Abstract
α-amylase which catalyzes the hydrolysis of α-1,4-glycosidic bonds in starch have frequently been cloned into various microbial workhorses to yield a higher recombinant titer. A thermostable SR74 α-amylase from Geobacillus stearothermophilus was found to have a huge potential in detergent industries due to its thermostability properties. The gene was cloned into a CTG-clade yeast Meyerozyma guilliermondii strain SO. However, the CUG ambiguity present in the strain SO has possibly altered the amino acid residues in SR74 amylase wild type (WT) encoded by CUG the codon from the leucine to serine. From the multiple sequence alignment, six mutations were found in recombinant SR74 α-amylase (rc). Their effects on SR74 α-amylase structure and function remain unknown. Herein, we predicted the structures of the SR74 amylases (WT and rc) using the template 6ag0.1.A (PDB ID: 6ag0). We sought to decipher the possible effects of CUG ambiguity in strain SO via in silico analysis. They are structurally identical, and the metal triad (CaI–CaIII) might contribute to the thermostability while CaIV was attributed to substrate specificity. Since the pairwise root mean square deviation (RMSD) between the WT and rc SR74 α-amylase was lower than the template, we suggest that the biochemical properties of rc SR74 α-amylase were better deduced from its WT, especially its thermostability.
1. Introduction
α-amylases (α-1,4-D-glucan-glucanohydrolase, EC 3.2.1.1) catalyse the endohydrolysis of starch into smaller moieties, encompassing maltodextrin, maltooligosaccharides and glucose, by cleaving α-1,4-glycosidic linkages [1]. Of the 167 glycoside hydrolase (GH) families created in the Carbohydrate-Active Enzymes database (CAZy) (as of June 2020) (http://www.cazy.org/), most α-amylases have been categorized into GH family 13 (GH13) with almost 95,000 available protein sequences [2]. A smaller number of α-amylases were also classified into GH57 and GH119 with 2791 and 27 available protein sequences, respectively. GH13, which belongs to a higher hierarchy of clan H (GH–H) in the CAZy database, shares the same catalytic machinery and possesses a similar structural fold to the catalytic domain i.e., the N-terminal (β/α)8-fold TIM barrel, with GH70 and GH77, although significant differences in overall the sequence are present [3].
In recent decades, bacteria and yeast have shown promising results in producing native and recombinant α-amylases such as Bacillus amyloliquefaciens [4], Bacillus stearothermophilus [5], Aspergillus flavus [6] and Pichia pastoris (recently known as Komagataella phaffii) [7]. This thermostable property of α-amylases is favourable in industrial application, especially in starch saccharification and liquefaction [1]. Several crystallographic and site-directed mutagenesis studies have shown that the presence of metal ions (Ca2+ and Na+), hydrophobic interactions, hydrogen linkages, salt bridges and N-glycosylation are attributed to the thermostability and longer half-life (t1/2) of most carbohydrate-active enzymes (CAZymes) [8,9,10,11,12,13].
To our knowledge, limited studies have been performed in predicting and elucidating the 3D structure of the recombinant thermostable α-amylases expressed in yeast, particularly in Meyerozyma guilliermondii via an in silico homology modelling strategy or protein crystallography. It is also noteworthy that none of the hits (Glucoside transferase family 48, GH5, GH18, GH32 and GH114) are in α-amylase families (GH13, GH57 and GH119) when a search of CAZymes-producing M. guilliermondii is performed against the CAZy database.
Recent studies have described the molecular cloning of the bacterial thermostable SR74 α-amylase gene from Geobacillus sp. SR74 into Escherichia coli BL21 (DE3) pLysS, K. phaffii GS115 and M. guilliermondii strain SO [7,14,15]. A comparable yield was observed when recombinant SR74 α-amylases were expressed in K. phaffii GS115 (28.6 U/mL) and M. guilliermondii strain SO (26 U/mL). The quantitative DNS assay was optimally performed at 60 °C for 30 min [7,15]. Moreover, the expression of SR74 α-amylase in strain SO was confirmed by observing a colourless halo around the yeast colonies on an iodine–starch screening plate [15]. In 2012, Oslan et al. [16] isolated the strain SO from spoiled orange, then the yeast was manipulated to express the recombinant thermostable bacterial lipase from Geobacillus zalihae strain T1 [17]. The result showed that the optimum temperature of the T1 lipase was reduced from 70 °C to 65 °C in K. phaffii and M. guilliermondii strain SO, respectively [17,18]. Changing the host with a similar gene and vector had reduced the thermostability of the enzyme itself, which could be attributed to the possible mutations (leucine to serine) in the amino acid sequence of recombinant T1 lipase expressed in strain SO.
