
Pre-Coking Strategy Strengthening Stability Performance of Supported Nickel Catalysts in Chloronitrobenzene Hydrogenation

Abstract
:1. Introduction
2. Results and Discussion
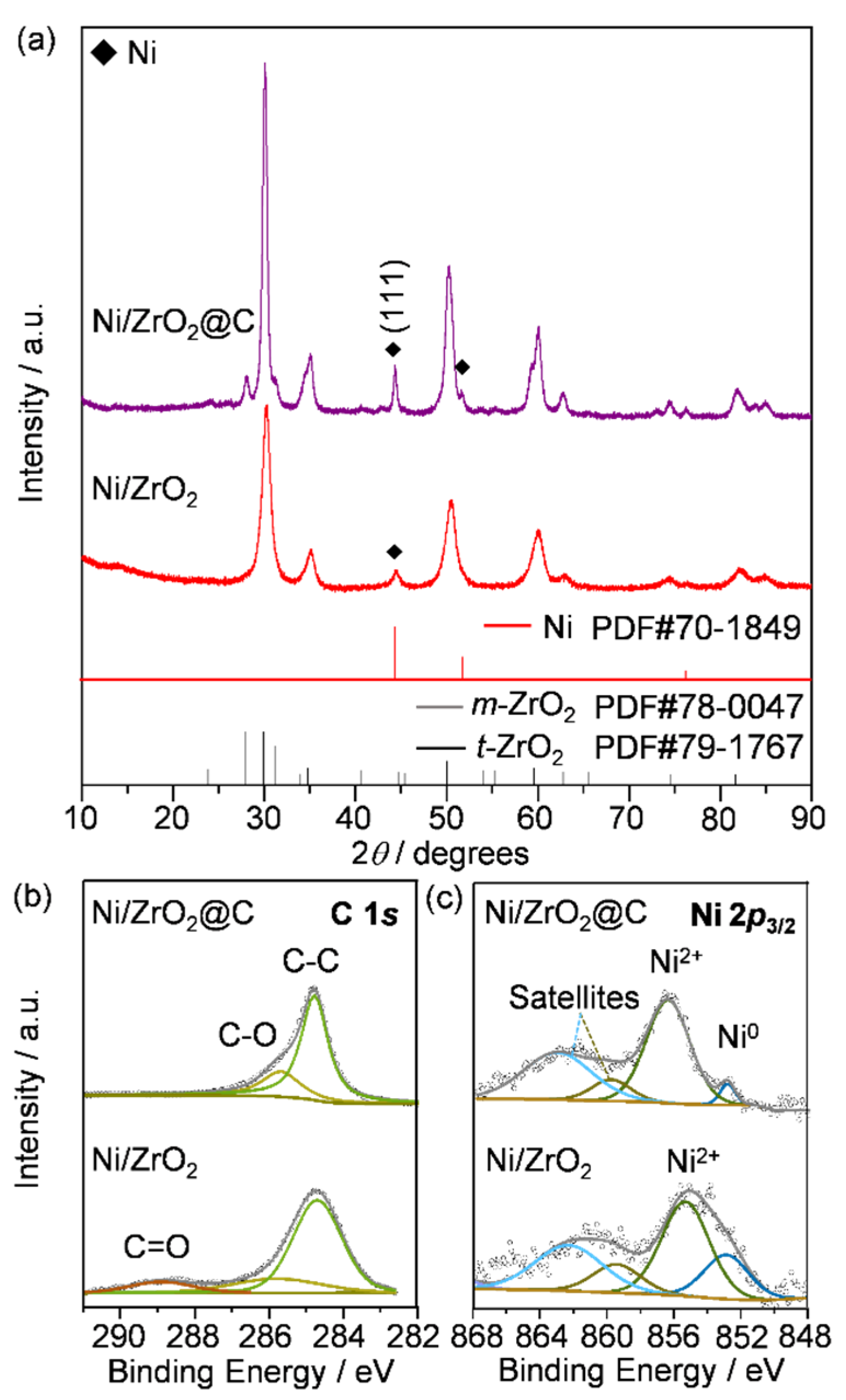
2.1. Synthesis and Characterization of Fresh Catalysts
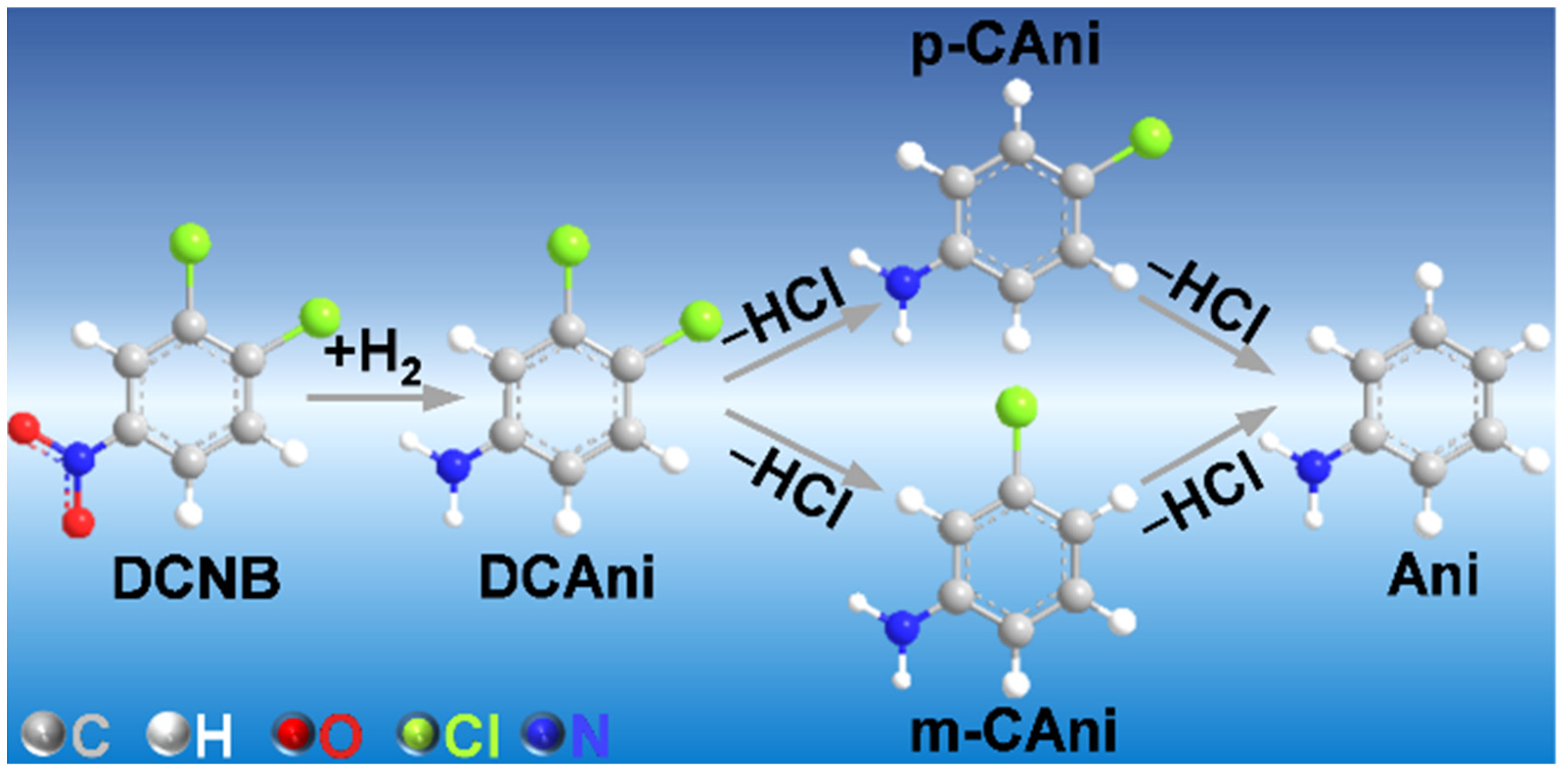
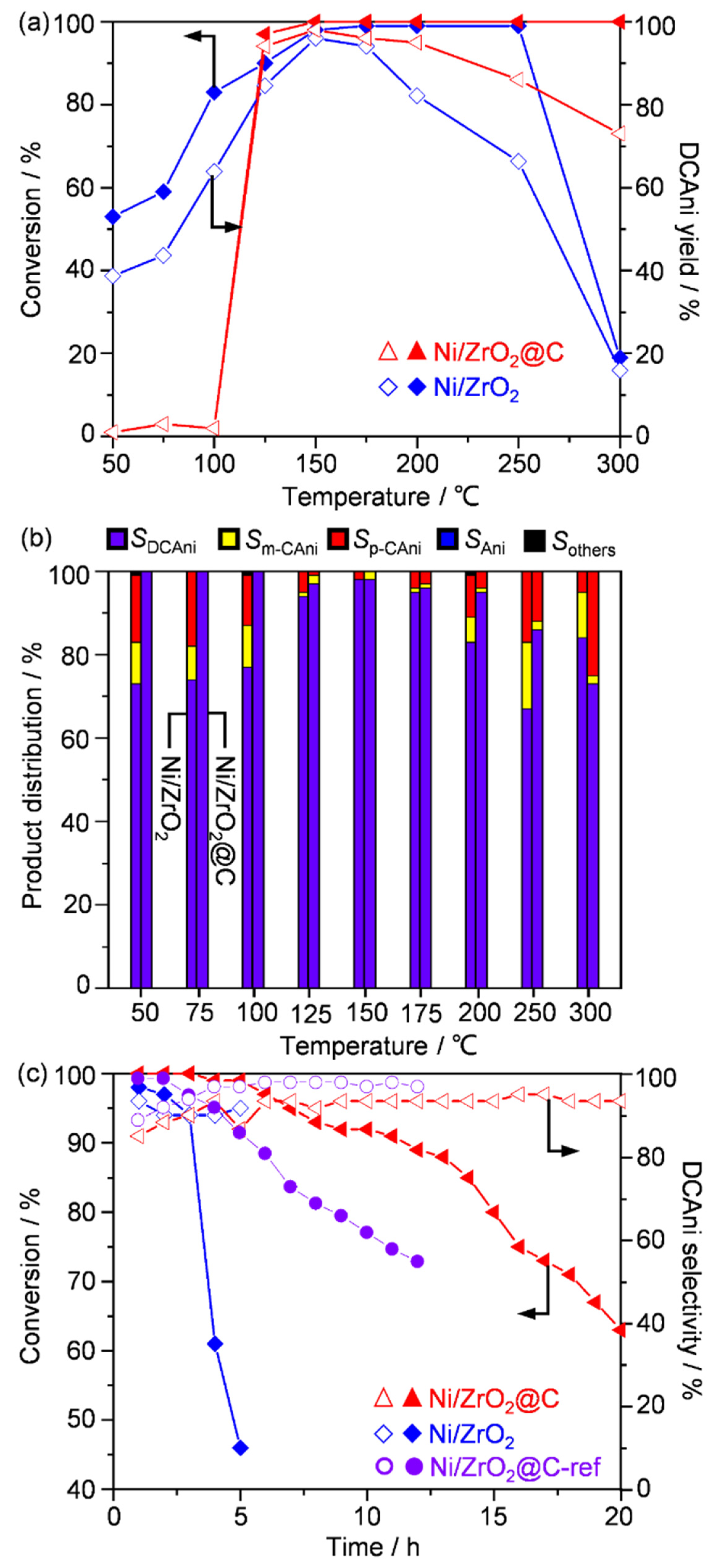
2.2. Catalytic Performance in 1,2-Dichloro-4-nitrobenzene Hydrogenation
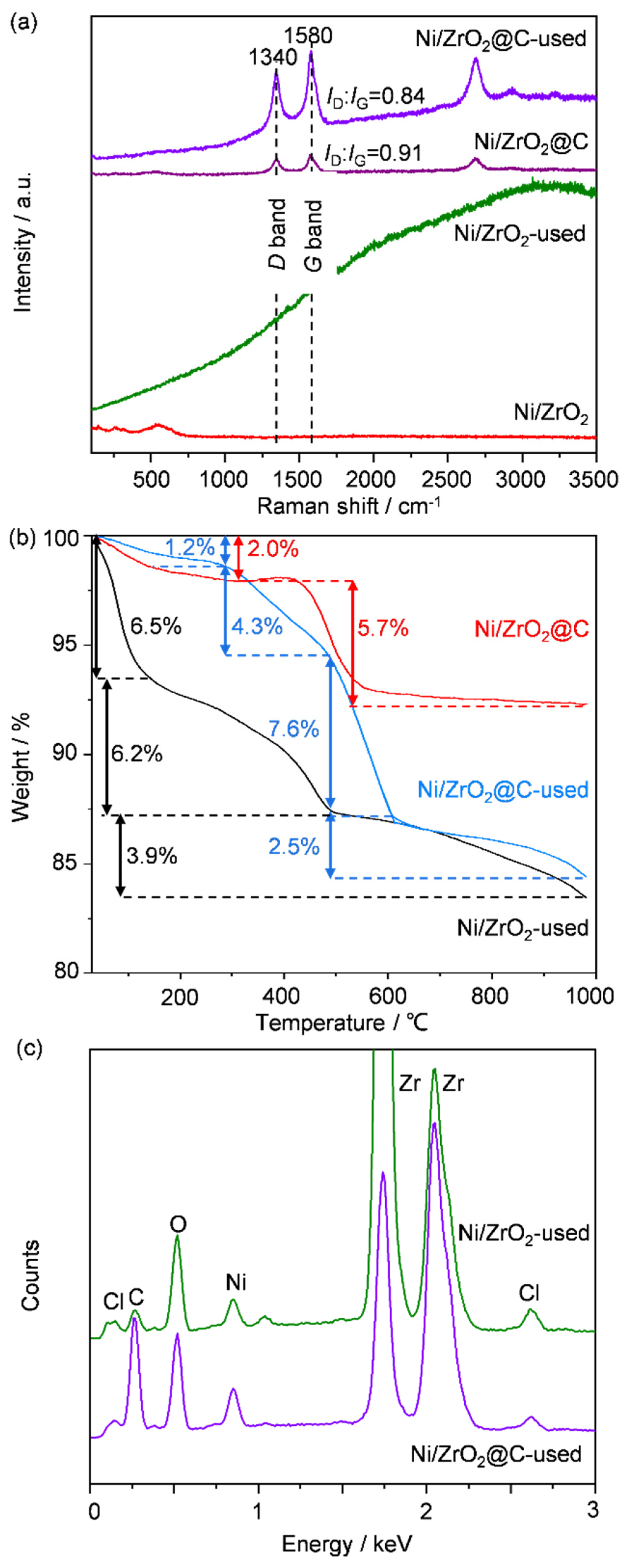
2.3. Interpretation of the Deactivation Mechanisms
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Pre-Coking Strategy for Supported Nickel Catalysts in Dry Reforming of Methane
3.3. Catalyst Characterization
3.4. Catalyst Evaluation in Chloronitrobenzene Hydrogenation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Loos, P.; Alex, H.; Hassfeld, J.; Lovis, K.; Platzek, J.; Steinfeldt, N.; Huebner, S. Selective hydrogenation of halogenated nitroaromatics to haloanilines in batch and flow. Org. Process Res. Dev. 2016, 20, 452–464. [Google Scholar] [CrossRef]
- Jaf, Z.N.; Altarawneh, M.; Miran, H.A.; Almatarneh, M.H.; Jiang, Z.-T.; Dlugogorski, B.Z. Catalytic hydrogenation of p-chloronitrobenzene to p-chloroaniline mediated by gamma-Mo2N. ACS Omega 2018, 3, 14380–14391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-Y.; Zhan, G.-P.; Wu, C.-D. In situ creation of multi-metallic species inside porous silicate materials with tunable catalytic roperties. Chem. Commun. 2021, 57, 6185–6188. [Google Scholar] [CrossRef] [PubMed]
