DMC-Mediated Copolymerization of CO2 and PO—Mechanistic Aspects Derived from Feed and Polymer Composition

Abstract
:
1. Introduction
2. Results
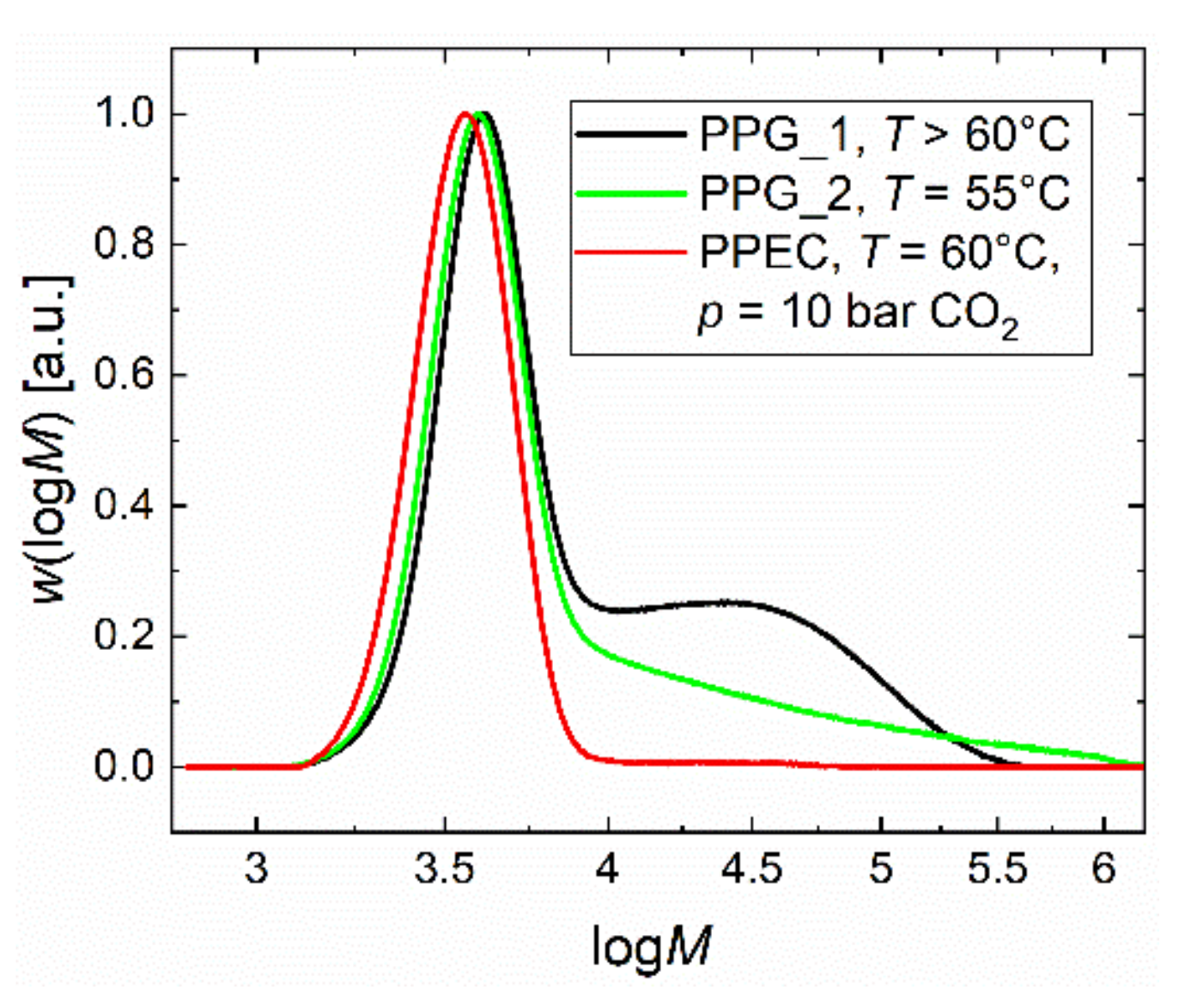
2.1. Homopolymerization of PO and Copolymerization with CO2 in Batch Processes
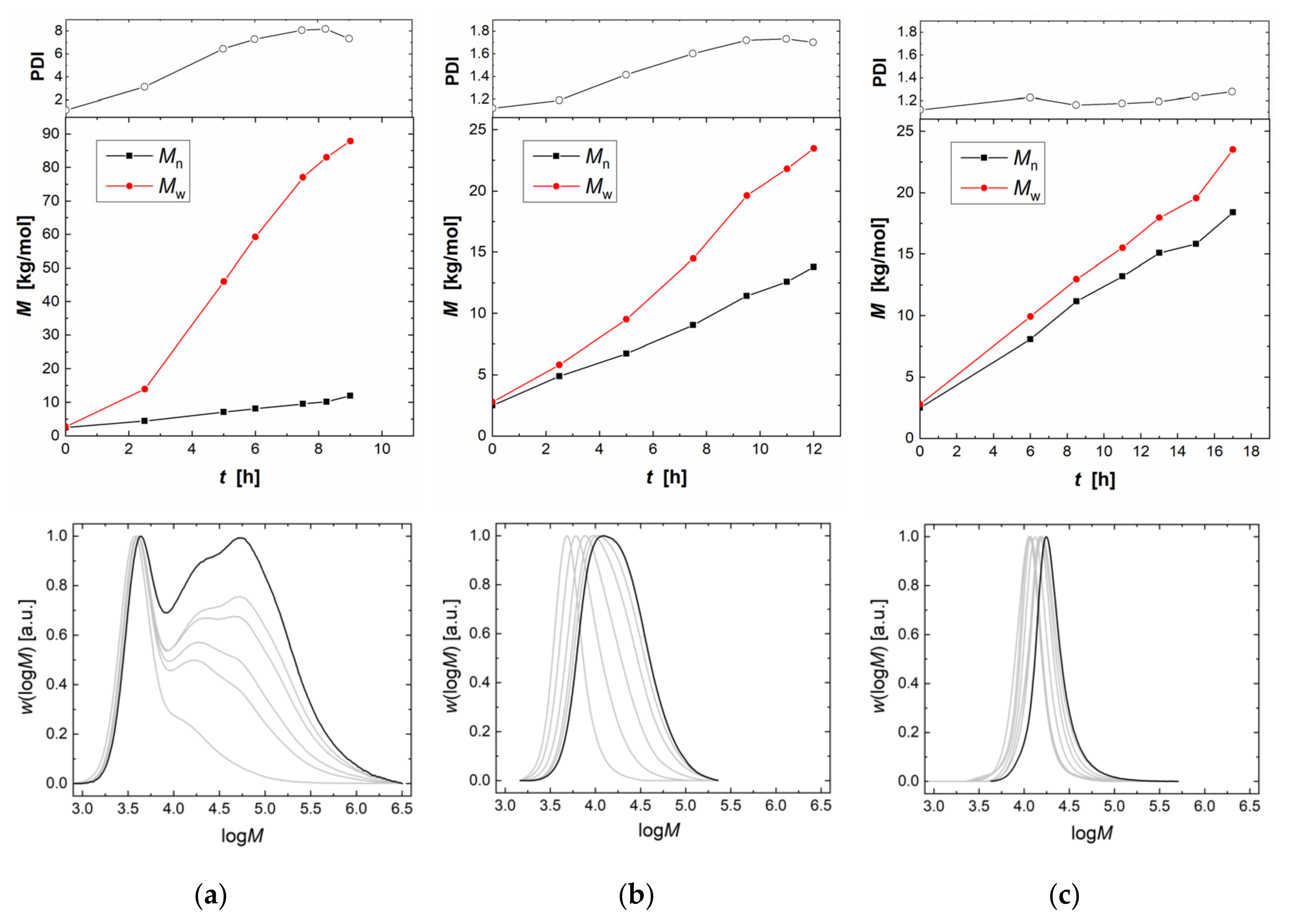
2.2. Copolymerization of CO2 and PO
2.3. Formation of Cyclic Propylene Carbonate
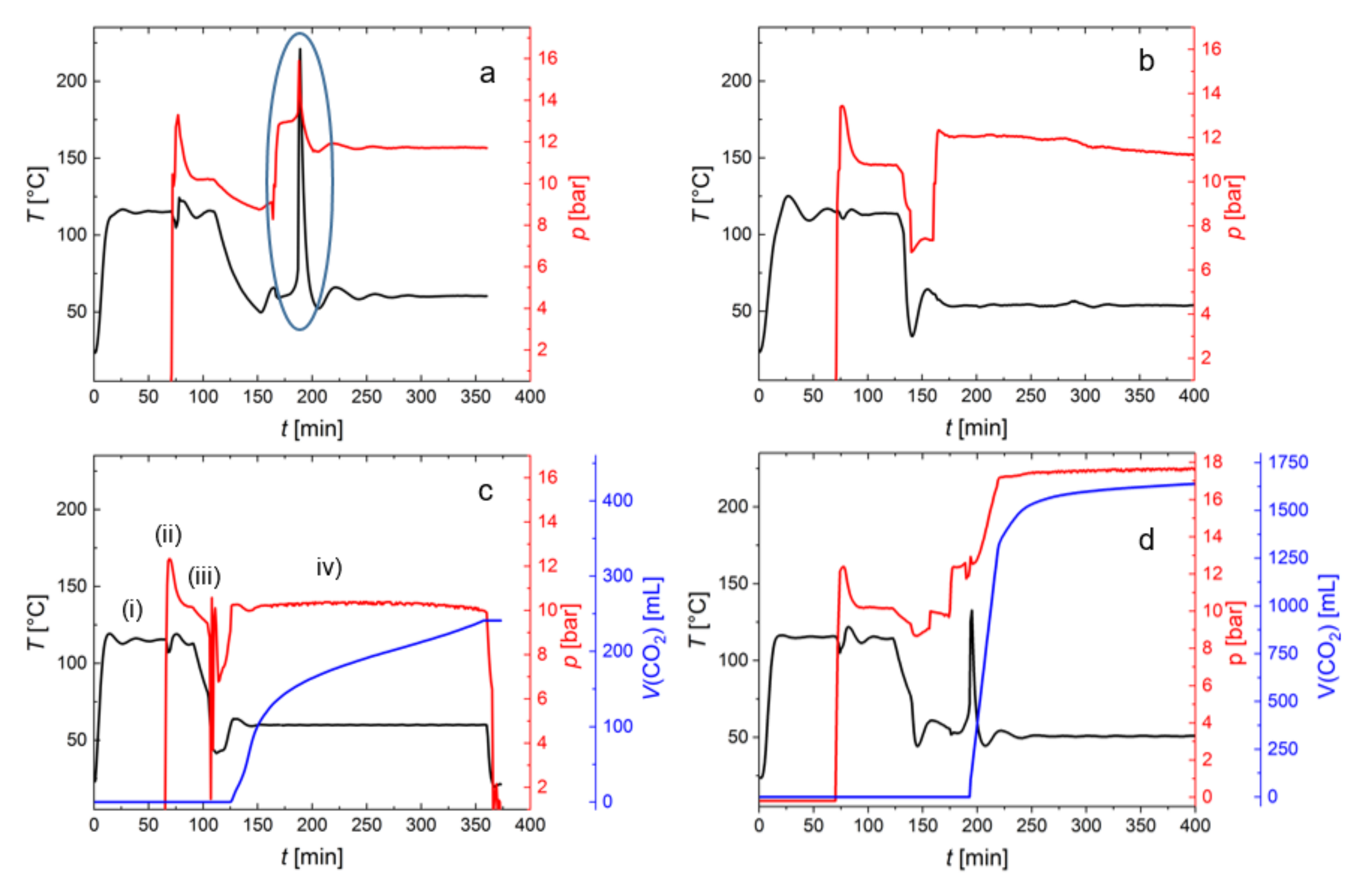
