
Asymmetric Henry Reaction of Nitromethane with Substituted Aldehydes Catalyzed by Novel In Situ Generated Chiral Bis(β-Amino Alcohol-Cu(OAc)2·H2O Complex

 ,
,  , ,
, ,
Abstract
:
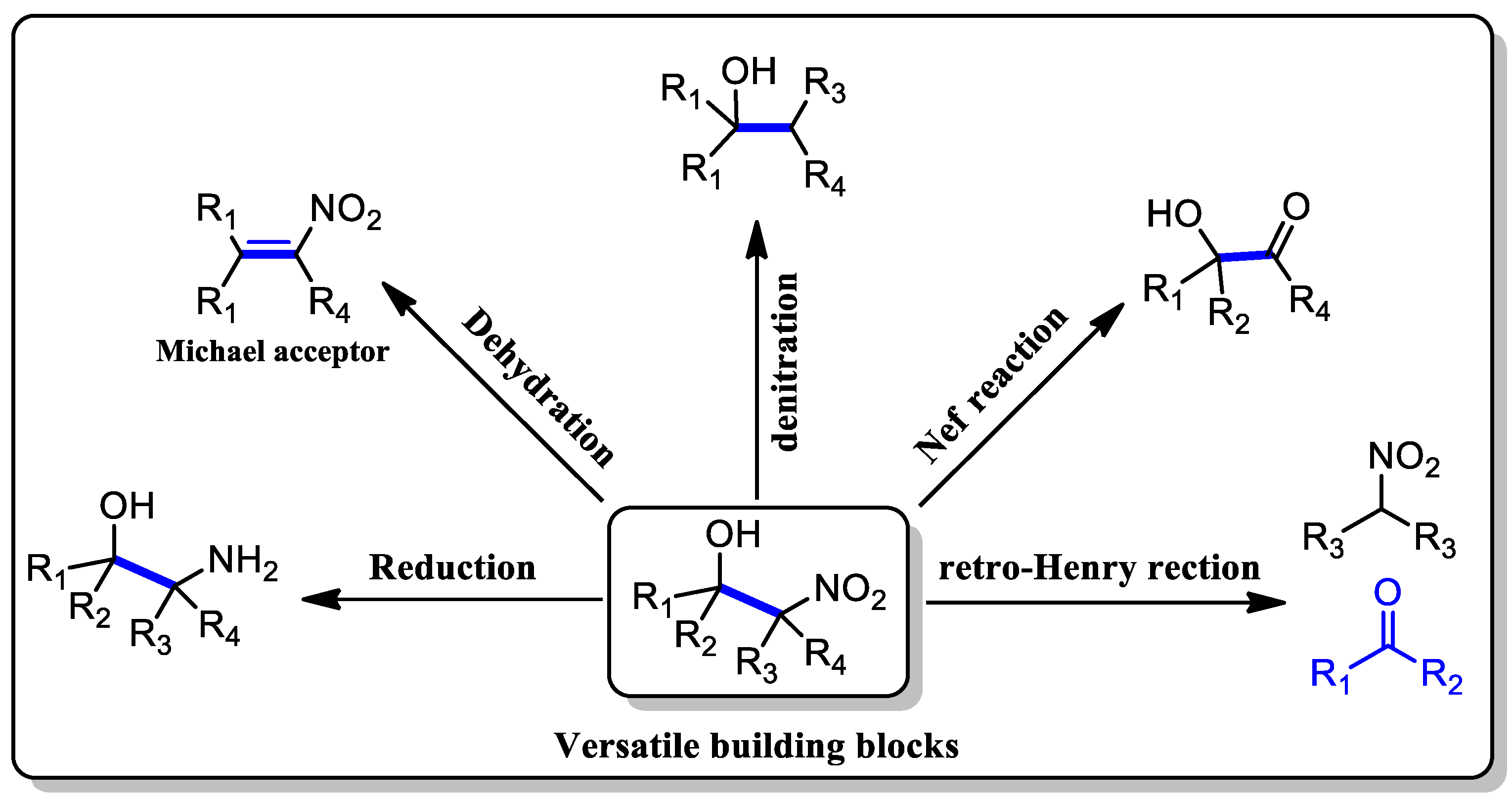
1. Introduction
2. Results and Discussion
2.1. Synthesis of Ligand L1–L5
2.2. Catalytic Studies of the Henry Reaction
3. Experiments
3.1. General
3.2. Synthesis of thiophene-2,5-diyldimethanol (2)
3.3. Synthesis of thiophene-2,5-dicarbaldehyde (3)
3.4. General Procedure for Synthesis of Chiral Diamine Alcohol Ligands (L1–L5)
3.4.1. ((2S,2′S)-2,2′-((Thiophene-2,5-diylbis(methylene))bis(azanediyl))bis(3-phenylpropan-1-ol) (L1)
3.4.2. (2S,2′S)-2,2′-((Thiophene-2,5-diylbis (methylene))bis(azanediyl))bis(2-phenylethan-1-ol) (L2)
3.4.3. (2S,2′S)-2,2′-((Thiophene-2,5-diylbis(methylene))bis(azanediyl))bis(3-methyl butan-1-ol) (L3)
3.4.4. (2S,2′S)-2,2′-((Thiophene-2,5-diylbis(methylene))bis(azanediyl))bis(3,3-dimethyl butan-1-ol) (L4)
3.4.5. (2S,2′S)-2,2′-((Thiophene-2,5-diylbis(methylene))bis(azanediyl) )bis(butan-1-ol) (L5)
3.5. General Procedures for the Synthesis of Racemic Nitroaldol Products (Rac 8a–m)
3.6. General Procedure for the Catalytic Asymmetric Henry Reaction (8a–m)
3.6.1. (R)-(+)-2-Nitro-1-(2-nitrophenyl)ethan-1-ol (8a)
3.6.2. (R)-(−)-2-Nitro-1-(4-nitrophenyl)ethan-1-ol (8b)
3.6.3. (R)-(−)-2-Nitro-1-(3-nitrophenyl)ethan-1-ol (8c)
3.6.4. (R)-(−)-1-(4-Bromophenyl)-2-nitroethan-1-ol (8d)
3.6.5. (R)-(−)-1-(Naphthalen-2-yl)-2-nitroethan-1-ol (8e)
3.6.6. (R)-(−)-2-Nitro-1-(4-(trifluoromethyl)phenyl)ethan-1-ol (8f)
3.6.7. (R)-(−)-1-(2,4-Dichlorophenyl)-2-nitroethan-1-ol (8g)
3.6.8. (R)-(−)-1-(4-Chlorophenyl)-2-nitroethan-1-ol (8h)
3.6.9. (R)-(−)-1-(4-Fluorophenyl)-2-nitroethan-1-ol (8i)
3.6.10. (R)-(−)-1-(4-Methoxyphenyl)-2-nitroethan-1-ol (8j)
3.6.11. (R)-(−)-2-Nitro-1-(p-tolyl)ethan-1-ol (8k)
3.6.12. (R)-(−)-2-Nitro-1-(m-tolyl)ethan-1-ol (8l)
3.6.13. (R)-(−)-2-Nitro-1-phenylethan-1-ol (8m)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mikami, K.; Lautens, M. New Frontiers in Asymmetric Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis: Supplement 1; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2003; Volume 1. [Google Scholar]
- Ojima, I. Catalytic Asymmetric Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Christmann, M.; Bräse, S. Asymmetric Synthesis ii: More Methods and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Nag, A. Asymmetric Synthesis of Drugs and Natural Products; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Blacker, J. Catalytic Asymmetric Synthesis, 2nd ed.; Ojima, I., Ed.; Wiley-VCH: New York, NY, USA, 2000; p. 864. ISBN 0-471-29805-0. [Google Scholar]