While the Meyerozyma species complex belongs to the Saccharomycotina CTG clade, the CUG codon is translated into serine (Ser) residue in M. guilliermondii (anamorph Candida guilliermondii) instead of leucine (Leu) residue in bacteria (Geobacillus sp.) and non-CTG clade yeast (K. phaffii) [19,20,21]. It is also noteworthy that Candida albicans (a CTG-clade yeast) mis-incorporates leucine at only 3% to 5% under normal and mild stress conditions, respectively. A similar study also that showed C. albicans can tolerate up to 28.1% of leucine mis-incorporation contributed by the CUG ambiguity present [22]. Moreover, such phenomenon in C. albicans can be extended to C. guilliermondii (teleomorph M. guilliermondii) since their introns in the Ser-tRNACGA are similar [23,24], further suggesting the very high occurrence rate of serine incorporation (95% to 97%) in a protein expressed in M. guilliermondii.
In recent years, studies have focused on the effect of CUG-encoded residue reversal from Ser back to Leu in human fungal pathogen C. albicans, employing the fact that the rate of leucine incorporation fluctuates to different stress conditions [25,26,27]. Although native proteins in CTG-clade yeast have a high tolerance towards CUG ambiguity [27], the effects towards the structural and physicochemical properties, especially on the thermostability of the recombinant bacterial protein still remains unclear. Here, we described the application of bioinformatic tools and the homology modelling platform in deciphering the potential alterations of the industrially important traits of recombinant bacterial thermostable SR74 α-amylase expressed in the CTG-clade yeast M. guilliermondii strain SO.
2. Results and Discussion
2.1. Structure and Sequence Analysis
Pairwise sequence alignment was performed to determine the sites of Leu to Ser mutation in recombinant (rc) SR74 α-amylase (Figure 1). Six mutation sites (L43S, L215S, L338S, L424S and L427S) were identified and the rc SR74 α-amylase shared a high identity (98.83%) to the wild-type (WT) SR74 α-amylase [14]. This Leu to Ser mutation was due to the CUG ambiguity possessed by the M. guilliermondii strain SO (previously known as P. guilliermondii strain SO) in the Saccharomycotina CTG clade [21,28]. While Leu and Ser are non-polar and polar amino acid residues, respectively, it is worth analysing the possible structural and physicochemical changes’ influence as there have been limited studies reporting on the impact of CUG ambiguity on the recombinant proteins.
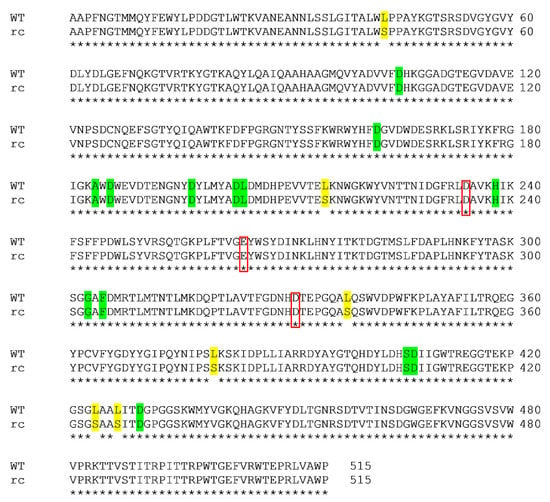
Figure 1.
Pairwise sequence alignment of wild-type (WT) and recombinant (rc) SR74 α-amylases. The alignment was performed using Clustal Omega. Six mutation (Leu to Ser) sites were identified and highlighted in yellow. The identity between both proteins was computed as 98.83%. The catalytic triad (D234, E264 and D331) was identified in red boxes. The calcium ion-interacting residues (D105, D162, A184, D186, D197, D203, L204, H238, G303, F305, S406, D407 and D430) were highlighted in green.
SR74 α-amylase has three domains, namely Domain A, B and C (Figure 2A). Domain A (residues 1–104 and 207–394) is known as the catalytic domain, which possesses a (β/α)8-fold TIM barrel. Such structural architecture of its catalytic domain has allowed the SR74 α-amylase to be categorized in GH13 [3]. Domain B (residues 105–206), which protrudes from Domain A, consists of five β-strands. These strands seem to interact with and be stabilized by the calcium ion metal triad (Figure 2B). Domain C (395–515), which consists of eight β-strands, has the so-called Greek key motif recently attributed to the substrate (starch) binding in G. thermoleovorans α-amylase [29]. Despite the metal triad (Ca2+-Ca2+-Ca2+) present in Domain A, another calcium ion also exists on the contact surface between Domains A and C. These calcium ions and their interactions with various amino acid residues are said to contribute largely to the structural stability [12,30].
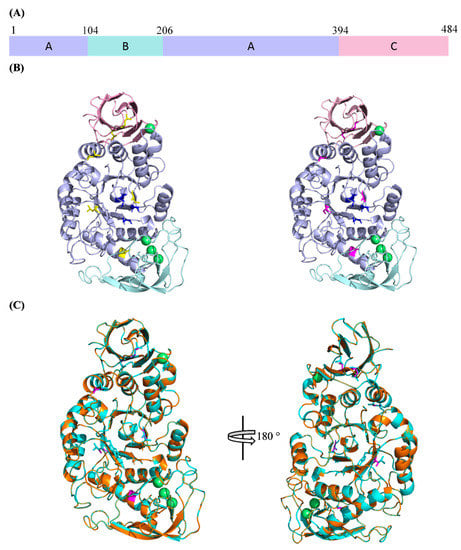
Figure 2.