- Tumma, M.; Srivastava, R. Transition metal nanoparticles supported on mesoporous polyaniline catalyzed reduction of nitroaromatics. Catal. Commun. 2013, 37, 64–68. [Google Scholar] [CrossRef]
- Zhang, B.; Asakura, H.; Zhang, J.; Zhang, J.; De, S.; Yan, N. Stabilizing a platinum (1) single-atom catalyst on supported phosphomolybdic acid without compromising hydrogenation activity. Angew Chem. Int. Ed. 2016, 55, 8319–8323. [Google Scholar] [CrossRef]
- Nie, S.; Yang, S.; Zhang, P. Solvent-free synthesis of mesoporous platinum-aluminum oxide via mechanochemistry: Toward selective hydrogenation of nitrobenzene to aniline. Chem. Eng. Sci. 2020, 220, 115619–115626. [Google Scholar] [CrossRef]
- Sun, Z.; Zhao, Y.; Xie, Y.; Tao, R.; Zhang, H.; Huang, C.; Liu, Z. The solvent-free selective hydrogenation of nitrobenzene to aniline: An unexpected catalytic activity of ultrafine Pt nanoparticles deposited on carbon nanotubes. Green Chem. 2010, 12, 1007–1011. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Ding, Y.; Wu, P. Pt nanoparticles entrapped in ordered mesoporous carbons: An efficient catalyst for the liquid phase hydrogenation of nitrobenzene and its derivatives. Chin. J. Catal. 2015, 36, 1995–2003. [Google Scholar] [CrossRef]
- Zhang, Q.; Bu, J.; Wang, J.; Sun, C.; Zhao, D.; Sheng, G.; Xie, X.; Sun, M.; Yu, L. Highly efficient hydrogenation of nitrobenzene to aniline over Pt/CeO2 catalysts: The shape effect of the support and key role of additional Ce3+ sites. ACS Catal. 2020, 10, 10350–10363. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Gao, R.; Liang, X.; Yu, Q.; Deng, Y.; Liu, J.; Peng, M.; Jiang, Z.; Li, S.; et al. A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 2019, 14, 354–362. [Google Scholar] [CrossRef]