- Babu, S.A. Exploitation of intramolecular glaser–eglinton–hay macrocyclization for the synthesis of new classes of optically active aza-oxo-thia polyether macrocycles from amino alcohol building blocks. Synlett 2017, 28, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Seebach, D.; Colvin, E.; Lehr, F.; Weller, T. Nitroaliphatic compounds-ideal intermediates in organic synthesis. Chimia 1979, 33, 1–18. [Google Scholar]
- Luzzio, F.A. The Henry reaction: Recent examples. Tetrahedron 2001, 57, 915–945. [Google Scholar] [CrossRef]
- Henry, L. Nitro-alcohols. Compt. Rend. Hebd. Seances Acad. Sci. 1895, 120, 1265–1268. [Google Scholar]
- Heaney, F. The nitro group in organic synthesis. Synthesis 2001, 2001, 2528. [Google Scholar] [CrossRef] [Green Version]
- White, J.D.; Shaw, S. A new catalyst for the asymmetric Henry reaction: Synthesis of β-nitroethanols in high enantiomeric excess. Org. Lett. 2012, 14, 6270–6273. [Google Scholar] [CrossRef] [PubMed]
- Sasai, H. The Henry (nitroaldol) reaction. In Comprehensive Organic Synthesis II, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 543–570. [Google Scholar]
- Ginesta, X.; Pastó, M.; Pericas, M.A.; Riera, A. New stereodivergent approach to 3-amino-2, 3, 6-trideoxysugars. Enantioselective synthesis of daunosamine, ristosamine, acosamine, and epi-daunosamine. Org. Lett. 2003, 5, 3001–3004. [Google Scholar] [CrossRef]
- Guo, Z.-L.; Deng, Y.-Q.; Zhong, S.; Lu, G. Enantioselective synthesis of (R)-salmeterol employing an asymmetric Henry reaction as the key step. Tetrahedron Asymmetry 2011, 22, 1395–1399. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, S.; Xue, F.; Lou, G.; Zhang, H.; Ma, S.; Duan, W.; Wang, W. Catalytic enantioselective henry reactions of isatins: Application in the concise synthesis of (S)-(−)-spirobrassinin. Chem. Eur. J. 2011, 17, 7791–7795. [Google Scholar] [CrossRef]
- Gogoi, N.; Boruwa, J.; Barua, N.C. A total synthesis of (−)-bestatin using Shibasaki’s asymmetric Henry reaction. Tetrahedron Lett. 2005, 46, 7581–7582. [Google Scholar] [CrossRef]
- Rao, D.S.; Shivani, K.; Padhi, S.K. Immobilized arabidopsis thaliana hydroxynitrile lyase-catalyzed retro-Henry reaction in the synthesis of (S)-β-nitroalcohols. Appl. Biochem. Biotechnol. 2021, 193, 560–576. [Google Scholar] [CrossRef]