The 3D structures of WT and rc SR74 α-amylases predicted using SWISS-MODEL. (A) Domain arrangement of SR74 α-amylases. Domains A, B and C are colour-coded and the domain boundaries are shown; (B) the predicted 3D structures of SR74 α-amylases. WT SR74 α-amylase with yellow-coloured leucine residue (left), rc SR74 α-amylase with magenta-coloured serine residue (right). Catalytic triad (D234, E264, D331) are blue-coloured and calcium ions (Ca2+) are in green. (C) The superimposition of both WT and rc SR74 α-amylases. Polypeptide chains of WT and rc SR74 α-amylases are coloured with cyan and orange, respectively. The views are related by a 180° rotation around the vertical axis. All the structural figures are viewed and prepared using the PyMOL Molecular Graphic System, Version 2.4 Schrödinger, LLC.
The catalytic residues of SR74 α-amylase are deduced to be two aspartic acids (D234 and D331) and one glutamic acid (E264) after the structural analysis and annotation inferred from UniProt. It is noteworthy that all these residues reside in different β-strands which make up the core (active cleft) in Domain A (Figure 2B). These residues are highly conserved and catalyse the hydrolysis of starch or maltooligosaccharides via α-retaining double displacement reaction [31]. Such conservation of the residues was uniformly observed throughout the multiple sequence alignment (MSA) of the Protein Basic Local Alignment Search Tool (BLASTP) results with >95% identity and 100% query coverage (Table S1). This further suggests that the site-directed mutagenesis of any strictly conserved catalytic residues will lead to irreversible loss in the amylolytic activity. Using ProtParam to estimate the theoretical physicochemical properties, the isoelectric point (pI) of both WT and rc SR74 α-amylases are similar at pH 5.61 while the molecular weight (MW) of WT SR74 α-amylase (58.55 kDa) is slightly higher than rc SR74 α-amylase (58.39 kDa).
2.2. Homology Modelling and Structure Validation
To predict the 3D structures of both WT and rc SR74 α-amylases in silico, SWISS-MODEL webserver was equipped. While the server searches template for evolutionary related protein structures against certain databases, 6ag0.1.A (chain A of PDB ID: 6ag0) was chosen according to the internal scoring matrices provided by the server. Although the template 4uzu.1.A (chain A of PDB ID: 4uzu) shares higher identity to both WT and rc SR74 α-amylases (98.83% and 97.66% respectively), 6ag0.1.A with slightly lower identities (98.45% and 97.28%, respectively) was chosen as the template to build and predict the protein structures. This was due to its higher primary global model quality estimate (GMQE) i.e., 0.99, where the GMQE value (nearer to 1) indicated that the 3D structures constructed have the highest expected accuracy since the GMQE value signifies the maximum joint distribution of several properties where the most likely structural similarity is achieved [32]. It is also noteworthy that the maximum BLASTP scores (1028) between 6ag0.1.A and both SR74 α-amylases were higher than that with 4uzu.1.A (1022). Although the Q qualitative model energy analyses (QMEANs) of the structures constructed using 4uzu.1.A as a template were slightly better (nearer to 0), but their scores were validated by Verify3D (except rc SR74 α-amylase), ERRAT and PROCHECK were comparatively lower than the structures constructed using 6ag0.1.A as the template (Table S2).
Upon constructing the structures of SR74 α-amylases using 6ag0.1.A as the template (Figure 2B), the protein structures were subjected to verification using Verify3D, ERRAT and PROCHECK. Both structures passed the verifications (Table S2) with scores higher than the structures constructed using other templates. Our choice on template selection was further justified when the 3D structures of both WT and rc SR74 α-amylases had 90.3% and 90.5% residues, respectively, in the most favoured region of the respective Ramachandran plots. While a Ramachandran plot has the ability to detect gross errors in the structures, the plot of residue φ-ψ torsion angles is considered the most telling and significant quality indicator of the protein structures [33,34]. Knowing that there were six mutation sites where Leu was replaced with Ser in the rc SR74 α-amylase, the 3D structures of both SR74 α-amylases were then superimposed to decipher the possible structural differences.