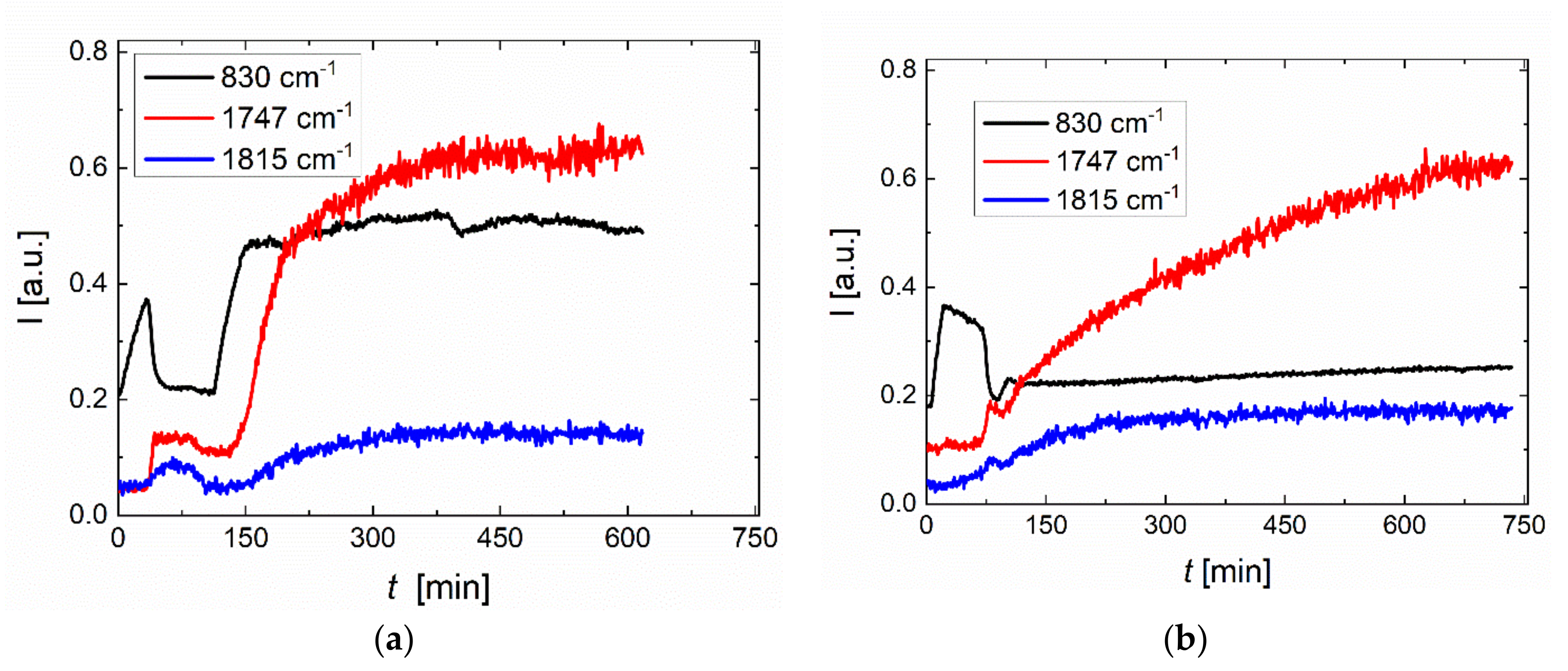
2.4. Process Monitoring of Copolymerization of PO with CO2
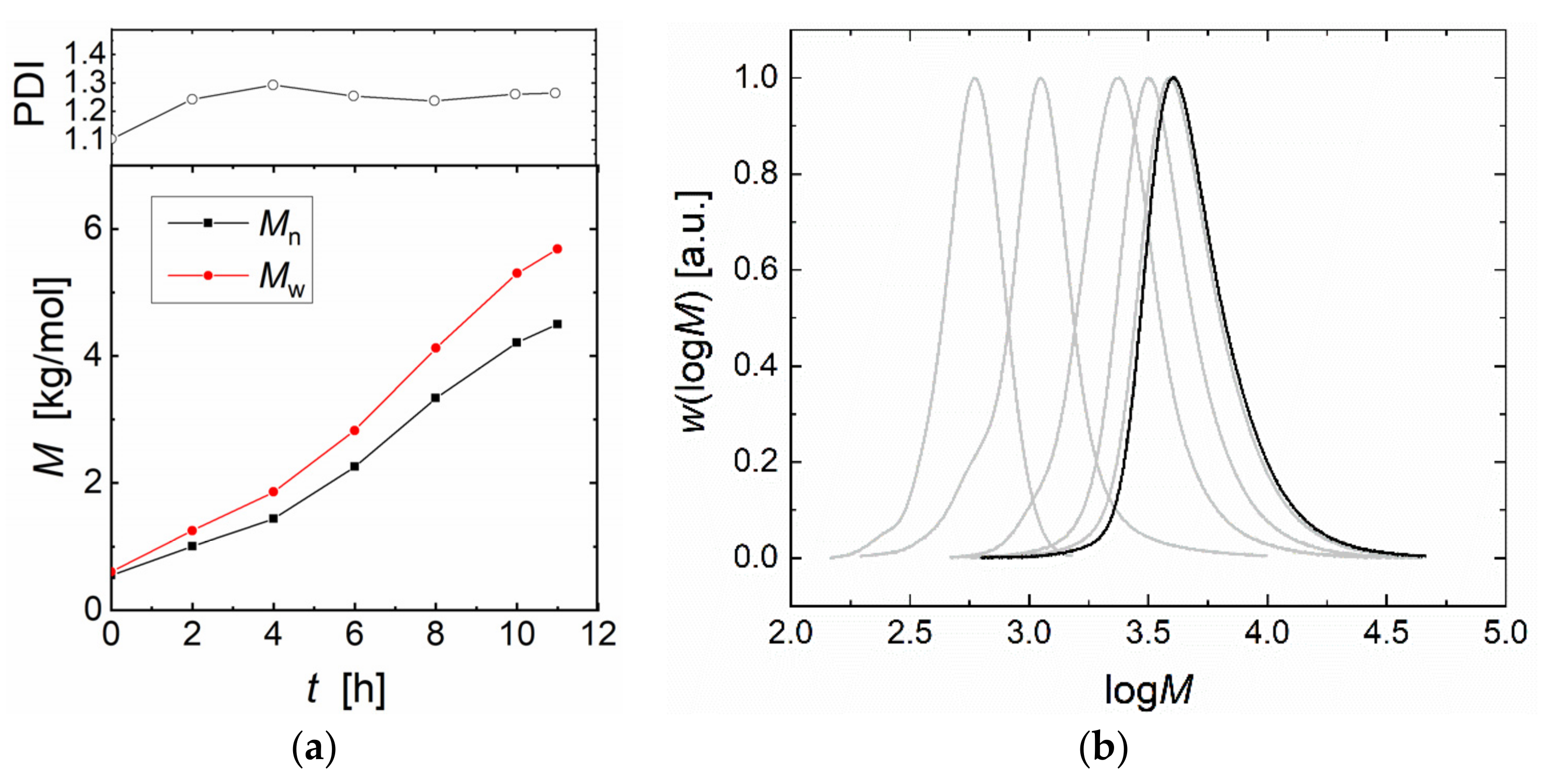
2.5. Amines as Cocatalyst/Conucleophile
2.6. CO2/PO Copolymerization with Ion-Exchanged DMC Surfaces
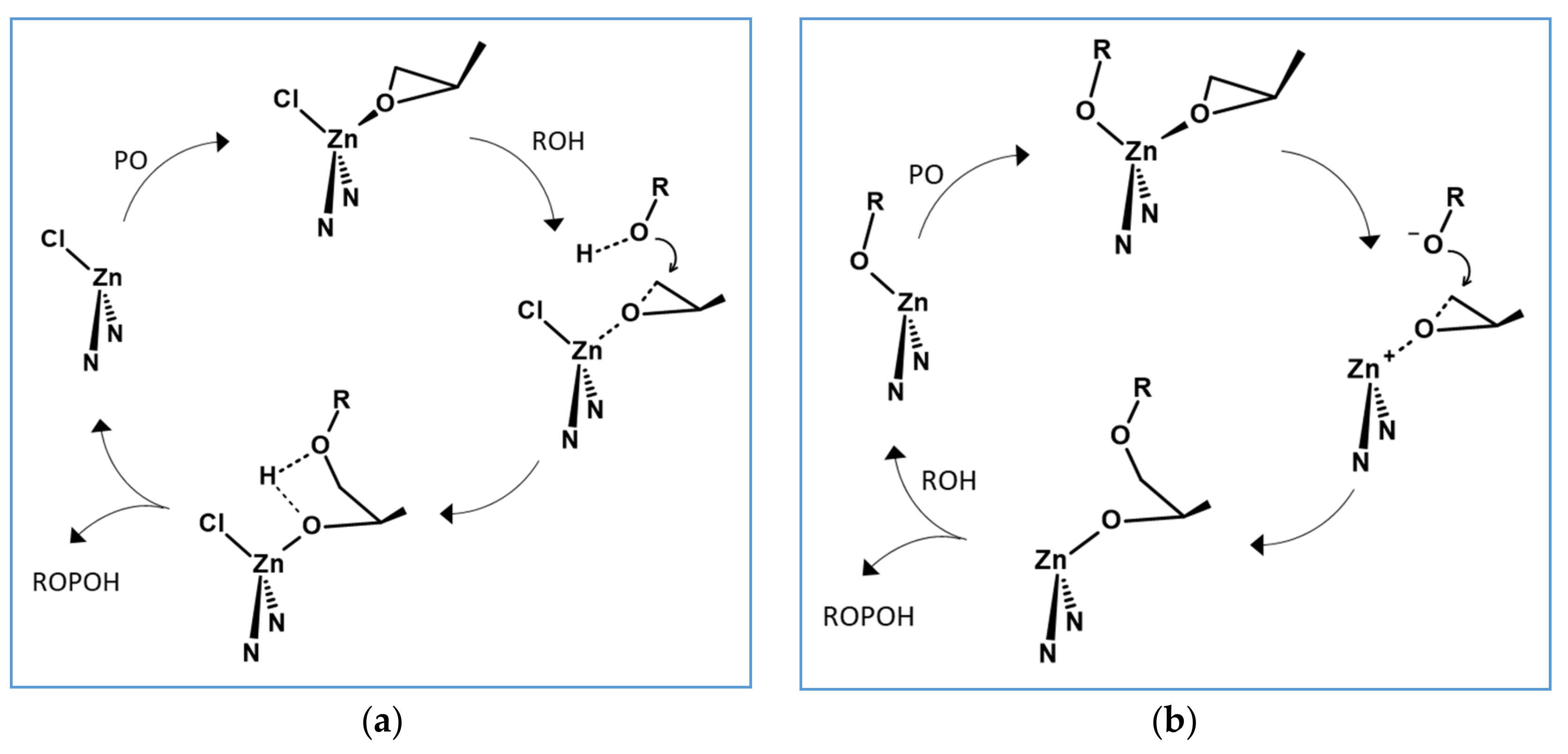
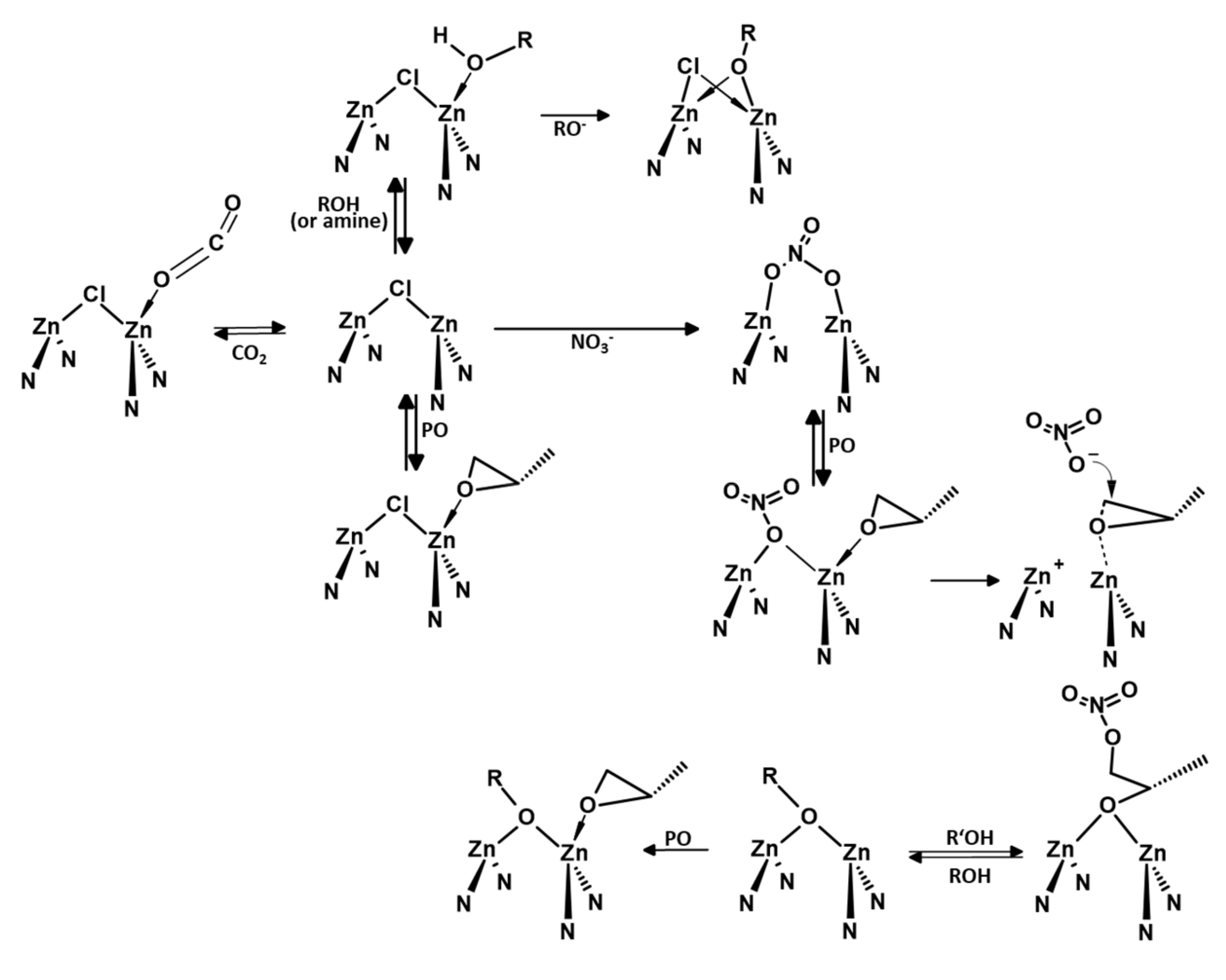
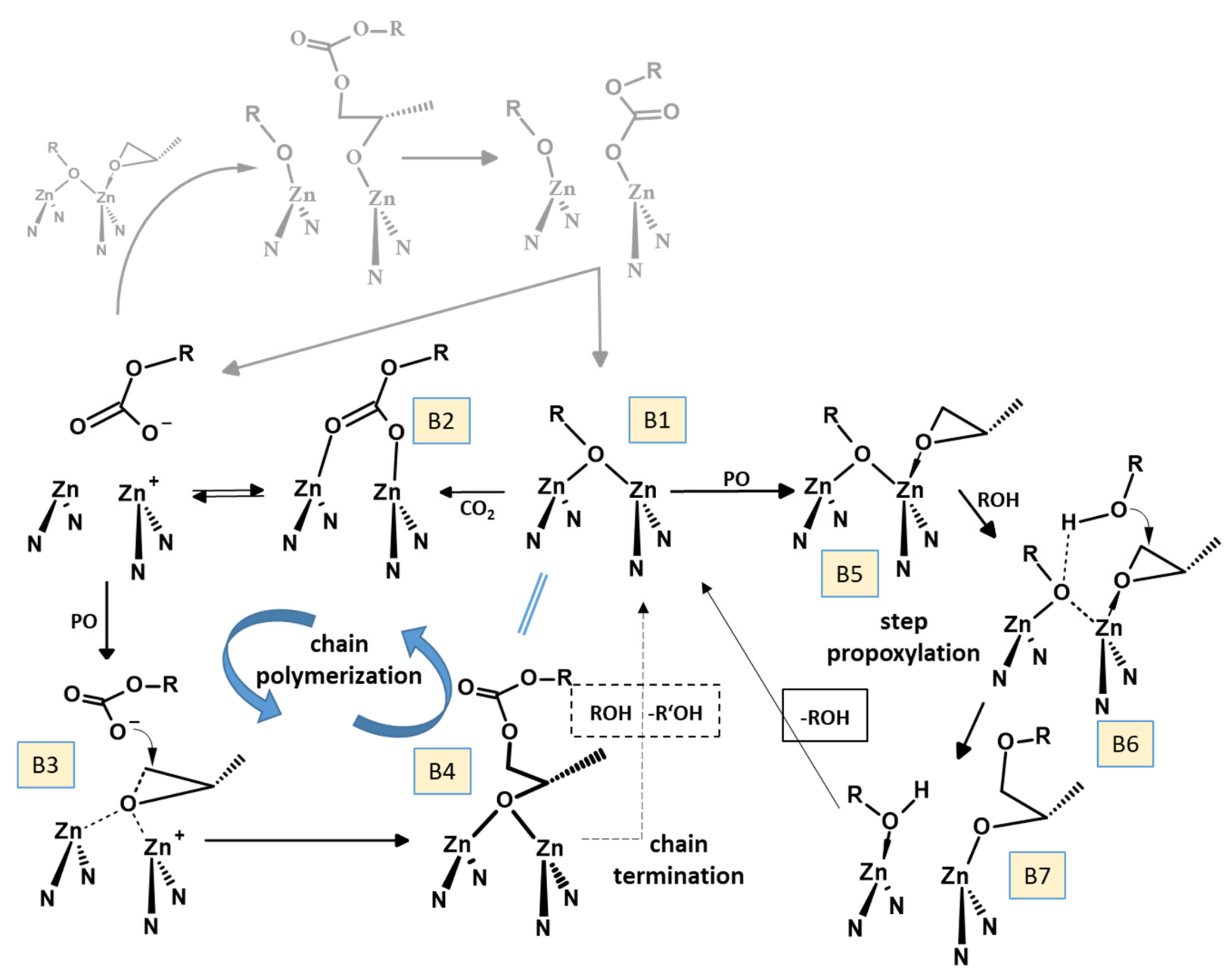
3. Mechanistic Discussion
4. Materials and Methods
4.1. Materials
4.2. Homopolymerization of PO
4.3. Copolymerization of CO2 and PO
4.4. Catalyst Characterization
4.5. Polymer Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Science to Enable Sustainable Plastics—A white paper from the 8th Chemical Sciences and Society Summit (CS3). Available online: https://www.rsc.org/globalassets/22-new-perspectives/sustainability/progressive-plastics/c19_tl_sustainability_cs3_whitepaper_a4_web_final.pdf (accessed on 15 September 2020).