- Suami, T.; Sasai, H.; Matsuno, K.; Suzuki, N.; Fukuda, Y.; Sakanaka, O. Synthetic approach toward antibiotic tunicamycins—VI total synthesis of tunicamycins. Tetrahedron Lett. 1984, 25, 4533–4536. [Google Scholar] [CrossRef]
- Sato, K.-I.; Akai, S.; Shoji, H.; Sugita, N.; Yoshida, S.; Nagai, Y.; Suzuki, K.; Nakamura, Y.; Kajihara, Y.; Funabashi, M. Stereoselective and efficient total synthesis of optically active tetrodotoxin from D-glucose. J. Org. Chem. 2008, 73, 1234–1242. [Google Scholar] [CrossRef]
- Jakubec, P.; Cockfield, D.M.; Dixon, D.J. Total synthesis of (−)-nakadomarin A. J. Am. Chem. Soc. 2009, 131, 16632–16633. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D.J. Total synthesis of manzamine A and related alkaloids. J. Am. Chem. Soc. 2012, 134, 17482–17485. [Google Scholar] [CrossRef] [PubMed]
- Sasai, H.; Suzuki, T.; Arai, S.; Arai, T.; Shibasaki, M. Basic character of rare earth metal alkoxides. Utilization in catalytic carbon-carbon bond-forming reactions and catalytic asymmetric nitroaldol reactions. J. Am. Chem. Soc. 1992, 114, 4418–4420. [Google Scholar] [CrossRef]
- Ananthi, N.; Velmathi, S. Asymmetric Henry reaction catalysed by transition metal complexes: A short review. ChemInform 2013, 44, 87–108. [Google Scholar]
- Alvarez-Casao, Y.; Marques-Lopez, E.; Herrera, R.P. Organocatalytic enantioselective Henry reactions. Symmetry 2011, 3, 220–245. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Wang, F.; Huang, X.; Wen, Y.; Feng, X. A new copper (I)—tetrahydrosalen-catalyzed asymmetric Henry reaction and its extension to the synthesis of (S)-Norphenylephrine. Chem. Eur. J. 2007, 13, 829–833. [Google Scholar] [CrossRef]
- Arai, T.; Watanabe, M.; Yanagisawa, A. Practical Asymmetric Henry reaction catalyzed by a chiral diamine-Cu (OAc) 2 complex. Org. Lett. 2007, 9, 3595–3597. [Google Scholar] [CrossRef]
- Evans, D.A.; Seidel, D.; Rueping, M.; Lam, H.W.; Shaw, J.T.; Downey, C.W. A new copper acetate-bis (oxazoline)-catalyzed, enantioselective Henry reaction. J. Am. Chem. Soc. 2003, 125, 12692–12693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasai, H.; Suzuki, T.; Itoh, N.; Shibasaki, M. Catalytic asymmetric nitroaldol reactions. A new practical method for the preparation of the optically active lanthanum complex. Tetrahedron Lett. 1993, 34, 851–854. [Google Scholar] [CrossRef]
