
Porous Hexacyanometallate(III) Complexes as Catalysts in the Ring-Opening Copolymerization of CO2 and Propylene Oxide


Abstract
:1. Introduction
2. Results
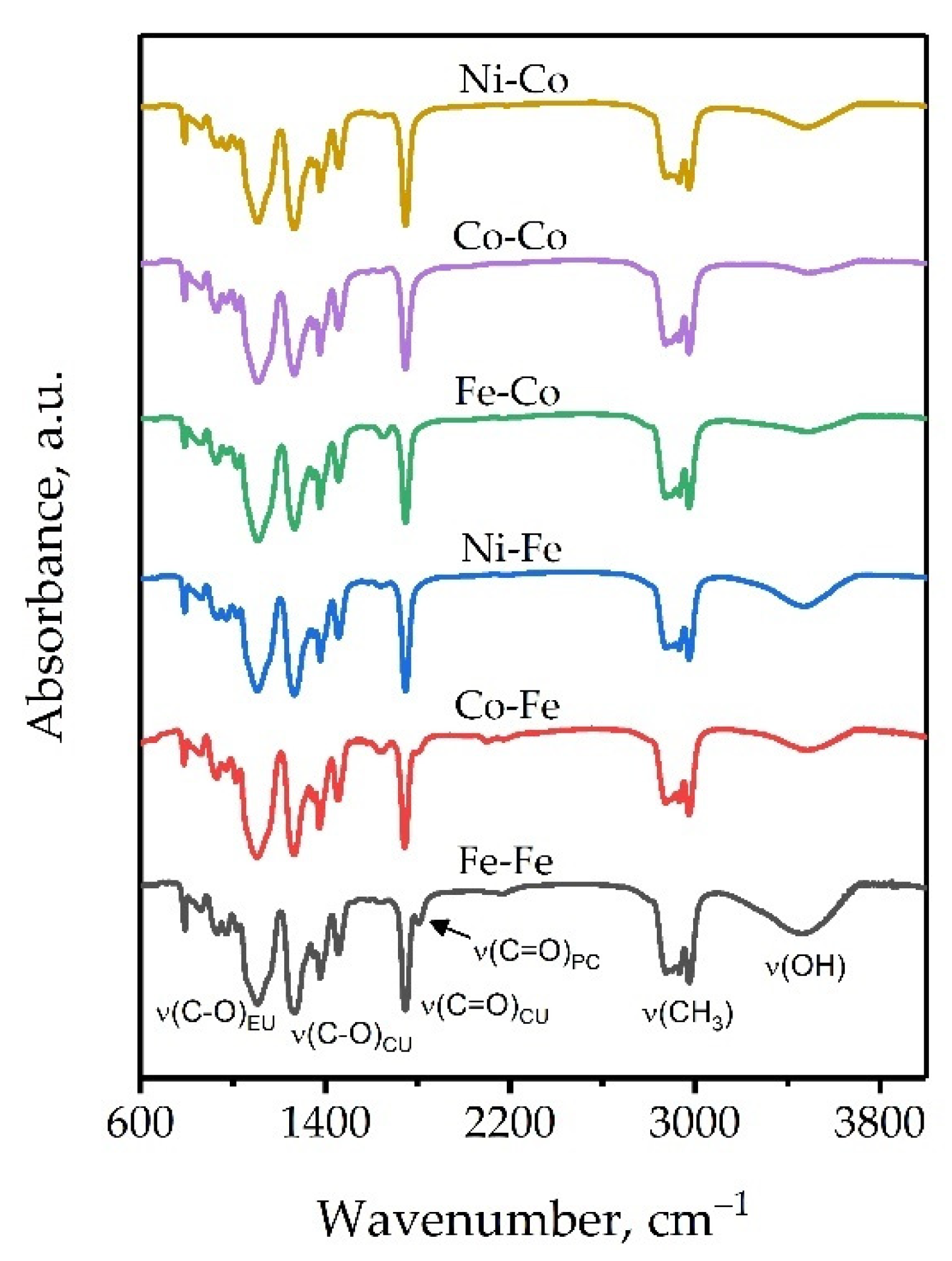
2.1. Porous Hexacyanometallates(III) Characterization
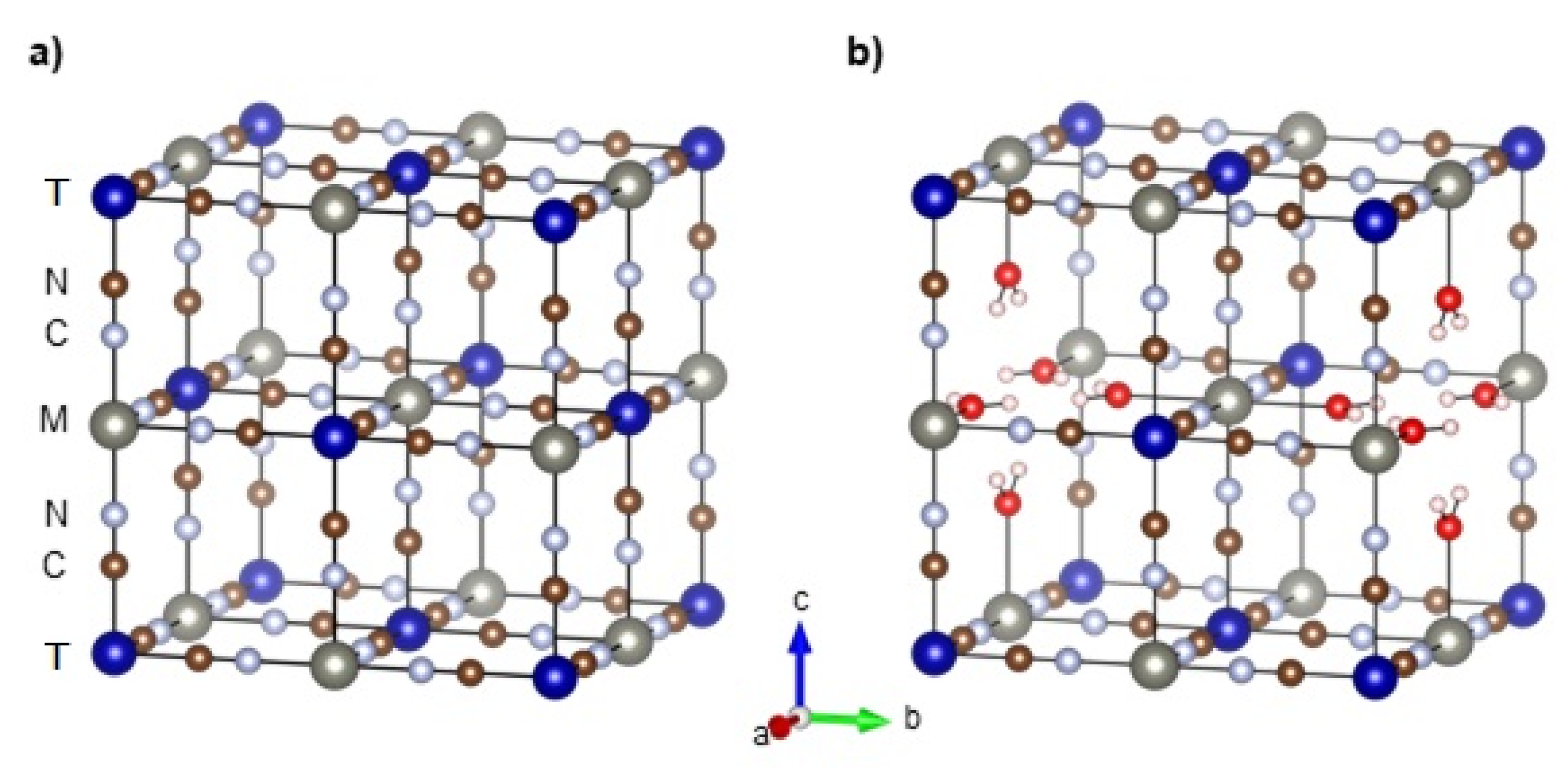
2.1.1. Crystal and Electronic Structure
2.1.2. Thermal Stability, Degree of Hydration, and Dehydration Process
2.1.3. Chemical Composition
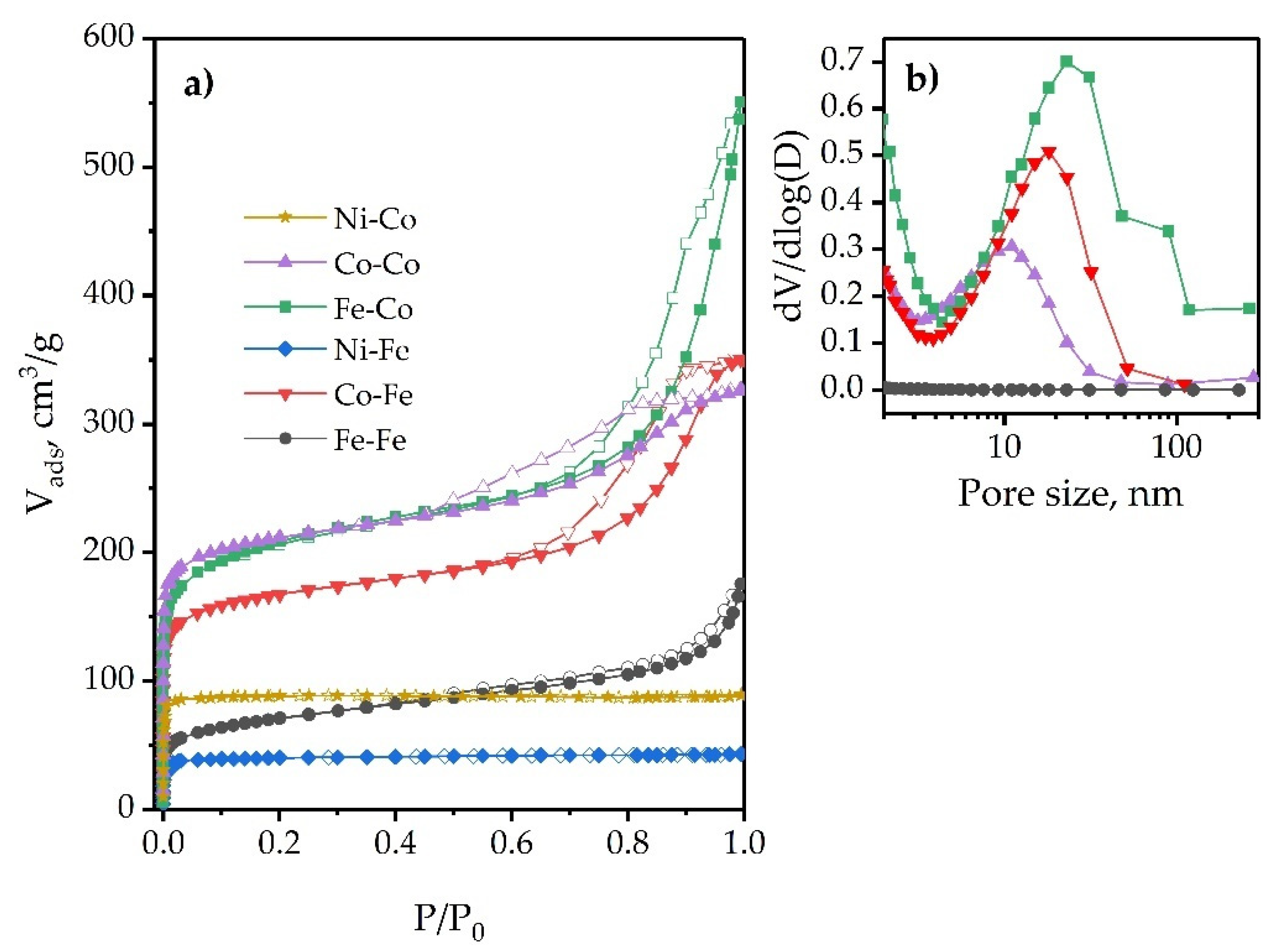
2.1.4. Morphology and Textural Properties
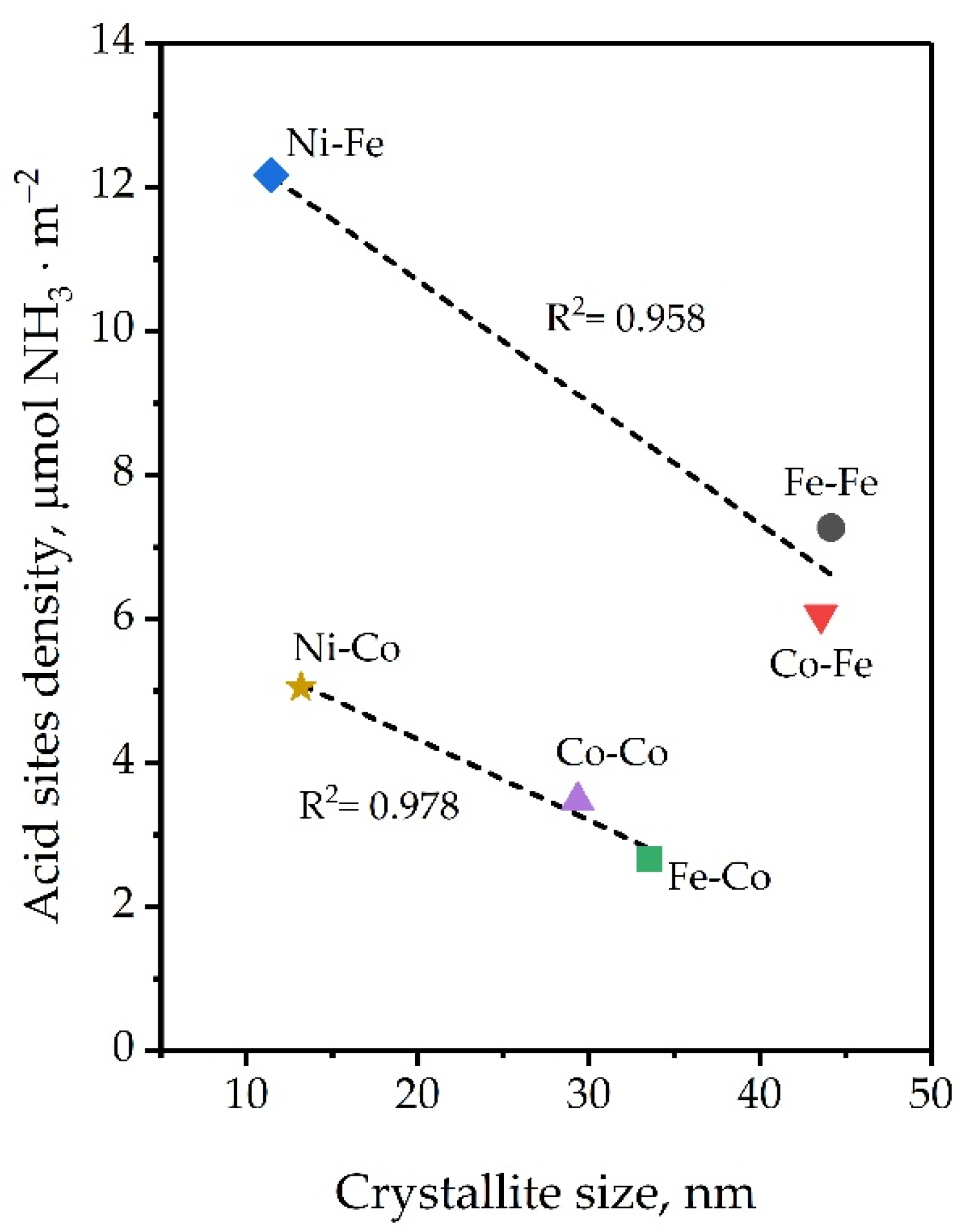
2.1.5. Density of Coordinatively Unsaturated Metal Sites (CUMSs)
2.2. Catalytic Activity Tests
Comparison of the Performance of the Studied Compounds with Other Cyanometallate Catalysts for CO2/PO Copolymerization
3. Materials and Methods
3.1. Materials