- Pietrobon, L.; Ronchin, L.; Sadraoui, C.; Pontello, R.; Tosetto, C.; Vavasori, A. Pd/C catalyzed selective hydrogenation of nitrobenzene to cyclohexanone oxime in the presence of NH2OH center dot HCl: Influence of the operative variables and insights on the reaction mechanism. Appl. Catal. A-Gen. 2020, 598, 117570–117582. [Google Scholar] [CrossRef]
- Sangeetha, P.; Shanthi, K.; Rao, K.S.R.; Viswanathan, B.; Selvam, P. Hydrogenation of nitrobenzene over palladium-supported catalysts-effect of support. Appl. Catal. A-Gen. 2009, 353, 160–165. [Google Scholar] [CrossRef]
- Zhao, M.; Feng, B.; Qiao, X.; Zhong, N.; Ge, X.; Ji, Y. Electronic modulation of palladium in metal phosphide nanoparticles for chemoselective reduction of halogenated nitrobenzenes. Chem.-Asian J. 2019, 14, 407–410. [Google Scholar] [CrossRef]
- Leng, F.; Gerber, I.C.; Lecante, P.; Moldovan, S.; Girleanu, M.; Axet, M.R.; Serp, P. Controlled and chemoselective hydrogenation of ntrobenzene over Ru@C-60 catalysts. ACS Catal. 2016, 6, 6018–6024. [Google Scholar] [CrossRef]
- Tomkins, P.; Gebauer-Henke, E.; Leitner, W.; Mueller, T.E. Concurrent hydrogenation of aromatic and nitro groups over carbon-supported ruthenium catalysts. ACS Catal. 2015, 5, 203–209. [Google Scholar] [CrossRef]
- Ma, L.; Wang, J.; Wang, H.; Zhang, Q.; Lu, C.; He, X.; Li, X. High halogenated nitrobenzene hydrogenation selectivity over nano Ir particles. Chin. J. Chem. Eng. 2017, 25, 306–312. [Google Scholar] [CrossRef]