- Trost, B.M.; Yeh, V.S. A dinuclear Zn catalyst for the asymmetric nitroaldol (Henry) reaction. Angew. Chem. 2002, 114, 889–891. [Google Scholar] [CrossRef]
- Li, H.; Wang, B.; Deng, L. Enantioselective nitroaldol reaction of α-ketoesters catalyzed by Cinchona alkaloids. J. Am. Chem. Soc. 2006, 128, 732–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-G.; Jiang, J.-J.; Shi, M. Development of axially chiral bis (arylthiourea)-based organocatalysts and their application in the enantioselective Henry reaction. Tetrahedron Asymmetry 2007, 18, 2773–2781. [Google Scholar] [CrossRef]
- Otevrel, J.; Bobal, P. Biphenyl-based bis (thiourea) organocatalyst for asymmetric and syn-selective Henry reaction. Synthesis 2017, 49, 593–603. [Google Scholar]
- Chinchilla, R.; Nájera, C.; Sánchez-Agulló, P. Enantiomerically pure guanidine-catalysed asymmetric nitroaldol reaction. Tetrahedron Asymmetry 1994, 5, 1393–1402. [Google Scholar] [CrossRef]
- Tetour, D.; Novotná, M.; Hodačová, J. Enantioselective Henry reaction catalyzed by copper(II) complex of bis (trans-cyclohexane-1, 2-diamine)-based ligand. Catalysts 2021, 11, 41. [Google Scholar] [CrossRef]
- Bandini, M.; Piccinelli, F.; Tommasi, S.; Umani-Ronchi, A.; Ventrici, C. Highly enantioselective nitroaldol reaction catalyzed by new chiral copper complexes. Chem. Commun. 2007, 6, 616–618. [Google Scholar] [CrossRef]
- Tanaka, K.; Hachiken, S. Enantioselective Henry reaction catalyzed by trianglamine–Cu(OAc)2 complex under solvent-free conditions. Tetrahedron Lett. 2008, 49, 2533–2536. [Google Scholar] [CrossRef]
- Kowalczyk, R.; Sidorowicz, Ł.; Skarżewski, J. Asymmetric Henry reaction catalyzed by chiral secondary diamine-copper(II) complexes. Tetrahedron Asymmetry 2008, 19, 2310–2315. [Google Scholar] [CrossRef]
- Kowalczyk, R.; Skarżewski, J. Asymmetric nitroaldol reaction catalyzed by copper–diamine complexes: Selective construction of two contiguous stereogenic centers. Tetrahedron Asymmetry 2009, 20, 2467–2473. [Google Scholar] [CrossRef]
- Jin, W.; Li, X.; Wan, B. A Highly diastereo-and enantioselective copper(I)-catalyzed Henry reaction using a bis (sulfonamide)—diamine ligand. J. Org. Chem. 2011, 76, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Chunhong, Z.; Liu, F.; Gou, S. Application of chiral N, N′-dialkyl-1, 2-cyclohexanediamine derivatives in asymmetric copper(II)-catalyzed Henry reactions. Tetrahedron Asymmetry 2014, 25, 278–283. [Google Scholar] [CrossRef]