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Alper, E.; Orhan, O.Y. CO2 utilization: Developments in conversion processes. Petroleum 2017, 3, 109–126. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chemie-Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef]
- Broecker, W.S. Thermohaline Circulation, the Achilles Heel of Our Climate System: Will Man-Made CO2 Upset the Current Balance? Science 1997, 278, 1582–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, H.; Folkert, R.J.M.; Hoekstra, J.; De Wolff, J.J. Identifying key sources of uncertainty in climate change projections. Clim. Chang. 2000, 45, 421–457. [Google Scholar] [CrossRef]
- Markewitz, P.; Kuckshinrichs, W.; Leitner, W.; Linssen, J.; Zapp, P.; Bongartz, R.; Schreiber, A.; Müller, T.E. Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 2012, 5, 7281–7305. [Google Scholar] [CrossRef] [Green Version]
- Lide, D.R. CRC Handbook of Chemistry and Physics; Internet V.; CRC Press: Boca Raton, FL, USA, 2005; ISBN 0849304792. [Google Scholar]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalt. Trans. 2007, 2975. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wilson, S.J. What’s new with CO2? Recent advances in its copolymerization with oxiranes. Green Chem. 2012, 14, 2665–2671. [Google Scholar] [CrossRef]
- Lu, X.B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef]
- Kim, G.; Ree, M.; Kim, H.; Kim, I.J.; Kim, J.R.; Lee, J.I. Biological affinity and biodegradability of poly(propylene carbonate) prepared from copolymerization of carbon dioxide with propylene oxide. Macromol. Res. 2008, 16, 473–480. [Google Scholar] [CrossRef]
- Ge, X.C.; Xu, Y.; Meng, Y.Z.; Li, R.K.Y. Thermal and mechanical properties of biodegradable composites of poly(propylene carbonate) and starch-poly(methyl acrylate) graft copolymer. Compos. Sci. Technol. 2005, 65, 2219–2225. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, X. Carbon dioxide-based copolymers: Environmental benefits of PPC, an industrially viable catalyst. Biotechnol. J. 2010, 5, 1164–1180. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Song, C.; Cao, M.; Hu, D.; Liu, L.; Liu, N.; Wang, S. Thermal properties and degradability of poly(propylene carbonate)/poly(hydroxybutyrate-co-hydroxyvalerate) (PPC/PHBV) blends. Polym. Degrad. Stab. 2009, 94, 575–583. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, L.; Wei, Y.; Wang, Y.; He, Y.; Xia, Q.; Zhang, X.; Liu, Z. A new route for the synthesis of propylene oxide from bio-glycerol derivated propylene glycol. Chem. Commun. 2009, 3934–3936. [Google Scholar] [CrossRef] [PubMed]
- Poland, S.J.; Darensbourg, D.J. A quest for polycarbonates provided: Via sustainable epoxide/CO2 copolymerization processes. Green Chem. 2017, 19, 4990–5011. [Google Scholar] [CrossRef]
- Gao, F.; Zhou, Q.; Dong, Y.; Qin, Y.; Wang, X.; Xiaojiang, Z.; Fosong, W. Ether linkage in poly(1,2-propylene carbonate), a key structure factor to tune its performances. J. Polym. Res. 2012, 19, 2–6. [Google Scholar] [CrossRef]
- Wang, D.; Yu, J.; Zhang, J.; He, J.; Zhang, J. Transparent bionanocomposites with improved properties from poly(propylene carbonate) (PPC) and cellulose nanowhiskers (CNWs). Compos. Sci. Technol. 2013, 85, 83–89. [Google Scholar] [CrossRef]
- Luinstra, G.A.; Borchardt, E. Material Properties of Poly(Propylene Carbonates). Adv. Polym. Sci. 2012, 245, 29–48. [Google Scholar] [CrossRef]
- Huihong, L.; Lisha, P.; Qiang, L.; Nai, X.; Lingbin, L.; Sujuan, P.; Songbao, F. Preparation and characterization of poly (propylene carbonate) hydroxy butyrate-co-hydroxyvalerate PHBv composite films by tubular film blowing process. e-Polymers 2010, 38. [Google Scholar] [CrossRef]
- DeBolt, M.; Kiziltas, A.; Mielewski, D.; Waddington, S.; Nagridge, M.J. Flexible polyurethane foams formulated with polyols derived from waste carbon dioxide. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Miao, Y.; Qiao, L.; Wang, X.; Wang, F. Waterborne polyurethanes from CO2 based polyols with comprehensive hydrolysis/oxidation resistance. Green Chem. 2016, 18, 524–530. [Google Scholar] [CrossRef]
- Alagi, P.; Ghorpade, R.; Choi, Y.J.; Patil, U.; Kim, I.; Baik, J.H.; Hong, S.C. Carbon Dioxide-Based Polyols as Sustainable Feedstock of Thermoplastic Polyurethane for Corrosion-Resistant Metal Coating. ACS Sustain. Chem. Eng. 2017, 5, 3871–3881. [Google Scholar] [CrossRef]
- Lee, S.H.; Cyriac, A.; Jeon, J.Y.; Lee, B.Y. Preparation of thermoplastic polyurethanes using in situ generated poly(propylene carbonate)-diols. Polym. Chem. 2012, 3, 1215. [Google Scholar] [CrossRef]
- Xu, C.; Cai, Z.; Xing, J.; Ren, Y.; Xu, W.; Shi, W. Synthesis of polypropylene carbonate polyol-based waterborne polyurethane modified with polysiloxane and its film properties. Fibers Polym. 2014, 15, 665–671. [Google Scholar] [CrossRef]
- Korashvili, R.; Nörnberg, B.; Bornholdt, N.; Borchardt, E.; Luinstra, G.A. Poly(propylencarbonat) aus Kohlendioxid: Herausforderungen für eine großtechnische Umsetzung. Chem. Ing. Tech. 2013, 85, 437–446. [Google Scholar] [CrossRef]
- Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M.A.; Müller, T.E.; Leitner, W.; Gürtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Luinstra, G. Poly(Propylene Carbonate), Old Copolymers of Propylene Oxide and Carbon Dioxide with New Interests: Catalysis and Material Properties. Polym. Rev. 2008, 48, 192–219. [Google Scholar] [CrossRef]
- Marbach, J.; Nörnberg, B.; Rahlf, A.F.; Luinstra, G.A. Zinc glutarate-mediated copolymerization of CO2 and PO—Parameter studies using design of experiments. Catal. Sci. Technol. 2017, 7, 2897–2905. [Google Scholar] [CrossRef]
- Sakharov, A.M.; Il’in, V.V.; Rusak, V.V.; Nysenko, Z.N.; Klimov, S.A. Copolymerization of propylene oxide with carbon dioxide catalyzed by zinc adipate. Russ. Chem. Bull. 2002, 51, 1451–1454. [Google Scholar] [CrossRef]
- Tan, C.; Hsu, T. Alternating Copolymerization of Carbon Dioxide and Propylene Oxide with a Rare-Earth-Metal Coordination Catalyst. Macromolecules 1997, 30, 3147–3150. [Google Scholar] [CrossRef]
- Soga, K.; Hyakkoku, K.; Izumi, K.; Ikeda, S. Alternating Copolymerization of Propylene Oxide and Carbon Dioxide With an Alumina Supported Diethylzinc Catalyst. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 2383–2392. [Google Scholar] [CrossRef]
- Jintang, D.; Jiajun, W.; Lianfang, F.; Long, W.; Xueping, G. Pressure dependence of the CO2/propylene oxide copolymerization catalyzed by zinc glutarate. J. Appl. Polym. Sci. 2010, 118, 366–371. [Google Scholar] [CrossRef]
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerisation of Carbon Dioxide and Epoxide. J. Polym. Sci. Part B Polym. Lett. 1969, 7, 287–292. [Google Scholar] [CrossRef]