2.3. Superimposition of WT and rc SR74 α-Amylases
The predicted 3D structures of the WT and rc SR74 α-amylases were superimposed and viewed using PyMOL (Figure 2C). Unexpectedly, both structures were superimposed very well and the root mean square deviation (RMSD) computed was only 0.001 when 650 outliers were excluded. However, it is also noteworthy that even when the outliers which occupied 15.90% of the total computed atoms in the structures were included, the RMSD was only 0.008 (Table S3). This result indicated that the impact of outliers on the structural deviation was limited. Although RMSD calculation possesses several disadvantages, most of its shortcomings are only subjected to the proteins with partial overlapping or completely different sequences [35]. The disadvantage is even more significant when a deviation of a position in a single loop is present, or a flexible terminus with a large global backbone RMSD [36]. To address these shortcomings, the secondary structures of both SR74 α-amylases were predicted and mapped using ENDscript 2.0 and ESPript 3.0, respectively (Figure S1), the numbers of α-helices (ten), β-strands (twenty-five) and their respective lengths were perfectly identical, further justifying the computed RMSD. A 180° rotation of the structure around the vertical axis allowed us to observe a clearer superimposition of the structures despite the RMSD (Figure 2C). The results were beyond our expectations since the polarity of Leu and Ser deviates significantly. Such findings prompted us to determine and analyse the protein—ligand interactions so that the possible alterations on the physicochemical properties encompassing thermostability and optimum pH could be understood.
2.4. Protein–Ligand Interactions
While most α-amylases are metalloenzymes, metal ions can either play the role of cofactors in enhancing the amylolytic activity of α-amylases, or act as the inhibitors to the enzymes [1]. Four calcium ions (Ca2+) were deduced and inferred from the template 6ag0.1.A (chain A of PDB ID: 6ag0) in both WT and rc SR74 α-amylases, namely from CaI to CaIV. CaIV resides in the surfaces between Domain A and Domain C, while others are located in the interior of Domain B. CaI binds to SR74 α-amylases through the carbonyl oxygen atom of H238 and the side-chain oxygen atoms of D105, D197 and D203 (Figure 3A). While CaI is located the nearest to the active cleft, this calcium ion is strictly conserved in α-amylases due to its ability to stabilize the region between α4 and β13 barrel (Figure S1). It is worth noting that D234 (one of the catalytic residues) is located in the stabilized architectural cleft, justifying the contribution of CaI in its thermostability and catalytic ability. A site-directed mutagenesis (A184D) study conducted has shown that the A184D mutant possessed a weakened positive charge on H158 towards the D182 carboxylate, resulting in a stronger interaction between D182 and CaI and an increase in the structural stability and amylolytic activity of truncated ASKA (TASKA) expressed in Anoxybacillus sp. [8]. While the CaI-interacting residues are invariantly conserved in the α-amylase families, these residues are sensitive to substitution and such mutation is detrimental to the protein’s amylolytic activity and thermostability [37,38].
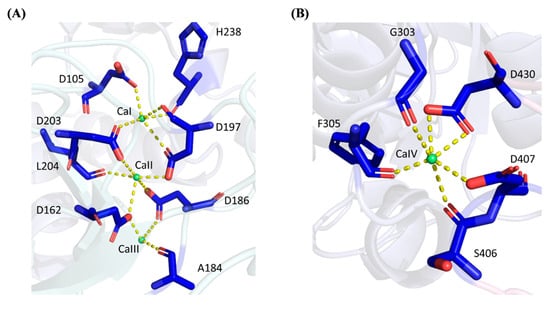
Figure 3.
Polar interactions between calcium ions and SR74 α-amylase. (A) CaI, CaII and CaIII interact with 8 residues via polar bonds. D197 and D203 form polar linkages with CaI and CaII while D162 and D186 interact with CaII and CaIII at the same time. (B) CaIV possesses polar interactions with 5 residues. Yellow dashed lines showed the polar interactions between residues and respective calcium ions.
CaII which is positioned between CaI and CaIII in the calcium metal triad, binds to SR74 α-amylases through Ca2+-O interactions with the carbonyl oxygen atom of L204, as well as the side-chain oxygen atoms of four aspartic acid residues (D162, D186, D197 and D203). Nevertheless, CaIII, which is the farthest from the active cleft, is ligated with the carbonyl oxygen atom of A184 and the side-chain oxygen atoms of D162 and D186. However, our results showed only three residues interacting with CaIII, while Xie et al. [12] reported an extra D205 which was bound to the template 6ag0.1.A with its side-chain oxygen atom. It is worth mentioning that while our study did not involve the elucidation of the crystallized structures of both WT and rc SR74 α-amylases, none of the water molecules were involved while inferring the protein–ligand interaction. Therefore, our observation without D205 could be attributed to this reason.
It is interesting to observe the cases where one amino acid residue (specifically aspartic acid) interacts with more than one calcium ion ligand in the predicted structures of SR74 α-amylases through side-chain oxygen atoms: (1) D197 and D203 are ligated with both CaI and CaII; (2) D162 and D186 interacts with CaII and CaIII at the same time. Such multiple interactions are hypothesized to stabilize the secondary structures nearby and eventually allow the SR74 α-amylases to function at a higher temperature (70 °C and 65 °C in Geobacillus sp. SR74 and K. phaffii GS115, respectively) [7,14]. Moreover, it was observed that most residues interacting with CaI–CaIII were made up of aspartic acids which are polar residues compared to H238 (which can be polar or non-polar depending on environmental pH) and A184. Such observation is justified by the amino acid composition in the enzymes where both WT and rc SR74 α-amylases recorded 8.0% (41 out of total 515 residues) of aspartic acid residues in the polypeptide, being the highest percentage after the fundamental glycine (9.7%) and threonine residues (8.5%). As reported by Xie et al. [12], the formation of linear metal triad by CaI–CaIII is unprecedented since the same region was previously reported to be occupied by the Ca2+-Na+-Ca2+ triad [30,39]. Such homology in the predicted model is worth proving using the protein crystallography method to decipher the most accurate triad identity and its interactions with the residues of the polypeptide chain.