- Kartusch, C.; Makosch, M.; Sa, J.; Hungerbuehler, K.; van Bokhoven, J.A. The dynamic structure of gold supported on ceria in the liquid phase hydrogenation of nitrobenzene. ChemCatChem 2012, 4, 236–242. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, K.; Wu, C.; Zhang, H.; Zhang, Q. In situ-formed cobalt embedded into N-doped carbon as highly efficient and selective catalysts for the hydrogenation of halogenated nitrobenzenes under mild conditions. Appl. Catal. A-Gen. 2020, 592, 117434–117442. [Google Scholar] [CrossRef]
- Sheng, Y.; Wang, X.; Yue, S.; Cheng, G.; Zou, X.; Lu, X. In situ synthesized silica-supported Co@N-doped carbon as highly efficient and reusable catalysts for selective reduction of halogenated nitroaromatics. ChemCatChem 2020, 12, 4632–4641. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, W.; Long, Y.; Wang, F.; Ma, J. Co-Ag alloy protected by nitrogen doped carbon as highly efficient and chemoselective catalysts for the hydrogenation of halogenated nitrobenzenes. J. Colloid Interf. Sci. 2018, 522, 217–227. [Google Scholar] [CrossRef]
- Zhang, X.; Li, P.; Xu, B.; Wang, J.; Fan, G.; Zhang, X.; Liu, X.; Zhang, K.; Jiang, W. In situ hydrogen activation inspiring efficient one-pot hydrogenation of halogenated nitrobenzenes over Ni-Co-based composites. Ind. Eng. Chem. Res. 2021, 60, 8312–8323. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, W.; Long, Y.; Qin, J.; Ma, J. Different transfer hydrogenation pathways of halogenated nitrobenzenes catalyzed by Fe-, Co- or Ni-based species confined in nitrogen doped carbon. Mol. Catal. 2020, 497, 111226–111235. [Google Scholar] [CrossRef]