- Khlebnikova, T.B.; Konev, V.N.; Pai, Z.P. Levopimaric acid derived 1, 2-diamines and their application in the copper-catalyzed asymmetric henry reaction. Tetrahedron 2018, 74, 260–267. [Google Scholar] [CrossRef]
- Lu, G.; Zheng, F.; Wang, L.; Guo, Y.; Li, X.; Cao, X.; Wang, C.; Chi, H.; Dong, Y.; Zhang, Z. Asymmetric henry reaction catalyzed by cu (ii)-based chiral amino alcohol complexes with c2-symmetry. Tetrahedron Asymmetry 2016, 27, 732–739. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M.; Gröger, H. Nitroaldol reaction. In Comprehensive Asymmetric Catalysis: Supplement; Springer: Berlin/Heidelberg, Germany, 2004; Volume 1. [Google Scholar]
- Palomo, C.; Oiarbide, M.; Mielgo, A. Unveiling reliable catalysts for the asymmetric nitroaldol (Henry) reaction. Angew. Chem. Int. Ed. 2004, 43, 5442–5444. [Google Scholar]
- Boruwa, J.; Gogoi, N.; Saikia, P.P.; Barua, N.C. Catalytic asymmetric Henry reaction. Tetrahedron Asymmetry 2006, 17, 3315–3326. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Laso, A. Recent advances in the catalytic asymmetric nitroaldol (Henry) reaction. Eur. J. Org. Chem. 2007, 2007, 2561–2574. [Google Scholar] [CrossRef]
- Blay, G.; Hernandez-Olmos, V.; Pedro, J.R. Development of new N, N-ligands for the enantioselective copper(II)-catalyzed Henry reaction. Synlett 2011, 2011, 1195–1211. [Google Scholar] [CrossRef]
- Shashank, A.B.; Ramachary, D.B. Organocatalytic diastereoselective synthesis of chiral decalines through the domino Claisen–Schmidt/Henry reaction. Org. Biomol. Chem. 2015, 13, 5110–5114. [Google Scholar] [CrossRef]
- Knudsen, K.R.; Risgaard, T.; Nishiwaki, N.; Gothelf, K.V.; Jørgensen, K.A. The first catalytic asymmetric aza-Henry reaction of nitronates with imines: A novel approach to optically active β-nitro-α-amino acid-and α, β-diamino acid derivatives. J. Am. Chem. Soc. 2001, 123, 5843–5844. [Google Scholar] [CrossRef] [PubMed]
- Kogami, Y.; Nakajima, T.; Ikeno, T.; Yamada, T. Enantioselective Henry reaction catalyzed by salen-cobalt complexes. Synthesis 2004, 2004, 1947–1950. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Laso, A. Enantioselective Henry reactions under dual Lewis acid/amine catalysis using chiral amino alcohol ligands. Angew. Chem. Int. Ed. 2005, 44, 3881–3884. [Google Scholar] [CrossRef]
- Farina, V.; Reeves, J.T.; Senanayake, C.H.; Song, J.J. Asymmetric synthesis of active pharmaceutical ingredients. Chem. Rev. 2006, 106, 2734–2793. [Google Scholar] [CrossRef]