Away from CaI–CaIII in Domain B, CaIV is located between Domain A and C. According to the coordination structure that surrounds CaIV (Figure 3B), it forms an approximately hexagonal geometry with six coordinated oxygens from G303, F305, S406, D407 and D430, of which two interactions are observed at the two carbonyl oxygen atoms in the side chain of aspartic acid residue (D430). Through the structural and coordination analysis of the residues involved, it is worth mentioning that CaIV was observed to bridge the loop extended from α6 in Domain B, with the region located between β18 and β19 in Domain C (Figure S1). While Domain C is known to possess the so-called Greek key motif, such coordination of CaIV between Domain B and C is hypothesized to contribute to the substrate specificity and catalytic activity [12,29].
Since a similar structure and protein–ligand interaction are observed in both the WT and rc SR74 α-amylases, we did not expect much difference in the physicochemical properties such as the optimum temperature and optimum pH. Moreover, it is worth mentioning that the six mutation sites observed in Figure 1 did not involve the catalytic triad (D234, E264 and D331) and the calcium ion-interacting residues. Therefore, it can be deduced that the physicochemical properties (especially optimum temperature) of rc SR74 α-amylase expressed in M. guilliermondii strain SO can be speculated from the WT SR74 α-amylase expressed in the previous yeast expression host, K. phaffii GS115 (optimum temperature at 65 °C), without any further biochemical tests. While the template 6ag0.1.A is a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04 (Bst-MFA), it is able to degrade maltoheptaose (G7), maltooctaose (G8) and maltonanoose (G9) into the maltohexaose (G6) as the major products, but is inactive towards maltopentaose (G5) and G6 due to its substrate and product specificities [12]. Therefore, we hypothesized that these physicochemical properties could be speculated from the template 6ag0.1.A (chain A of PDB ID: 6ag0) through the superimposition and computation of RMSD.
2.5. Superimposition of 6ag0.1.A, WT and rc SR74 α-Amylases
The template 6ag0.1.A, WT and rc SR74 α-amylases were superimposed and the pairwise RMSD was computed using PyMOL (Figure 4). The pairwise RMSD (excluding outliers) of 0.066 and 0.067 was recorded when 6ag0.1.A was superimposed with WT and rc SR74 α-amylases, respectively. These results were justified when a few residues in 6ag0.1.A simultaneously variated from the SR74 α-amylases in the multiple sequence alignment (MSA) among three polypeptides (Figure S2). The identities of 6ag0.1.A with WT and rc SR74 α-amylases were computed to be 98.45% and 97.28%, respectively, reasoned from both the variant and mutation sites. However, it is noteworthy that the RMSD increased when outliers were included in its computation (Table S3). Moreover, based on the predicted secondary structures mapped onto the MSA of three proteins (Figure S2), the β10, β11 and β23 of the template 6ag0.1.A were slightly shorter than our proteins of interest. Therefore, without the need to perform in vitro biochemical tests, the physicochemical properties (thermostability) of rc SR74 α-amylase were better inferred from WT SR74 α-amylase compared to the template 6ag0.1.A due to its lower RMSD (0.001) computed and discussed earlier. However, the substrate and product specificities of rc SR74 α-amylase can possibly be deduced from the template 6ag0.1.A since the non-conserved residues between our enzyme and the template did not involve any catalytic triad and calcium ion-interacting residues (Figure S2).
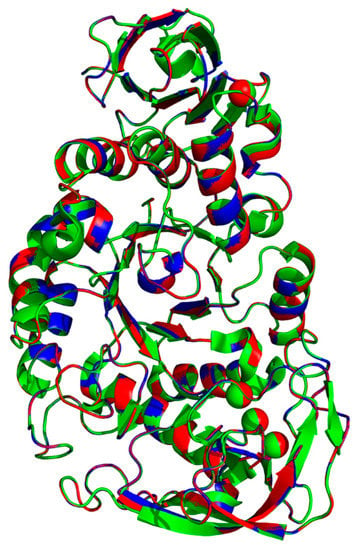
Figure 4.
Superimposition of template 6ag0.1.A, WT and rc SR74 α-amylases. All polypeptides are colour-coded: 6ag0.1.A (green), WT (blue) and rc SR74 α-amylases (red). The pairwise root mean square deviation (RMSD) among three polypeptides were computed with and without the elimination of outliers.