- Tian, M.; Cui, X.; Yuan, M.; Yang, J.; Ma, J.; Dong, Z. Efficient chemoselective hydrogenation of halogenated nitrobenzenes over an easily prepared gamma-Fe2O3-modified mesoporous carbon catalyst. Green Chem. 2017, 19, 1548–1554. [Google Scholar] [CrossRef]
- Tian, S.; Hu, M.; Xu, Q.; Gong, W.; Chen, W.; Yang, J.; Zhu, Y.; Chen, C.; He, J.; Liu, Q.; et al. Single-atom Fe with Fe1N3 structure showing superior performances for both hydrogenation and transfer hydrogenation of nitrobenzene. Sci. China Mater. 2021, 64, 642–650. [Google Scholar] [CrossRef]
- Xiong, W.; Wang, Z.; He, S.; Hao, F.; Yang, Y.; Lv, Y.; Zhang, W.; Liu, P.; Luo, H. Nitrogen-doped carbon nanotubes as a highly active metal-free catalyst for nitrobenzene hydrogenation. Appl. Catal. B-Environ. 2020, 260, 118105–118115. [Google Scholar] [CrossRef]
- Lv, J.; Zheng, Y.; Zhu, Y.; Yuan, M.; Chang, Y.; Dong, Z. Renewable soybean pulpderived N-doped carbon materials for efficient chemoselective hydrogenation of halogenated nitrobenzenes. ChemistrySelect 2019, 4, 4083–4091. [Google Scholar] [CrossRef]
- Sheng, Y.; Wang, X.; Xing, Z.; Chen, X.; Zou, X.; Lu, X. Highly active and chemoselective reduction of halogenated nitroarenes catalyzed by ordered mesoporous carbon supported platinum nanoparticles. ACS Sustain. Chem. Eng. 2019, 7, 8908–8916. [Google Scholar] [CrossRef]