- S, S.; Min, J.K.; Seong, J.E.; Na, S.J.; Lee, B.Y. A highly active and recyclable catalytic system for CO2/propylene oxide copolymerization. Angew. Chem.-Int. Ed. 2008, 47, 7306–7309. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-M.; Liu, Y.; Wu, G.-P.; Liu, J.; Lu, X.-B. Stereoregular polycarbonate synthesis: Alternating copolymerization of CO2 with aliphatic terminal epoxides catalyzed by multichiral cobalt(III) complexes. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 4894–4901. [Google Scholar] [CrossRef]
- Konieczynska, M.D.; Lin, X.; Zhang, H.; Grinstaff, M.W. Synthesis of Aliphatic Poly(ether 1,2-glycerol carbonate)s via Copolymerization of CO 2 with Glycidyl Ethers Using a Cobalt Salen Catalyst and Study of a Thermally Stable Solid Polymer Electrolyte. ACS Macro Lett. 2015, 533–537. [Google Scholar] [CrossRef]
- Allen, S.D.; Moore, D.R.; Lobkovsky, E.B.; Coates, G.W. High-activity, single-site catalysts for the alternating copolymerization of CO2 and propylene oxide. J. Am. Chem. Soc. 2002, 124, 14284–14285. [Google Scholar] [CrossRef]
- Piesik, D.F.J.; Range, S.; Harder, S. Bimetallic calcium and zinc complexes with bridged β-diketiminate ligands: Investigations on epoxide/CO2 copolymerization. Organometallics 2008, 27, 6178–6187. [Google Scholar] [CrossRef]
- Cheng, M.; Moore, D.R.; Reczek, J.J.; Chamberlain, B.M.; Lobkovsky, E.B.; Coates, G.W. Single-site β-diiminate zinc catalysts for the alternating copolymerization of CO2 and epoxides: Catalyst synthesis and unprecedented polymerization activity. J. Am. Chem. Soc. 2001, 123, 8738–8749. [Google Scholar] [CrossRef]
- Moore, D.R.; Cheng, M.; Lobkovsky, E.B.; Coates, G.W. Mechanism of the alternating copolymerization of epoxides and CO2 using β-diiminate zinc catalysts: Evidence for a bimetallic epoxide enchainment. J. Am. Chem. Soc. 2003, 125, 11911–11924. [Google Scholar] [CrossRef]
- Sheng, X.; Wu, W.; Qin, Y.; Wang, X.; Wang, F. Efficient synthesis and stabilization of poly(propylene carbonate) from delicately designed bifunctional aluminum porphyrin complexes. Polym. Chem. 2015, 6, 4719–4724. [Google Scholar] [CrossRef]
- Anderson, C.E.; Vagin, S.I.; Xia, W.; Jin, H.; Rieger, B. Cobaltoporphyrin-Catalyzed CO2/Epoxide Copolymerization: Selectivity Control by Molecular Design. Macromolecules 2012, 45, 6840–6849. [Google Scholar] [CrossRef]
- Chatterjee, C.; Chisholm, M.H. The Influence of the Metal (Al, Cr, and Co) and the Substituents of the Porphyrin in Controlling the Reactions Involved in the Copolymerization of Propylene Oxide and Carbon Dioxide by Porphyrin Metal (III) Complexes. 1. Aluminum Chemistry. Inorg. Chem. 2011, 50, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Kuroda, K. The Cobalt Porphyrin−Lewis Base System: A Highly Selective Catalyst for Alternating Copolymerization of CO2 and Epoxide under Mild Conditions. Macromolecules 2008, 41, 312–317. [Google Scholar] [CrossRef]
- Luinstra, G.A.; Haas, G.R.; Molnar, F.; Bernhart, V.; Eberhardt, R.; Rieger, B. On the formation of aliphatic polycarbonates from epoxides with chromium(III) and aluminum(III) metal-salen complexes. Chem.-A Eur. J. 2005, 11, 6298–6314. [Google Scholar] [CrossRef]
- Kember, M.R.; Buchard, A.; Williams, C.K. Catalysts for CO2/epoxide copolymerisation. Chem. Commun. 2011, 47, 141–163. [Google Scholar] [CrossRef] [Green Version]
- Trott, G.; Saini, P.K.; Williams, C.K. Catalysts for CO2/epoxide copolymerization. Phil. Trans. R. Soc. A 2016, 374–392. [Google Scholar] [CrossRef] [Green Version]
- Asano, S.; Aida, T.; Inoue, S. “Immortal” polymerization. Polymerization of epoxide catalysed by an aluminium porphyrin-alcohol system. J. Chem. Soc. Chem. Commun. 1985, 1148–1149. [Google Scholar] [CrossRef]
- Inoue, S. Immortal polymerization: The outset, development, and application. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2861–2871. [Google Scholar] [CrossRef]
- Liu, S.; Qin, Y.; Chen, X.; Wang, X.; Wang, F. One-pot controllable synthesis of oligo(carbonate-ether) triol using a Zn-Co-DMC catalyst: The special role of trimesic acid as an initiation-transfer agent. Polym. Chem. 2014, 5, 6171–6179. [Google Scholar] [CrossRef]
- Ma, K.; Bai, Q.; Zhang, L.; Liu, B. Synthesis of flame-retarding oligo(carbonate-ether) diols: Via double metal cyanide complex-catalyzed copolymerization of PO and CO2 using bisphenol A as a chain transfer agent. RSC Adv. 2016, 6, 48405–48410. [Google Scholar] [CrossRef]
- Liu, S.; Miao, Y.; Qiao, L.; Qin, Y.; Wang, X.; Chen, X.; Wang, F. Controllable synthesis of a narrow polydispersity CO2-based oligo(carbonate-ether) tetraol. Polym. Chem. 2015, 6, 7580–7585. [Google Scholar] [CrossRef]
- Clayman, N.E.; Morris, L.S.; LaPointe, A.M.; Keresztes, I.; Waymouth, R.M.; Coates, G.W. Dual catalysis for the copolymerisation of epoxides and lactones. Chem. Commun. 2019, 55, 6914–6917. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Chain transfer agents utilized in epoxide and CO2 copolymerization processes. Green Chem. 2019, 21, 2214–2223. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.Y.; Hu, L.F.; Zhang, X.H.; Du, B.Y.; Xu, J.T. Carbon dioxide-based copolymers with various architectures. Prog. Polym. Sci. 2018, 82, 120–157. [Google Scholar] [CrossRef]
- Sudakar, P.; Sivanesan, D.; Yoon, S. Copolymerization of Epichlorohydrin and CO2 Using Zinc Glutarate: An Additional Application of ZnGA in Polycarbonate Synthesis. Macromol. Rapid Commun. 2016, 37, 788–793. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kim, H.; Yoon, J.; Heo, K.; Ree, M. Synthesis of zinc glutarates with various morphologies using an amphiphilic template and their catalytic activities in the copolymerization of carbon dioxide and propylene oxide. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 4079–4088. [Google Scholar] [CrossRef]
- Ree, M.; Hwang, Y.; Kim, J.-S.; Kim, H.; Kim, G.; Kim, H. New findings in the catalytic activity of zinc glutarate and its application in the chemical fixation of CO2 into polycarbonates and their derivatives. Catal. Today 2006, 115, 134–145. [Google Scholar] [CrossRef]
- Klaus, S.; Lehenmeier, M.W.; Herdtweck, E.; Deglmann, P.; Ott, A.K.; Rieger, B. Mechanistic Insights into Heterogeneous Zinc Dicarboxylates and Theoretical Considerations for CO2–Epoxide Copolymerization. J. Am. Chem. Soc. 2011, 133, 13151–13161. [Google Scholar] [CrossRef]
- Pan, X.; Liu, Z.; Cheng, R.; He, X.; Liu, B. Insight into the reaction mechanisms between CO2 and epoxides over Zn(II) phenoxide catalytic system—A DFT study. J. Organomet. Chem. 2015, 775, 67–75. [Google Scholar] [CrossRef]
- Chen, S.; Xiao, Z.; Ma, M. Copolymerization of carbon dioxide and epoxides with a novel effective Zn-Ni double-metal cyanide complex. J. Appl. Polym. Sci. 2008, 107, 3874–3877. [Google Scholar] [CrossRef]
- Zhang, X.H.; Wei, R.J.; Zhang, Y.Y.; Du, B.Y.; Fan, Z.Q. Carbon dioxide/epoxide copolymerization via a nanosized zinc-cobalt(III) double metal cyanide complex: Substituent effects of epoxides on polycarbonate selectivity, regioselectivity and glass transition temperatures. Macromolecules 2015, 48, 536–544. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Lin, F.; Qi, G. Preparation of double metal cyanide compleces from water-insoluble zinc compounds and their catalytic performance for copolymerization of epoxide and CO2. React. Kinet. Catal. Lett. 2007, 91, 69–75. [Google Scholar] [CrossRef]