3. Materials and Methods
3.1. Acquisition of Nucleotide and Amino Acid Sequences of SR74 α-Amylases
Nucleotide information of Geobacillus sp. SR74 amylase was retrieved from GenBank (accession number: FJ997644.1). Nucleotide sequences were translated into amino acid sequences using ExPASy server (web.expasy.org/translate/). “Standard” and “Alternative yeast nuclear” genetic codes were applied for SR74 α-amylases expressed from the wild-type Geobacillus sp. SR74 (WT) and CTG clade yeast Meyerozyma guilliermondii strain SO (rc), respectively. SignalP 5.0 server (www.cbs.dtu.dk/services/SignalP/) was used to identify the signal peptide in SR74 α-amylase. The physicochemical parameters of SR74 α-amylases (WT and rc) were estimated and computed using ProtParam tool (web.expasy.org/protparam/). Pairwise alignment of the WT versus rc was conducted using Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo/) to determine the mutation (Leu to Ser) sites.
3.2. Sequence and Structural Analysis of SR74 α-Amylases
Functional annotations on sequences of SR74 α-amylases were searched against and inferred from UniProtKB (www.uniprot.org/uniprot/). Multiple sequence alignment (MSA) of SR74 α-amylases (WT and rc) and template 6ag0.1.A (chain A of PDB ID: 6ag0) [12] was generated using Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo/). Secondary structure information was predicted and mapped preliminary onto the alignment using ENDscript 2.0 (endscript.ibcp.fr/ESPript/ENDscript/) and ESPript 3.0 (espript.ibcp.fr/ESPript/ESPript/), respectively. Polar interactions between calcium ions were detected and viewed using PyMOL with the reference of template 6ag0.1.A [12].
3.3. In Silico 3D Structure Prediction of SR74 α-Amylases
Mature amino acid sequences of WT and rc SR74 α-amylases were used to predict and construct 3D structure in silico via SWISS-MODEL webserver (swissmodel.expasy.org/). The webserver [40,41] searched the template for evolutionary related protein structures against the SWISS-MODEL template library (SMTL) via BLAST [42] and HHblits [43]. Structures with the best primary global model quality estimate (GMQE) with a compromised qualitative model energy analysis (QMEAN) score were validated using Verify3D (servicesn.mbi.ucla.edu/Verify3D/) [44,45], ERRAT (servicesn.mbi.ucla.edu/ERRAT/) [46] and PROCHECK (servicesn.mbi.ucla.edu/PROCHECK/) [47,48]. All the predicted structure models were viewed using PyMOL Molecular Graphic System, Version 2.4 Schrödinger, LLC.
3.4. Superimposition of SR74 α-Amylases with Template 6ag0.1.A
Superimposition of SR74 α-amylases (WT and rc) with template 6ag0.1.A [12] was performed using PyMOL. Root mean square deviation (RMSD) between the possible pairs of proteins was computed in PyMOL with or without the elimination of outliers (cycles = 0, transform = 0).
4. Conclusions
While the M. guilliermondii strain SO is a CTG-clade yeast with CUG ambiguity, six possible mutations (L43S, L215S, L338S, L424S and L427S) were revealed in rc SR74 α-amylase. However, there was no significant structural difference observed between the WT and rc SR74 α-amylases. These results have been further justified with similar protein–ligand interactions in both structures. The superimposition among the SR74 α-amylase (WT and rc) and the template also suggested that the biochemical properties (especially thermostability) of rc SR74 α-amylase are preferably deduced from its WT without the needs to perform in vitro biochemical studies. To conclude, this in silico study has preliminarily proven that the CUG ambiguity in CTG-clade yeast M. guilliermondii strain SO has very limited effects on its functions and physicochemical properties, compared to its wild-type (Geobacillus sp. SR74). Future work involving molecular docking and dynamics simulation are proposed to elucidate and confirm its catalytic specificities (substrates and products) and time-dependent behaviours (fluctuations and conformation changes), respectively. These essential studies and the large-scale optimization of the recombinant enzyme production in strain SO will allow rc SR74 α-amylase to function optimally for industrial application, especially in the food and detergent industries. It is also recommended to perform codon optimization prior to gene cloning in the future to maintain the nature of the enzyme if CUG codons are observed in the gene sequences.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/9/1059/s1, Figure S1: Secondary structure map of WT and rc SR74 α-amylases, Figure S2: Multiple sequence alignment (MSA) of the template 6ag0.1.A, WT and rc SR74 α-amylases, Table S1: Multiple sequence alignment (MSA) of the BLASTP results, Table S2: Validation of SR74 α-amylases 3D structures using Verify3D, ERRAT and PROCHECK, Table S3: Pairwise RMSD within template 6ag0.1.A, WT and rc SR74 α-amylases.
