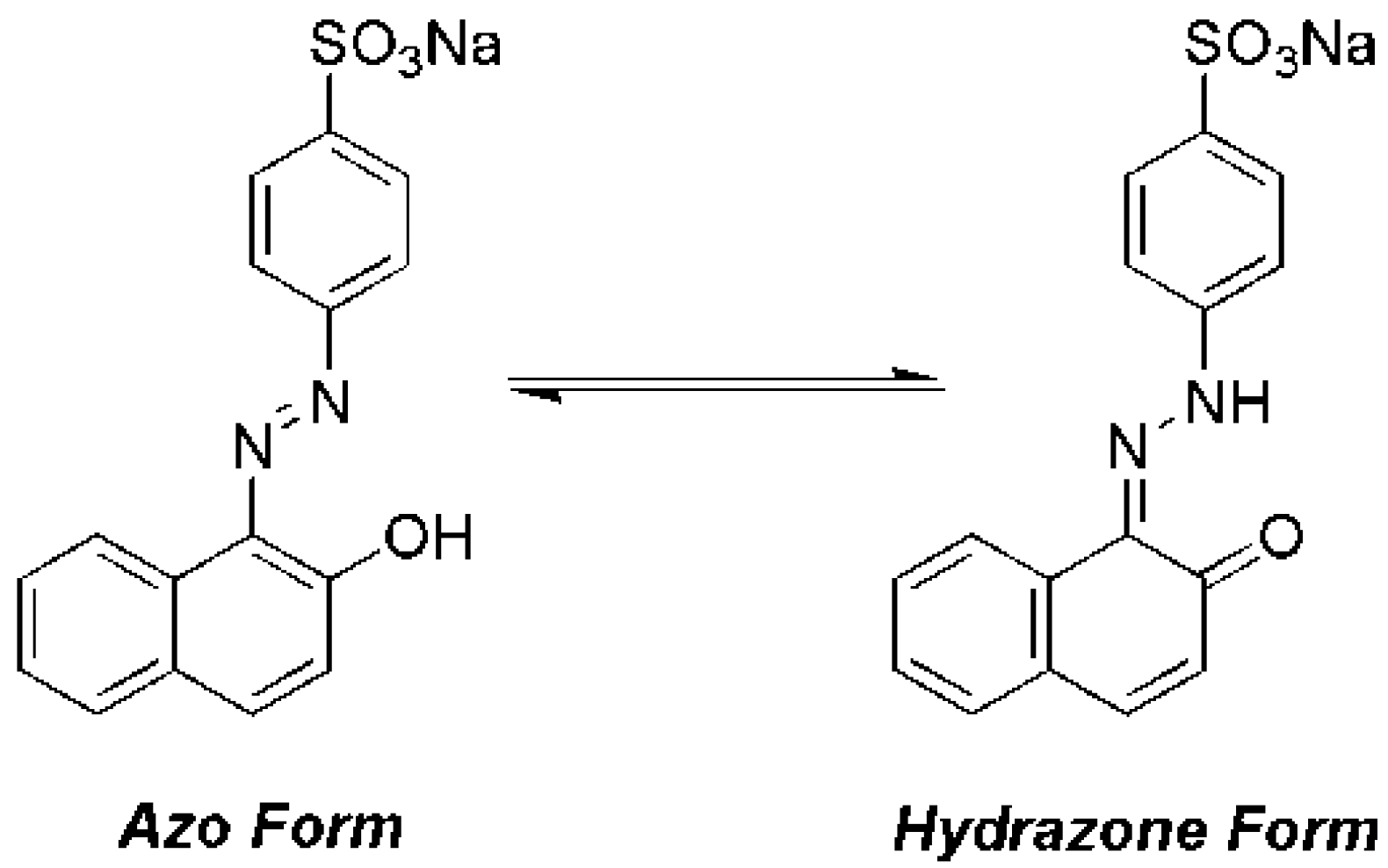{kind=link}
| Catalysts | Au particle Size (nm) | Surface Area (m2/g) | Bandgap (eV) |
|---|---|---|---|
| Au/TiO2 | 3.9 | 50 | 3.12 |
| Au-Fe(1:0.5)/TiO2 | 4.3 | 51 | 2.79 |
| Au-Fe(1:1)/TiO2 | 4.4 | 57 | 2.40 |
| Au-Fe(1:2)/TiO2 | 5.1 | 58 | 2.23 |
| Catalyst | Fe2+2p3/2 | Fe3+2p3/2 | OL | OV | OC |
|---|---|---|---|---|---|
| Au-Fe(1:0.5)/TiO2 | 709.8 (35%) | 713.9 (65%) | 529.3 (64%) | 530.6 (33%) | 531.2 (3%) |
| Au-Fe(1:1)/TiO2 | 709.5 (44%) | 713.5 (56%) | 529.1 (52%) | 529.9 (26%) | 531.2 (22%) |
| Au-Fe(1:2)/TiO2 | 709.8 (50%) | 713.5 (50%) | 529.2 (45%) | 530.0 (25%) | 531.3 (30%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waheed, A.; Shi, Q.; Maeda, N.; Meier, D.M.; Qin, Z.; Li, G.; Baiker, A. Strong Activity Enhancement of the Photocatalytic Degradation of an Azo Dye on Au/TiO2 Doped with FeOx. Catalysts 2020, 10, 933. https://doi.org/10.3390/catal10080933
Waheed A, Shi Q, Maeda N, Meier DM, Qin Z, Li G, Baiker A. Strong Activity Enhancement of the Photocatalytic Degradation of an Azo Dye on Au/TiO2 Doped with FeOx. Catalysts. 2020; 10(8):933. https://doi.org/10.3390/catal10080933
Chicago/Turabian StyleWaheed, Ammara, Quanquan Shi, Nobutaka Maeda, Daniel M. Meier, Zhaoxian Qin, Gao Li, and Alfons Baiker. 2020. "Strong Activity Enhancement of the Photocatalytic Degradation of an Azo Dye on Au/TiO2 Doped with FeOx" Catalysts 10, no. 8: 933. https://doi.org/10.3390/catal10080933
APA StyleWaheed, A., Shi, Q., Maeda, N., Meier, D. M., Qin, Z., Li, G., & Baiker, A. (2020). Strong Activity Enhancement of the Photocatalytic Degradation of an Azo Dye on Au/TiO2 Doped with FeOx. Catalysts, 10(8), 933. https://doi.org/10.3390/catal10080933