- Gao, Y.; Gu, L.; Qin, Y.; Wang, X.; Wang, F. Dicarboxylic acid promoted immortal copolymerization for controllable synthesis of low-molecular weight oligo(carbonate-ether) diols with tunable carbonate unit content. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 5177–5184. [Google Scholar] [CrossRef]
- Meng, Q.; Cheng, R.; Li, J.; Wang, T.; Liu, B. Copolymerization of CO2 and propylene oxide using ZnGA/DMC composite catalyst for high molecular weight poly(propylene carbonate). J. CO2 Util. 2016, 16, 86–96. [Google Scholar] [CrossRef]
- Marbach, J.; Höfer, T.; Bornholdt, N.; Luinstra, G.A. Catalytic Chain Transfer Copolymerization of Propylene Oxide and CO2 using Zinc Glutarate Catalyst. ChemistryOpen 2019, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Wojdeł, J.C.; Bromley, S.T.; Illas, F.; Jansen, J.C. Development of realistic models for Double Metal Cyanide catalyst active sites. J. Mol. Model. 2007, 13, 751–756. [Google Scholar] [CrossRef]
- Almora-Barrios, N.; Pogodin, S.; Bellarosa, L.; García-Melchor, M.; Revilla-Lõpez, G.; García-Ratés, M.; Vázquez-García, A.B.; Hernández-Ariznavarreta, P.; Lõpez, N. Structure, activity, and deactivation mechanisms in double metal cyanide catalysts for the production of polyols. ChemCatChem 2015, 7, 928–935. [Google Scholar] [CrossRef]
- Lawniczak-Jablonska, K.; Dynowska, E.; Lisowski, W.; Sobczak, J.W.; Chruściel, A.; Hreczuch, W.; Libera, J.; Reszka, A. Structural properties and chemical bonds in double metal cyanide catalysts. X-Ray Spectrom. 2015, 44, 330–338. [Google Scholar] [CrossRef]
- Robertson, N.J.; Qin, Z.; Dallinger, G.C.; Lobkovsky, E.B.; Lee, S.; Coates, G.W. Two-dimensional double metal cyanide complexes: Highly active catalysts for the homopolymerization of propylene oxide and copolymerization of propylene oxide and carbon dioxide. Dalt. Trans. 2006, 5390–5395. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Moore, D.R. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: Discovery, reactivity, optimization, and mechanism. Angew. Chem.-Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ahn, J.T.; Lee, S.H.; Ha, C.S.; Park, D.W. Preparation of multi-metal cyanide catalysts and ring-opening polymerization of propylene oxide. Catal. Today 2004, 93–95, 511–516. [Google Scholar] [CrossRef]
- McDaniel, K.G. High Productivity Alkoxylation Processes. EP 1942126 A1, 9 July 2008. [Google Scholar]
- Shakhova, S.F.; Rutenberg, O.L.; Targanskaya, M.N. Liquid-gas equilibrium in the Epoxypropane-Carbon Dioxide system. Russ. J. Phys. Chem. 1973, 47, 895–896. [Google Scholar]
- Wang, J.; Duan, J.; Wang, L.; Gu, X.; Feng, L. Measurement and Modeling of the High-Pressure Phase Behavior of the Carbon Dioxide + Propene Oxide Binary System. J. Chem. Eng. Data 2010, 55, 3379–3382. [Google Scholar] [CrossRef]
- Peng, D.Y.; Robinson, D.B. A New Two-Constant Equation of State. Ind. Eng. Chem. Fundam. 1976, 15, 59–64. [Google Scholar] [CrossRef]
- Li, X.H.; Meng, Y.Z.; Zhu, Q.; Tjong, S.C. Thermal decomposition characteristics of poly(propylene carbonate) using TG/IR and Py-GC/MS techniques. Polym. Degrad. Stab. 2003, 81, 157–165. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yeung, A.D. Thermodynamics of the carbon dioxide-epoxide copolymerization and kinetics of the metal-free degradation: A computational study. Macromolecules 2013, 46, 83–95. [Google Scholar] [CrossRef]
- Nörnberg, B.; Spottog, C.; Rahlf, A.; Korashvili, R.; Berlin, C.; Luinstra, G.A. Semi-batch copolymerization of propylene oxide and carbon dioxide. Macromol. Symp. 2013, 333, 190–196. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yarbrough, J.C.; Ortiz, C.; Fang, C.C. Comparative kinetic studies of the copolymerization of cyclohexene oxide and propylene oxide with carbon dioxide in the presence of chromium salen derivatives. In situ FTIR measurements of copolymer vs cyclic carbonate production. J. Am. Chem. Soc. 2003, 125, 7586–7591. [Google Scholar] [CrossRef]
- Dixon, D.D.; Ford, M.E.; Mantell, G.J. Thermal stabilization of poly(alkylene carbonate)s. J. Polym. Sci. Polym. Lett. Ed. 1980, 18, 131. [Google Scholar] [CrossRef]
- Nörnberg, B.; Luinstra, G.A. Influence of norbornene dicarboxylic anhydride on the copolymerization of carbon dioxide and propylene oxide. Eur. Polym. J. 2015, 73, 297–307. [Google Scholar] [CrossRef]
- Barreto, C.; Hansen, E.; Fredriksen, S. Novel solventless purification of poly(propylene carbonate): Tailoring the composition and thermal properties of PPC. Polym. Degrad. Stab. 2012, 97, 893–904. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Holtcamp, M.W.; Struck, G.E.; Zimmer, M.S.; Niezgoda, S.A.; Rainey, P.; Robertson, J.B.; Draper, J.D.; Reibenspies, J.H. Catalytic activity of a series of Zn(II) phenoxides for the copolymerization of epoxides and carbon dioxide. J. Am. Chem. Soc. 1999, 121, 107–116. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wildeson, J.R.; Yarbrough, J.C.; Reibenspies, J.H. Bis 2,6-difluorophenoxide dimeric complexes of zinc and cadmium and their phosphine adducts: Lessons learned relative to carbon dioxide/cyclohexene oxide alternating copolymerization processes catalyzed by zinc phenoxides. J. Am. Chem. Soc. 2000, 122, 12487–12496. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wildeson, J.R.; Yarbrough, J.C. Solid-state structures of zinc(II) benzoate complexes. Catalyst precursors for the coupling of carbon dioxide and epoxides. Inorg. Chem. 2002, 41, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.; McDaniel, K.; Lenahan, R.; Gastinger, R.; Morrison, M. IMPACT Technology—A Greener Polyether Process. In Proceedings of the 13th Annual Green Chemistry & Engineering Conference, College Park, MD, USA, 23–25 June 2009; Bayer Material Science: Leverkusen, Germany, 2009. [Google Scholar]
- Pohl, M. Tailor-made Polyols for Sustainable Polyurethanes by Catalyzed Copolymerization of CO2 and Epoxides; RWTH Aachen: Aachen, Germany, 2017. [Google Scholar]
- Pazos, J.; Browne, E. Catch-up kinetics: Selectivity based on equivalent weight in the polymerization of alkylene oxides by double metal cyanide catalysts. Polym. Prepr. 2008, 49, 434–435. [Google Scholar]
- Lee, S.H.; Lee, I.K.; Ha, J.Y.; Jo, J.K.; Park, I.; Ha, C.S.; Suh, H.; Kim, I. Tuning of the activity and induction period of the polymerization of propylene oxide catalyzed by double metal cyanide complexes bearing β-alkoxy alcohols as complexing agents. Ind. Eng. Chem. Res. 2010, 49, 4107–4116. [Google Scholar] [CrossRef]
- Ionescu, M. Chemistry and Technology of Polyols for Polyurethane; Rapra Technology Limited: Shawbury, Shrewsbury, Shropshire, UK, 2005; ISBN 1859575013. [Google Scholar]