Author Contributions
Conceptualization, S.J.L. and S.N.O.; methodology, S.J.L.; investigation, S.J.L. and S.N.O.; writing—original draft preparation, S.J.L.; writing—review and editing, S.J.L., N.D.M.N., A.B.S., and S.N.O.; visualization, S.J.L.; supervision, S.N.O.; project administration, S.J.L. and S.N.O.; funding acquisition, S.N.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Universiti Putra Malaysia, Putra-IPS grant number GP-IPS/2017/9516700.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lim, S.J.; Hazwani-Oslan, S.N.; Oslan, S.N. Purification and Characterisation of Thermostable α-Amylases from Microbial Sources. BioResources 2020, 15, 2005–2029. [Google Scholar]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Janeček, Š.; Svensson, B.; MacGregor, E.A. α-Amylase: An enzyme specificity found in various families of glycoside hydrolases. Cell. Mol. Life Sci. 2014, 71, 1149–1170. [Google Scholar] [CrossRef]
- Du, R.; Song, Q.; Zhang, Q.; Zhao, F.; Kim, R.C.; Zhou, Z.; Han, Y. Purification and characterization of novel thermostable and Ca-independent α-amylase produced by Bacillus amyloliquefaciens BH072. Int. J. Biol. Macromol. 2018, 115, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Ban, X.; Gu, Z.; Li, C.; Hong, Y.; Cheng, L.; Li, Z. Structure-Based Engineering of a Maltooligosaccharide-Forming Amylase to Enhance Product Specificity. J. Agric. Food Chem. 2020, 68, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Karim, K.M.R.; Husaini, A.; Sing, N.N.; Sinang, F.M.; Roslan, H.A.; Hussain, H. Purification of an alpha amylase from Aspergillus flavus NSH9 and molecular characterization of its nucleotide gene sequence. 3 Biotech 2018, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Salleh, A.B.; Rahman, R.N.Z.R.A.; Chor Leow, T.; Oslan, S.N. Expression and characterization of geobacillus stearothermophilus sr74 recombinant α -amylase in pichia pastoris. Biomed. Res. Int. 2015, 2015, 529059. [Google Scholar] [CrossRef]
- Chai, K.P.; Othman, N.F.B.; Teh, A.H.; Ho, K.L.; Chan, K.G.; Shamsir, M.S.; Goh, K.M.; Ng, C.L. Crystal structure of Anoxybacillus α-amylase provides insights into maltose binding of a new glycosyl hydrolase subclass. Sci. Rep. 2016, 6, 23126. [Google Scholar] [CrossRef]
- Mehta, D.; Satyanarayana, T. Structural elements of thermostability in the maltogenic amylase of Geobacillus thermoleovorans. Int. J. Biol. Macromol. 2015, 79, 570–576. [Google Scholar] [CrossRef]
- Mehta, D.; Satyanarayana, T. Bacterial and archaeal α-amylases: Diversity and amelioration of the desirable characteristics for industrial applications. Front. Microbiol. 2016, 7, 1129. [Google Scholar] [CrossRef]
- Liao, S.-M.; Liang, G.; Zhu, J.; Lu, B.; Peng, L.-X.; Wang, Q.-Y.; Wei, Y.-T.; Zhou, G.-P.; Huang, R.-B. Influence of Calcium Ions on the Thermal Characteristics of α-amylase from Thermophilic Anoxybacillus sp. GXS-BL. Protein Pept. Lett. 2019, 26, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, Y.; Ban, X.; Zhang, Z.; Gu, Z.; Li, C.; Hong, Y.; Cheng, L.; Jin, T.; Li, Z. Crystal structure of a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04. Int. J. Biol. Macromol. 2019, 138, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Ban, X.; Gu, Z.; Li, C.; Hong, Y.; Cheng, L.; Li, Z. Insights into the thermostability and product specificity of a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04. Biotechnol. Lett. 2020, 42, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Kassaye, E.K. Molecular Cloning and Expression of a Thermostable α-Amylase From Geobacillus sp. Master’s Thesis, Universiti Putra Malaysia, Seri Kembangan, Malaysia, 2009. [Google Scholar]
- Nasir, N.S.M.; Leow, C.T.; Oslan, S.N.H.; Salleh, A.B.; Oslan, S.N. Molecular expression of a recombinant thermostable bacterial amylase from Geobacillus stearothermophilus SR74 using methanol-free Meyerozyma guilliermondii strain SO yeast system. BioResources 2020, 15, 3161–3172. [Google Scholar]
- Oslan, S.N.; Salleh, A.B.; Rahman, R.R.A.; Basri, M.; Leow, T.C. Locally isolated yeasts from Malaysia: Identification, phylogenetic study and characterization. Acta Bochim. Pol. 2012, 59, 225–229. [Google Scholar] [CrossRef]
- Oslan, S.N.; Salleh, A.B.; Rahman, R.N.Z.R.A.; Leow, T.C.; Sukamat, H.; Basri, M. A newly isolated yeast as an expression host for recombinant lipase. Cell. Mol. Biol. Lett. 2015, 20, 279–293. [Google Scholar] [CrossRef]