- Ichikawa, S.; Tada, M.; Iwasawea, Y.; Ikariya, T. The role of carbon dioxide in chemoselective hydrogenation of halonitroaromatics over supported noble metal catalysts in supercritical carbon dioxide. Chem. Commun. 2005, 36, 924–926. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Gu, H.; Yan, X. A novel transfer hydrogenation with high hydrogen utilization for the hydrogenation of halogenated nitrobenzene without hydrodehalogenation. Catal. Lett. 2009, 132, 16–21. [Google Scholar] [CrossRef]
- Kondeboina, M.; Enumula, S.S.; Muppala, A.R.; Madduluri, V.R.; Burri, D.R.; Kamaraju, S.R.R. Carbon coating on SiO2 in Co/C-SiO2 catalysts for high and stable activity in nitrobenzene hydrogenation. ChemistrySelect 2017, 2, 5716–5722. [Google Scholar] [CrossRef]
- Qin, Z.; Chen, J.; Xie, X.; Luo, X.; Su, T.; Ji, H. CO2 reforming of CH4 to syngas over nickel-based catalysts. Environ. Chem. Lett. 2020, 18, 997–1017. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Chen, M.; Liang, D.; Yang, Z.; Cheng, W.; Tang, Z.; Wang, J.; Zhang, H. Recent advances during CH4 dry reforming for syngas production: A mini review. Int. J. Hydrogen Energy 2021, 46, 5852–5874. [Google Scholar] [CrossRef]
- Duan, X.; Yin, J.; Feng, A.; Huang, M.; Fu, W.; Xu, W.; Huang, Z.; Zhang, J. Continuous hydrogenation of halogenated nitroaromatic compounds in a micropacked bed reactor. J. Flow Chem. 2021. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dNi(111)a | SBETb | Vpc | C d,e | Ni d,e | Zr d,e | Cl d,e | O d,e | |
|---|---|---|---|---|---|---|---|---|
| Catalysts | /nm | /m2 g−1 | /cm3 g−1 | /at.% | /at.% | /at.% | /at.% | /at.% |