- Hayes, J.E.; Langsdorf, L.J.; Isaacs, B.H.; Armellini, F.J. Process for Rapid Activation of Double Metal Cyanide Catalysts. U.S. Patent 5,844,070, 1 December 1998. [Google Scholar]
- Pazos, J.F. Preparation of Double Metal Cyanide-catalyzed Polyols by Continous Addition of Starter. U.S. Patent 5,777,177, 7 July 1998. [Google Scholar]
- O’Connor, J.M.; Lickei, D.L.; Grieve, R.L. Preparing Polyether Polyols with DMC Catalysts. U.S. Patent 6,359,101, 19 March 2002. [Google Scholar]
- Pazos, J.F.; Shih, T.T. Continuous Preparation of Low Unsaturation Polyoxyalkylene Polyether Polyols with Continuos Addition of Starter. U.S. Patent 5,689,012, 18 November 1997. [Google Scholar]
- Zhang, W.; Lu, L.; Cheng, Y.; Xu, N.; Pan, L.; Lin, Q.; Wang, Y. Clean and rapid synthesis of double metal cyanide complexes using mechanochemistry. Green Chem. 2011, 13, 2701–2703. [Google Scholar] [CrossRef]
- Kuyper, J.; Boxhoorn, G. Hexacyanometallate salts used as alkene-oxide polymerization catalysts and molecular sieves. J. Catal. 1987, 105, 163–174. [Google Scholar] [CrossRef]
- Zhang, X.H.; Hua, Z.J.; Chen, S.; Liu, F.; Sun, X.K.; Qi, G.R. Role of zinc chloride and complexing agents in highly active double metal cyanide catalysts for ring-opening polymerization of propylene oxide. Appl. Catal. A Gen. 2007, 325, 91–98. [Google Scholar] [CrossRef]
- Huang, Y.J.; Qi, G.R.; Chen, L.S. Effects of morphology and composition on catalytic performance of double metal cyanide complex catalyst. Appl. Catal. A Gen. 2003, 240, 263–271. [Google Scholar] [CrossRef]
- Ionescu, M. Synthesis of High Molecular Weight Polyether Polyols with Double Metal Cyanide Catalysts (DMC Catalysts). In Chemistry and Technology of Polyols for Polyurethanes; Rapra Technology Limited: Shawbury, Shrewsbury, Shropshire, UK, 2005; pp. 177–194. ISBN 9781859575017. [Google Scholar]
- Kim, I.; Anas, K.; Lee, S.; Ha, C.S.; Park, D.W. Tuning of the activity and induction period of double metal cyanide catalyzed ring-opening polymerizations of propylene oxide by using ionic liquids. Catal. Today 2008, 131, 541–547. [Google Scholar] [CrossRef]
- Lee, S.; Baek, S.T.; Anas, K.; Ha, C.S.; Park, D.W.; Lee, J.W.; Kim, I. Tuning of activity, induction period and polymer properties of double metal cyanide catalyzed ring-opening polymerizations of propylene oxide by using quaternary ammonium salts. Polymer (Guildf.) 2007, 48, 4361–4367. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, J.T.; Ha, C.S.; Yang, C.S.; Park, I. Polymerization of propylene oxide by using double metal cyanide catalysts and the application to polyurethane elastomer. Polymer (Guildf.) 2003, 44, 3417–3428. [Google Scholar] [CrossRef]
- Shechter, L.; Wynstra, J.; Kurkjy, R.P. Glycidyl Ether Reactions with Amines. Ind. Eng. Chem. 1956, 48, 94–97. [Google Scholar] [CrossRef]
- Cyriac, A.; Lee, S.H.; Varghese, J.K.; Park, E.S.; Park, J.H.; Lee, B.Y. Immortal CO2/Propylene Oxide Copolymerization: Precise Control of Molecular Weight and Architecture of Various Block Copolymers. Macromolecules 2010, 43, 7398–7401. [Google Scholar] [CrossRef]
- Sebastian, J.; Srinivas, D. Structure-induced catalytic activity of Co-Zn double-metal cyanide complexes for terpolymerization of propylene oxide, cyclohexene oxide and CO2. RSC Adv. 2015, 5, 18196–18203. [Google Scholar] [CrossRef]
- Sun, X.-K.; Zhang, X.-H.; Liu, F.; Chen, S.; Du, B.-Y.; Wang, Q.; Fan, Z.-Q.; Qi, G.-R. Alternating copolymerization of carbon dioxide and cyclohexene oxide catalyzed by silicon dioxide/Zn-Co’’’ double metal cyanide complex hybrid catalysts with a nanolamellar structure. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3128–3139. [Google Scholar] [CrossRef]
- Pinilla, M.; Andrés-Iglesias, C.; Fernández, A.; Salmi, T.; Galdámez, J.R.; García-Serna, J. Effect of Zn/Co initial preparation ratio in the activity of double metal cyanide catalysts for propylene oxide and CO2 copolymerization. Eur. Polym. J. 2017, 88, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Liu, Z.; Cheng, R.; Jin, D.; He, X.; Liu, B. Experimental and theoretical studies on CO2 and propylene oxide (PO) copolymerization catalyzed by ZnEt2-glycerine-Y(CCl3COO)3 ternary catalyst. J. Organomet. Chem. 2014, 753, 63–71. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Xia, W.; Vagin, S.I.; Rieger, B. Regarding Initial Ring Opening of Propylene Oxide in its Copolymerization with CO2 Catalyzed by a Cobalt(III) Porphyrin Complex. Chem. Eur. J. 2014, 20, 15499–15504. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.T.; Thomas, C.M.; Peretti, K.L.; Lobkovsky, E.B.; Coates, G.W. Copolymerization of cyclohexene oxide and carbon dioxide using (salen)Co(iii) complexes: Synthesis and characterization of syndiotactic poly(cyclohexene carbonate). J. Chem. Soc. Dalt. Trans. 2006, 60, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, J.; Srinivas, D. Effects of method of preparation on catalytic activity of Co-Zn double-metal cyanide catalysts for copolymerization of CO2 and epoxide. Appl. Catal. A Gen. 2014, 482, 300–308. [Google Scholar] [CrossRef]
- Lee, I.K.; Ha, J.Y.; Cao, C.; Park, D.W.; Ha, C.S.; Kim, I. Effect of complexing agents of double metal cyanide catalyst on the copolymerizations of cyclohexene oxide and carbon dioxide. Catal. Today 2009, 148, 389–397. [Google Scholar] [CrossRef]
- Sebastian, J.; Srinivas, D. Solid, double-metal cyanide catalysts for synthesis of hyperbranched polyesters and aliphatic polycarbonates. J. Chem. Sci. 2014, 126, 499–509. [Google Scholar] [CrossRef]
- Varghese, J.K.; Park, D.S.; Jeon, J.Y.; Lee, B.Y. Double metal cyanide catalyst prepared using H3Co(CN)6 for high carbonate fraction and molecular weight control in carbon dioxide/propylene oxide copolymerization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4811–4818. [Google Scholar] [CrossRef]
- Kim, I.; Yi, M.J.; Lee, K.J.; Park, D.W.; Kim, B.U.; Ha, C.S. Aliphatic polycarbonate synthesis by copolymerization of carbon dioxide with epoxides over double metal cyanide catalysts prepared by using ZnX 2 (X = F, Cl, Br, I). Catal. Today 2006, 111, 292–296. [Google Scholar] [CrossRef]
- Sebastian, J.; Srinivas, D. Double-Metal Cyanide Catalyst Design in CO2/Epoxide Copolymerization. In Sustainable Polymers from Biomass; Tang, C., Ryu, C.Y., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; pp. 315–344. [Google Scholar]
- Chen, S.; Chen, L. Fe/Zn double metal cyanide catalyzed ring-opening polymerization of propylene oxide: 2. Characterization of active structure of double metal cyanide catalysts. Colloid Polym. Sci. 2004, 282, 1033–1038. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, Y.; Chen, L. Preparation of crown ether complexing highly active double metal cyanide catalysts and copolymerization of CO2 and propylene oxide. Chin. J. Catal. 2015, 36, 1304–1311. [Google Scholar] [CrossRef]