3.2. Preparation of Porous Hexacyanometallates(III)
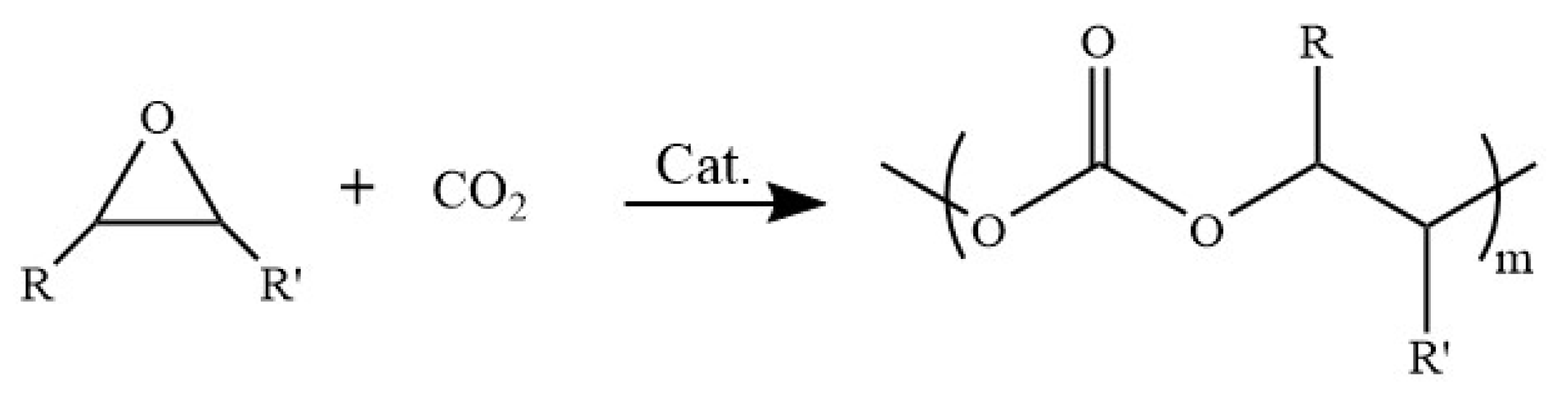
3.3. CO2/PO Copolymerization Procedure
3.4. Characterization
3.4.1. Porous Hexacyanometallate(III) Characterization
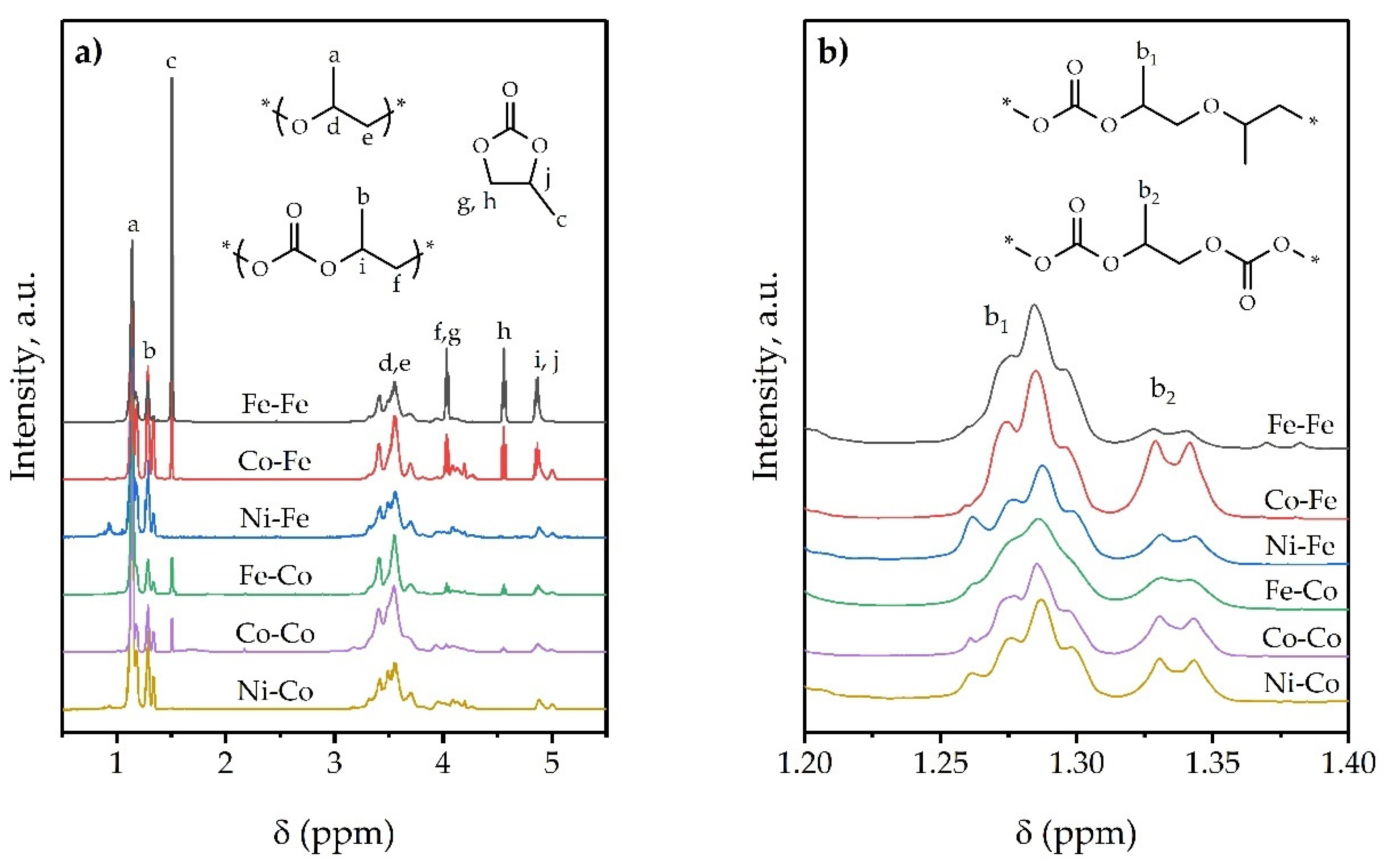
3.4.2. Polymer Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piernas-Muñoz, M.J.; Castillo-Martínez, E. Prussian Blue-Based Batteries; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Wojdel, J.C.; Bromley, S.T.; Illas, F.; Jansen, J.C. Development of realistic models for Double Metal Cyanide catalyst active sites. J. Mol. Model. 2007, 13, 751–756. [Google Scholar] [CrossRef]
- Imanishi, N.; Morikawa, T.; Kondo, J.; Yamane, R.; Takeda, Y.; Yamamoto, O.; Sakaebe, H.; Tabuchi, M. Lithoum intercalation behavior of iron cyanometallates. J. Power Source 1999, 81, 530–534. [Google Scholar] [CrossRef]