| NiO/ZrO2 | - | 74 | 0.25 | 22.24 | 2.65 | 18.08 | 0 | 57.03 |
| Ni/ZrO2 | 8 | 94 | 0.20 | 25.37 (22.14) | 1.74 (7.45) | 19.38 (23.37) | 0 (0.05) | 53.51 (47.00) |
| Ni/ZrO2-used | 12 | 98 | 0.25 | 30.70 (24.78) | 4.00 (5.71) | 12.28 (19.46) | 15.78 (3.33) | 37.24 (46.72) |
| Ni/ZrO2@C | 19 | 41 | 0.23 | 55.45 (40.76) | 1.58 (6.02) | 10.61 (17.46) | 0 (0.01) | 32.36 (35.57) |
| Ni/ZrO2@C-used | 23 | 60 | 0.25 | 67.10 (63.11) | 1.97 (3.58) | 5.36 (9.86) | 6.40 (0.96) | 19.16 (22.49) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Wang, S.; Lin, R.; Mou, X.; Ding, Y. Pre-Coking Strategy Strengthening Stability Performance of Supported Nickel Catalysts in Chloronitrobenzene Hydrogenation. Catalysts 2021, 11, 1156. https://doi.org/10.3390/catal11101156
Wang P, Wang S, Lin R, Mou X, Ding Y. Pre-Coking Strategy Strengthening Stability Performance of Supported Nickel Catalysts in Chloronitrobenzene Hydrogenation. Catalysts. 2021; 11(10):1156. https://doi.org/10.3390/catal11101156
Chicago/Turabian StyleWang, Ping, Shiyi Wang, Ronghe Lin, Xiaoling Mou, and Yunjie Ding. 2021. "Pre-Coking Strategy Strengthening Stability Performance of Supported Nickel Catalysts in Chloronitrobenzene Hydrogenation" Catalysts 11, no. 10: 1156. https://doi.org/10.3390/catal11101156
APA StyleWang, P., Wang, S., Lin, R., Mou, X., & Ding, Y. (2021). Pre-Coking Strategy Strengthening Stability Performance of Supported Nickel Catalysts in Chloronitrobenzene Hydrogenation. Catalysts, 11(10), 1156. https://doi.org/10.3390/catal11101156