- Dienes, Y.; Leitner, W.; Müller, M.G.J.; Offermans, W.K.; Reier, T.; Reinholdt, A.; Weirich, T.E.; Müller, T.E. Hybrid sol–gel double metal cyanide catalysts for the copolymerisation of styrene oxide and CO2. Green Chem. 2012, 14, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Subhani, M.A.; Gürtler, C.; Leitner, W.; Müller, T.E. Nanoparticulate TiO2-Supported Double Metal Cyanide Catalyst for the Copolymerization of CO2 with Propylene Oxide. Eur. J. Inorg. Chem. 2016, 1944–1949. [Google Scholar] [CrossRef]
- An, N.; Li, Q.; Yin, N.; Kang, M.; Wang, J. Effects of addition mode on Zn–Co double metal cyanide catalyst for synthesis of oligo(propylene-carbonate) diols. Appl. Organomet. Chem. 2018, 32, 1–13. [Google Scholar] [CrossRef]
- Bohres, E.; Hill, F.; Ruppel, R.; Baum, E. Verfahren zur Herstellung von Multimetallcyanidkatalysatoren. WO2004/022227 A1, 18 March 2014. [Google Scholar]
- Le-Khac, B. Double Metal Cyanide Complex Catalysts. U.S. Patent 5,731,407, 24 March 1998. [Google Scholar]
- Li, S.; Qi, W.; Han, Y.; Chen, C.; Hu, B.; Yuan, M.; Ding, G. The Continuous Method to Prepare Complex Catalysts Containing Two or More Double Metal Cyanides. CN 1457928A1, 26 November 2003. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Polymer | Mn [kg/mol] | Mw [kg/mol] | PDI |
|---|---|---|---|---|
| 1 | PPG_1 | 6.7 | 26.4 | 4.0 |
| 2 | PPG_2 | 5.5 | 46.3 | 8.4 |
| 3 | PPEC | 3.5 | 4.1 | 1.2 |
| 4 | PPG/PPEC | 4.8 | 8.6 | 1.8 |
| Entry | N | T [°C] | p (bar) | Yield [g] | Mn [g/mol] | PDI | fcPC | F |
|---|---|---|---|---|---|---|---|---|
| 5 | 0.173 | 60 | 18.9 | 5.7 | 630 | 1.2 | 0.00 | 0.13 |
| 6 | 80 | 25.3 | 6.4 | 660 | 1.1 | 0.02 | 0.13 | |
| 7 | 100 | 32.6 | 31.5 | 1000 | 1.4 | 0.11 | 0.16 | |
| 8 | 120 | 40.0 | 32.8 | 1080 | 1.2 | 0.08 | 0.14 | |
| 9 | 0.115 | 60 | 13.6 | 5.8 | 630 | 1.1 | 0.00 | 0.12 |
| 10 | 80 | 18.2 | 6.9 | 660 | 1.1 | 0.02 | 0.15 | |
| 11 | 100 | 23.6 | 33.3 | 1060 | 1.5 | 0.08 | 0.16 | |
| 12 | 120 | 30.9 | 33.0 | 1080 | 1.4 | 0.07 | 0.12 | |
| 13 | 0.058 | 60 | 8.3 | 5.4 | 570 | 1.2 | 0.00 | 0.17 |
| 14 | 80 | 11.0 | 7.7 | 660 | 1.2 | 0.02 | 0.15 | |
| 15 | 100 | 14.2 | 33.5 | 1070 | 1.5 | 0.08 | 0.13 | |
| 16 | 120 | 19.7 | 32.5 | 1100 | 1.3 | 0.06 | 0.11 |
| Entry | Reaction Time [h] | Yield [g] | Mn [g/mol] | PDI | fcarb | fether | fcPC | F |
|---|---|---|---|---|---|---|---|---|
| 17 | 6 | 5.7 | 600 | 1.1 | 0.16 | 0.84 | <0.01 | 0.19 |
| 18 | 12 | 7.8 | 610 | 1.1 | 0.16 | 0.84 | <0.01 | 0.19 |
| 19 | 24 | 8.9 | 650 | 1.1 | 0.15 | 0.85 | <0.01 | 0.18 |
| Entry | Polymer | CTA | T [°C] | p [bar] | Mn [kg/mol] | PDI | fcPC | F |
|---|---|---|---|---|---|---|---|---|
| 20 | PPEC | Lupranol | 120 | 10 | 5.5 | 1.3 | 0.03 | 0.11 |
| 21 | PPEC_1 | Caradol | 90 | 10 | 12.0 | 7.3 | * | * |
| 22 | PPEC_2 | Caradol | 120 | 10 | 13.8 | 1.7 | 0.02 | 0.17 |
| 23 | PPG | Caradol | 120 | Ar | 18.4 | 1.3 |
| PPEC_1 2.5 h | PPEC_1 5 h | PPEC_1 6 h | PPEC_1 7.5 h | PPEC_1 8.25 h | PPEC_1 9 h | ||
|---|---|---|---|---|---|---|---|
| Peak 1 | Mn_1 [kg/mol] | 3.4 | 3.6 | 3.6 | 3.7 | 3.7 | 3.9 |
| Mw_1 [kg/mol] | 3.9 | 4.0 | 4.1 | 4.1 | 4.1 | 4.4 | |
| PDI_1 | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 | |
| Peak 2 | Mn_2 [kg/mol] | 8.1 | 8.4 | 9.0 | 9.3 | 9.4 | |
| Mw_2 [kg/mol] | 13.8 | 13.4 | 12.5 | 12.3 | 12.4 | ||
| PDI_2 | 1.7 | 1.6 | 1.4 | 1.3 | 1.3 | ||
| Rel. I (vs. 1) | 0.52 | 0.47 | 0.36 | 0.36 | 0.52 | ||
| Peak 3 | Mn_3 [kg/mol] | 6.7 | 15.4 | 16.9 | 19.2 | 21.9 | 23.8 |
| Mw_3 [kg/mol] | 16.4 | 69.5 | 80.2 | 97.2 | 105.7 | 106.2 | |
| PDI_3 | 2.5 | 4.5 | 4.7 | 5.1 | 4.8 | 4.5 | |
| Rel. I (vs. 1) | 0.46 | 1.69 | 2.28 | 3.12 | 3.36 | 4.43 |
| Sample | fcarb | fether | fcPC | F |
|---|---|---|---|---|
| PPEC_2_2.5h | 0.12 | 0.84 | 0.03 | 0.15 |
| PPEC_2_5h | 0.13 | 0.84 | 0.03 | 0.15 |
| PPEC_2_7.5h | 0.15 | 0.82 | 0.03 | 0.18 |
| PPEC_2_9.5h | 0.14 | 0.84 | 0.03 | 0.16 |
| PPEC_2_11h | 0.14 | 0.84 | 0.02 | 0.17 |
| PPEC_2_12h | 0.14 | 0.83 | 0.02 | 0.17 |
| Entry | namine [10−2 mol%] | VTEA [µL] | tind [min] | tend [min] | ΔT [°C] | Entry | mDDA [mg] | tind [min] | tend [min] | ΔT [°C] |
|---|---|---|---|---|---|---|---|---|---|---|
| 24 | 0 | - | 5 | 45 | 14 | |||||
| 25 | 0.66 | 0.5 | 13 | 63 | 6 | 30 | 0.6 | 8 | 57 | 6 |
| 26 | 1.31 | 1 | 11 | 65 | 6 | 31 | 1.3 | 9 | 57 | 4 |
| 27 | 2.62 | 2 | 5 | 75 | 6 | 32 | 2.6 | 6 | >51 # | 7 |
| 28 | 6.58 | 5 | 5 | 190 | 2 | 33 | 6.6 | 8 | 63 | 4 |
| 29 | 13.1 | 10 | 5 | >23 h * | n.d. | 34 | n.d. | n.d. | n.d. | n.d. |
| N | Entry | VTEA [µL] | Mn [g/mol] | PDI | fcPC | F | Entry | mDDA [mg] | Mn [g/mol] | PDI | fcPC | F |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.115 | 35 | 0 | 1000 | 1.5 | 0.07 | 0.14 | ||||||
| 36 | 0.5 | 960 | 1.8 | 0.06 | 0.16 | 45 | 0.6 | 1160 | 1.4 | 0.06 | 0.12 | |
| 37 | 1 | n.d. | n.d. | n.d. | n.d. | 46 | 1.3 | 1210 | 1.2 | 0.06 | 0.09 | |
| 38 | 2 | 1020 | 1.6 | 0.06 | 0.15 | 47 | 2.6 | 1090 | 1.7 | 0.05 | 0.16 | |
| 39 | 5 | 1070 | 1.6 | 0.06 | 0.14 | 48 | 6.6 | 1190 | 1.5 | 0.06 | 0.15 | |
| 0.058 | 40 | 0 | 1100 | 1.3 | 0.03 | 0.16 | ||||||
| 41 | 0.5 | 940 | 1.8 | 0.05 | 0.08 | 49 | 0.6 | 1220 | 1.4 | 0.03 | 0.08 | |
| 42 | 1 | 1080 | 1.2 | 0.04 | 0.07 | 50 | 1.3 | 1230 | 1.2 | 0.04 | 0.06 | |
| 43 | 2 | 1090 | 1.5 | 0.04 | 0.10 | 51 | 2.6 | 1200 | 1.3 | 0.04 | 0.08 | |
| 44 | 5 | 1090 | 1.6 | 0.04 | 0.11 | 52 | 6.6 | 1230 | 1.2 | 0.04 | 0.07 |
| Element | DMC-Cl a | DMC-NO3 a | DMC-Cl b | DMC-NO3 b |
|---|---|---|---|---|
| C [wt.%] | 28.2 ± 2.2 | 30.2 ± 0.4 | 22.9 ± 0.1 | 24.4 ± 1.1 |
| H [wt.%] | 2.1 ± 0.1 | 1.7 ± 0.2 | ||
| N [wt.%] | 8.3 ± 4.5 | 31.2 ± 2.6 | 17.6 ± 0.3 | 21.8 ± 0.7 |
| O [wt.%] | 1.5 ± 0.1 | 5.7 ± 1.4 | 5.7 ± 0.1 | 11.9 ± 0.1 |
| Zn [wt.%] | 39.1 ± 1.9 | 21.9 ± 2.4 | 30.2 ± 0.5 | 29.8 ± 0.9 |
| Co [wt.%] | 17.6 ± 1.7 | 11.0 ± 1.4 | 12.6 ± 0.2 | 15.7 ± 0.6 |
| Na [wt.%] | 0 ± 0.0 | 0 ± 0.0 | 0.3 ± 0.1 | |
| Cl [wt.%] | 5.3 ± 1.3 | 0 ± 0.0 | 7.4 ± 0.1 | 0.1 ± 0.0 |
| Entry | Experiment | T [°C] | p [bar] | N | Yield [g] | Mn [g/mol] | PDI | fcPC | F |
|---|---|---|---|---|---|---|---|---|---|
| 53 | DMC-NO3_1 | 100 | 14.2 | 0.058 | 28.2 | 1000 | 2.5 | 0.01 | 0.25 |
| 54 | DMC-NO3_2 | 100 | 23.6 | 0.115 | 26.6 | 960 | 2.2 | 0.02 | 0.29 |
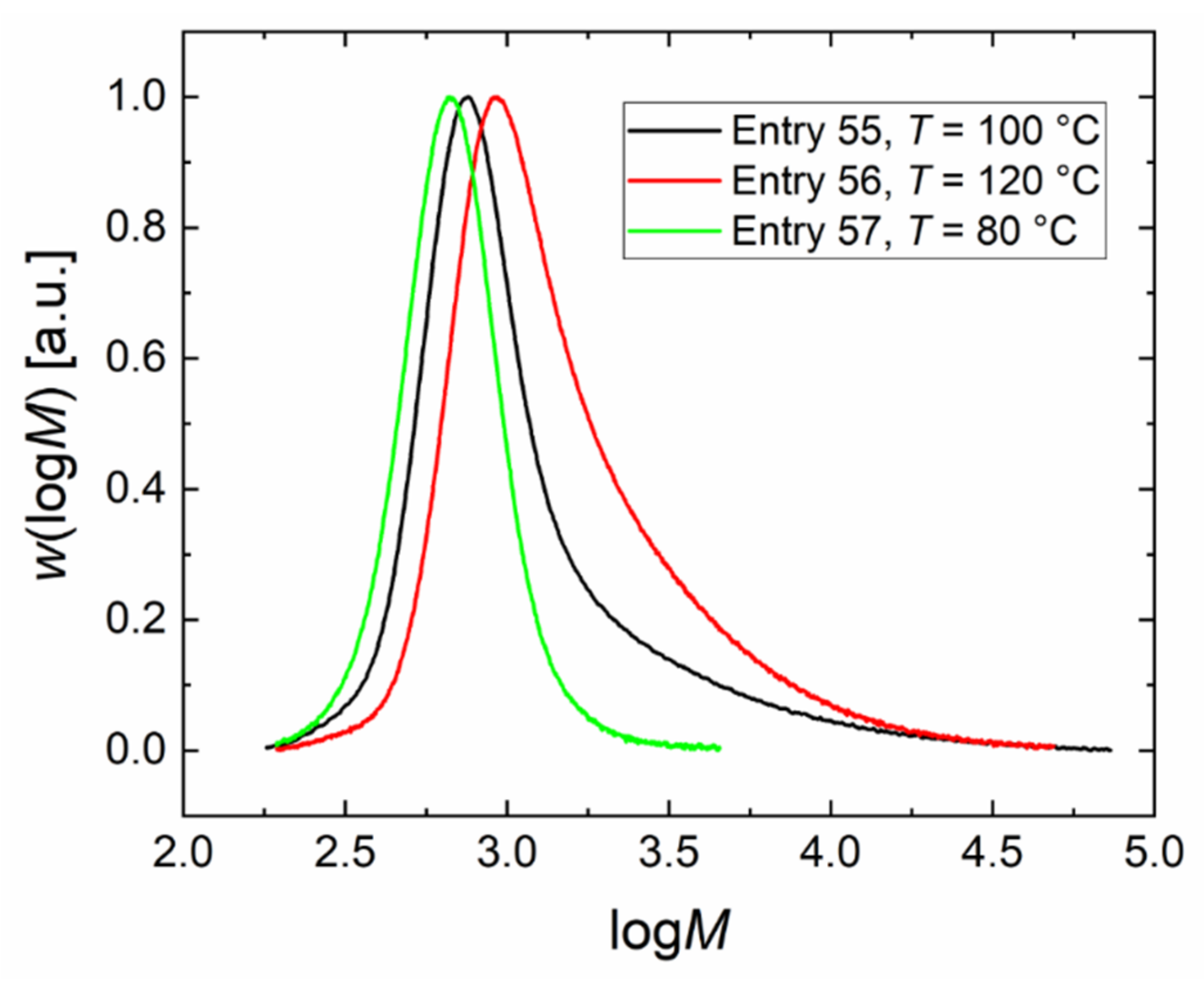
| 55 | DMC-NO3_3 | 100 | 32.6 | 0.173 | 22.0 | 880 | 2.3 | 0.02 | 0.45 |
| 56 | DMC-NO3_4 | 120 | 40 | 0.173 | 36.1 | 1170 | 2.0 | 0.03 | 0.43 |
| 57 | DMC-NO3_5 | 80 | 25.3 | 0.173 | 7.7 | 640 | 1.2 | 0.05 | 0.42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stahl, S.-F.; Luinstra, G.A. DMC-Mediated Copolymerization of CO2 and PO—Mechanistic Aspects Derived from Feed and Polymer Composition. Catalysts 2020, 10, 1066. https://doi.org/10.3390/catal10091066
Stahl S-F, Luinstra GA. DMC-Mediated Copolymerization of CO2 and PO—Mechanistic Aspects Derived from Feed and Polymer Composition. Catalysts. 2020; 10(9):1066. https://doi.org/10.3390/catal10091066
Chicago/Turabian StyleStahl, Sarah-Franziska, and Gerrit A. Luinstra. 2020. "DMC-Mediated Copolymerization of CO2 and PO—Mechanistic Aspects Derived from Feed and Polymer Composition" Catalysts 10, no. 9: 1066. https://doi.org/10.3390/catal10091066