- Hanusa, T.P. Cyanide complexes of the transition metals. In Encyclopedia of Inorganic Chemistry and Bioinorganic Chemistry, 1st ed.; King, R.B., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Hibble, S.J.; Cheyne, S.M.; Hannon, A.C.; Eversfield, S.G. CuCN: A polymorphic material. Structure of one form determine from total neutron diffraction. Inorg. Chem. 2002, 41, 4990–4992. [Google Scholar] [CrossRef] [PubMed]
- Hibble, S.J.; Wood, G.B.; Bilbé, E.J.; Pohl, A.H.; Tucker, M.G.; Hannon, A.C.; Chippindale, A.M. Structures and negative thermal expansion properties of the one-dimensional cyanides, CuCN, AgCN and AuCN. Z. Kristallogr. 2010, 225, 457–462. [Google Scholar] [CrossRef]
- Hibble, S.J.; Cheyne, S.M.; Hannon, A.C.; Eversfield, S.G. Beyond bragg scattering: The structure of AgCN determined from total neutron diffraction. Inorg. Chem. 2002, 41, 1042–1044. [Google Scholar] [CrossRef]
- Zhang, K.; Lee, T.H.; Bubach, B.; Ostadhassan, M.; Jang, H.W.; Choi, J.; Shokouhimehr, M. Layered metal-organic framework based on tetracyanonickelate as a cathode material for in situ Li-ion storage. RSC Adv. 2019, 9, 21363–21370. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, T.; Fukunaga, M.; Takahashi, M.; Takeda, M. Study by X-ray crystallography and Mössbauer spectroscopy of the layered compounds with two-dimensional metal complex iron(II) tetracyanonicketale(II). Mol. Cryst. Liq. Cryst. 1994, 244, 331–336. [Google Scholar] [CrossRef]
- Hibble, S.J.; Chippindale, A.M.; Bilbé, E.J.; Marelli, E.; Harris, P.J.F.; Hannon, A.C. Structures of Pd(CN)2 and Pt(CN)2: Intrinsically nanocrystalline materials? Inorg. Chem. 2011, 50, 104–113. [Google Scholar] [CrossRef]