- Periyasamy, N.A. Purification and Characterization of Recombinant Thermostable T1 Lipase Expressed from Pichia pastoris. Bachelor’s Thesis, Universiti Putra Malaysia, Seri Kembangan, Malaysia, 2015. [Google Scholar]
- De Marco, L.; Epis, S.; Capone, A.; Martin, E.; Bozic, J.; Crotti, E.; Ricci, I.; Sassera, D. The genomes of four Meyerozyma caribbica isolates and novel insights into the Meyerozyma guilliermondii species complex. G3 Genes Genomes Genet. 2018, 8, 755–759. [Google Scholar] [CrossRef]
- Krassowski, T.; Coughlan, A.Y.; Shen, X.X.; Zhou, X.; Kominek, J.; Opulente, D.A.; Riley, R.; Grigoriev, I.V.; Maheshwari, N.; Shields, D.C.; et al. Evolutionary instability of CUG-Leu in the genetic code of budding yeasts. Nat. Commun. 2018, 9, 1997. [Google Scholar] [CrossRef]
- Romi, W.; Keisam, S.; Ahmed, G.; Jeyaram, K. Reliable differentiation of Meyerozyma guilliermondii from Meyerozyma caribbica by internal transcribed spacer restriction fingerprinting. BMC Microbiol. 2014, 14, 52. [Google Scholar] [CrossRef]
- Gomes, A.C.; Miranda, I.; Silva, R.M.; Moura, G.R.; Thomas, B.; Akoulitchev, A.; Santos, M.A.S. A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans. Genome Biol. 2007, 8, R206. [Google Scholar] [CrossRef]
- Ueda, T.; Suzuki, T.; Tokogawa, T.; Nishikawa, K.; Watanabe, K. Unique structure of new serine tRNAs responsible for decoding leucine codon CUG in various Candida species and their putative ancestral tRNA genes. Biochimie 1994, 76, 1217–1222. [Google Scholar] [CrossRef]
- Massey, S.E.; Moura, G.; Beltrão, P.; Almeida, R.; Garey, J.R.; Tuite, M.F.; Santos, M.A.S. Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG condon in Candida spp. Genome Res. 2003, 13, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, A.R.; Simões, J.; Lee, W.; Rung, J.; Weil, T.; Gut, I.G.; Gut, M.; Bayés, M.; Rizzetto, L.; Cavalieri, D.; et al. Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc. Natl. Acad. Sci. USA 2013, 110, 11079–11084. [Google Scholar] [CrossRef] [PubMed]
- Fraga, J.S.; Sárkány, Z.; Silva, A.; Correia, I.; Pereira, P.J.B.; Macedo-Ribeiro, S. Genetic code ambiguity modulates the activity of a C. albicans MAP kinase linked to cell wall remodeling. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Simões, J.; Bezerra, A.R.; Moura, G.R.; Araújo, H.; Gut, I.; Bayes, M.; Santos, M.A.S. The fungus Candida albicans tolerates ambiguity at multiple codons. Front. Microbiol. 2016, 7, 401. [Google Scholar] [CrossRef] [PubMed]
- Dujon, B. Yeast evolutionary genomics. Nat. Rev. Genet. 2010, 11, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Satyanarayana, T. Domain C of thermostable α-amylase of Geobacillus thermoleovorans mediates raw starch adsorption. Appl. Microbiol. Biotechnol. 2014, 98, 4503–4519. [Google Scholar] [CrossRef]
- Pan, S.; Gu, Z.; Ding, N.; Zhang, Z.; Chen, D.; Li, C.; Hong, Y.; Cheng, L.; Li, Z. Calcium and sodium ions synergistically enhance the thermostability of a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04. Food Chem. 2019, 15, 170–176. [Google Scholar] [CrossRef]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Jones, T.A. Phi/Psi-chology: Ramachandran revisited. Structure 1996, 4, 1395–1400. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. In International Tables for Crystallography; Arnold, E., Himmel, D.M., Rossmann, M.G., Eds.; Wiley: Hoboken, NJ, USA, 2012; pp. 684–687. [Google Scholar]
- Rueda, M.; Orozco, M.; Totrov, M.; Abagyan, R. BioSuper: A web tool for the superimposition of biomolecules and assemblies with rotational symmetry. BMC Struct. Biol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed]
- Chen, Y.H.; Chuang, L.Y.; Lo, H.F.; Hu, H.Y.; Wu, T.J.; Lin, L.L.; Chi, M.C. Mutational analysis of the proposed calcium-binding aspartates of a truncated a-amylase from Bacillus sp. strain TS-23. Ann. Microbiol. 2010, 60, 307–315. [Google Scholar] [CrossRef]
- Priyadharshini, R.; Gunasekaran, P. Site-directed mutagenesis of the calcium-binding site of α-amylase of Bacillus licheniformis. Biotechnol. Lett. 2007, 29, 1493–1499. [Google Scholar] [CrossRef]
- Offen, W.A.; Viksoe-Nielsen, A.; Borchert, T.V.; Wilson, K.S.; Davies, G.J. Three-dimensional structure of a variant “Termamyl-like” Geobacillus stearothermophilus α-amylase at 1.9Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 66–70. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).