- Mellah, M.; Voituriez, A.; Schulz, E. Chiral sulfur ligands for asymmetric catalysis. Chem. Rev. 2007, 107, 5133–5209. [Google Scholar] [CrossRef]
- Trippé, G.; Canevet, D.; Le Derf, F.; Frère, P.; Sallé, M. An extended tetrathiafulvalene redox-ligand incorporating a thiophene spacer. Tetrahedron Lett. 2008, 49, 5452–5454. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, R.P.; Parvez, M.; Sutherland, T.C.; Viccars, J. Synthesis and Optical and Electronic Properties of Thiophene Derivatives; Wiley Online Library: Hoboken, NJ, USA, 2009. [Google Scholar]
- Chen, X.; Elsenbaumer, R.L. Synthesis of thieno [3, 4-d]-1, 3-dithiol-2-one derivatives. Tetrahedron Lett. 2009, 50, 3750–3752. [Google Scholar] [CrossRef]
- Ley, S.V.; Smith, S.C.; Woodward, P.R. Further reactions of t-butyl 3-oxobutanthioate and t-butyl 4-diethyl-phosphono-3-oxobutanthioate: Carbonyl coupling reactions, amination, use in the preparation of 3-acyltetramic acids and application to the total synthesis of fuligorubin A. Tetrahedron 1992, 48, 1145–1174. [Google Scholar] [CrossRef]
- Fondo, M.; Corredoira-Vázquez, J.; GarcíaDeibe, A.M.; Sanmartín-Matalobos, J. An easy approach to obtain alcohol-amines by reduction of alcohol functionalized imines. Proceedings 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Sreenath, K.; Yuan, Z.; Macias-Contreras, M.; Ramachandran, V.; Clark, R.J.; Zhu, L. Dual role of acetate in copper (II) acetate catalyzed dehydrogenation of chelating aromatic secondary amines: A kinetic case study of copper-catalyzed oxidation reactions. Eur. J. Inorg. Chem. 2016, 2016, 3728–3743. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, X.; Li, Z.; Huang, Z.; Chen, F. Ketoreductase catalyzed stereoselective bioreduction of α-nitro ketones. Org. Biomol. Chem. 2019, 17, 3575–3580. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.D.; Lai, W.H.; Hu, D.; Chen, Z.; Wu, A.A.; Ruan, Y.P.; Zhou, Z.H.; Chen, H.B. Highly enantioselective henry reactions of aromatic aldehydes catalyzed by an amino alcohol–copper(II) complex. Chem. Eur. J. 2012, 18, 10515–10518. [Google Scholar] [CrossRef] [PubMed] [Green Version]





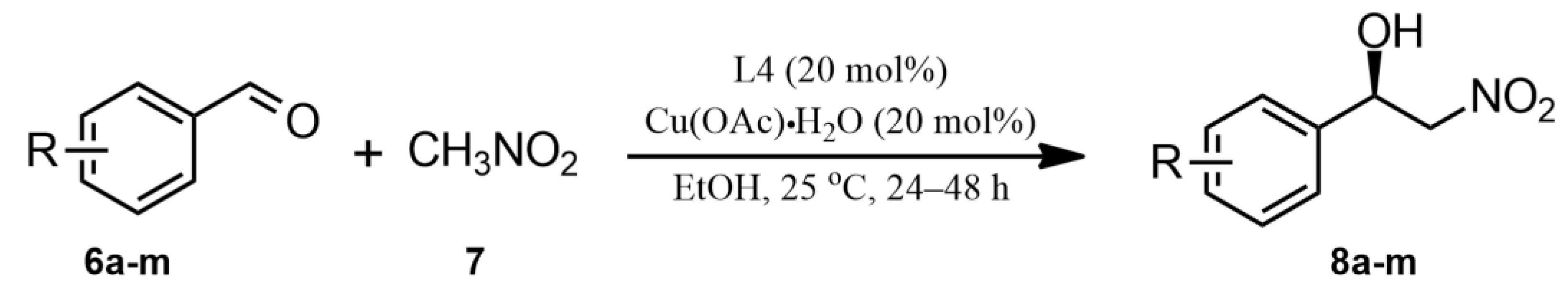

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry [a] | Ligands | L:Cu(OAc)2·H2O (mol%) | Temp [t] | Time [T/h] | Yield (%) [b] | ee % [c] |
|---|---|---|---|---|---|---|
| 1. | L1 | 20 | 25 °C | 24 | 99 | 92.3 |
| 2. | L2 | 20 | 25 °C | 24 | 90 | 94.0 |
| 3. | L3 | 20 | 25 °C | 48 | 91 | 89.9 |
| 4. | L4 | 20 | 25 °C | 24 | 99 | 94.6 |
| 5. | L5 | 20 | 25 °C | 48 | 89 | 90.0 |
| 6. | L4 | 20 | 10 °C | 48 | 87 | 90.4 |
| 7. | L4 | 5 | 25 °C | 24 | 80 | 87.1 |
| 8. | L4 | 10 | 25 °C | 24 | 88 | 85.2 |
| 9. | L4 | 25 | 25 °C | 24 | 99 | 94.0 |
| Entry [a] | Solvent | Time (h) | Yield (%) [b] | ee % [c] |
|---|---|---|---|---|
| 1. | MeOH | 24 | 88 | 85.3 |
| 2. | i-PrOH | 48 | 80 | 86.6 |
| 3. | t-BuOH | 48 | 99 | 82.6 |
| 4. | THF | 48 | 79 | 68.3 |
| Entry [a] | Metal Salt | Time [T/h] | Yield (%) [b] | ee (%) [c] |
|---|---|---|---|---|
| 1. | Cu(OAc)2·H2O | 24 | 99 | 94.6 |
| 2. | Cu(OAc)2·nH2O | 24 | 97 | 94.3 |
| 3. | Zn(OTf)2 | 48 | 99 | 6.6 |
| 4. | Cu(OTf)2 | 72 | 20 | 1 |
| 5. | CuBr2 | 72 | - | - |
| 6. | CuCl2 | 72 | - | - |
| 7. | Zn(OAc)2·2H2O | 72 | 50 | 19.0 |
| Entry [a] | R (6a–m) | Products | Time (h) | Yield (%) [b] | ee % [c] |
|---|---|---|---|---|---|
| 1. | 2-NO2 | 8a | 24 | 99 | 94.6 (R) |
| 2. | 4-NO2 | 8b | 24 | 96 | 81.3 (R) |
| 3. | 3-NO2 | 8c | 48 | 91 | 81.2 (R) |
| 4. | 4-Br | 8d | 48 | 80 | 76.4 (R) |
| 5. | Naph | 8e | 48 | 66 | 75.1 (R) |
| 6. | 4-CF3 | 8f | 24 | 82 | 58.9 (R) |
| 7. | 2,4-Cl | 8g | 48 | 86 | 53.0 (R) |
| 8. | 4-Cl | 8h | 48 | 66 | 73.1 (R) |
| 9. | 4-F | 8i | 48 | 75 | 60.9 (R) [d] |
| 10. | 4-OCH3 | 8j | 24 | 77 | 63.9 (R) [d] |
| 11. | 4-CH3 | 8k | 24 | 85 | 81.3 (R) |
| 12. | 3-CH3 | 8l | 48 | 88 | 60.8 (R) [d] |
| 13. | H | 8m | 48 | 82 | 89.2 (R) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alammari, A.S.; Al-Majid, A.M.; Barakat, A.; Alshahrani, S.; Ali, M.; Islam, M.S. Asymmetric Henry Reaction of Nitromethane with Substituted Aldehydes Catalyzed by Novel In Situ Generated Chiral Bis(β-Amino Alcohol-Cu(OAc)2·H2O Complex. Catalysts 2021, 11, 1208. https://doi.org/10.3390/catal11101208
Alammari AS, Al-Majid AM, Barakat A, Alshahrani S, Ali M, Islam MS. Asymmetric Henry Reaction of Nitromethane with Substituted Aldehydes Catalyzed by Novel In Situ Generated Chiral Bis(β-Amino Alcohol-Cu(OAc)2·H2O Complex. Catalysts. 2021; 11(10):1208. https://doi.org/10.3390/catal11101208
Chicago/Turabian StyleAlammari, Abdullah Saleh, Abdullah Mohammed Al-Majid, Assem Barakat, Saeed Alshahrani, Mohammad Ali, and Mohammad Shahidul Islam. 2021. "Asymmetric Henry Reaction of Nitromethane with Substituted Aldehydes Catalyzed by Novel In Situ Generated Chiral Bis(β-Amino Alcohol-Cu(OAc)2·H2O Complex" Catalysts 11, no. 10: 1208. https://doi.org/10.3390/catal11101208