- Weiser, H.B.; Milligan, W.O.; Bates, J.B. X-Ray diffraction studies on heavy-metal iron-cyanides. J. Phys. Chem. 1942, 46, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, F.; Samain, L.; Long, G.J. Characterization and utilization of Prussian blue and its pigments. Dalton Trans. 2016, 45, 18018–18044. [Google Scholar] [CrossRef]
- Müller, H.; Müller, W.; Wehner, M.; Liewald, H. Artists’ colors. In Ullman’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012; Volume 4, pp. 241–254. [Google Scholar]
- Sato, O.; Iyoda, T.; Fujishima, A.; Hashimoto, K. Photoinduced magnetization of cobalt-iron cyanide. Science 1996, 272, 704–705. [Google Scholar] [CrossRef]
- Fornasieri, G.; Bordage, A.; Bleuzen, A. Magnetism and photomagnetism of Prussian blue analogue nanoparticles. Eur. J. Inorg. Chem. 2018, 259–271. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/full/10.1002/ejic.201700819 (accessed on 24 November 2021). [CrossRef] [Green Version]
- Yuan-Fu, X.; Zhi, Y.; Yong, S.; Hong-Bo, H.; Xiao-Zeng, Y.; Zhou, X.Z.; Li, Z.W.; Kunkel, H.P.; Williams, G. Mössbauer evidence for ferromagnetic ordering in copper-iron cyanometallates. Chin. Phys. Lett. 2002, 19, 595–598. [Google Scholar] [CrossRef]
- Ferlay, S.; Mallah, T.; Ouahès, R.; Veillet, P.; Verdaguer, M. A room-temperature organometallic magnet based on Prussian blue. Nature 1995, 378, 701–703. [Google Scholar] [CrossRef]
- Rogez, G.; Marvilliers, A.; Sarr, P.; Parsons, S.; Teat, S.J.; Ricard, L.; Mallah, T. Tuning the optical properties of Prussian blue-like complexes. Chem. Commun. 2002, 14, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Singh, R.P.; Bala, C. Solid membranes for copper hexacyanoferrate(II) as thallium(I) sensitive electrode. Anal. Lett. 1982, 15, 1557–1563. [Google Scholar] [CrossRef]
- Tani, Y.; Eun, H.; Umezawa, Y. A cation selective electrode based on copper(II) and nickel(II) hexacyanoferrates: Dual response mechanism, selective uptake or adsorption of analyte cations. Electrochim. Acta 1998, 43, 3431–3441. [Google Scholar] [CrossRef]
- Li, W.; Han, C.; Cheng, G.; Chou, S.; Liu, H.; Dou, S. Chemical properties, structural properties, and energy storage applications of Prussian blue analogues. Small 2019, 15, 1900470. [Google Scholar] [CrossRef]
- Hurlbutt, K.; Wheeler, S.; Capone, I.; Pasta, M. Prussian blue analogs as battery materials. Joule 2018, 2, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Kaye, S.S.; Long, J.R. The role of vacancies in the hydrogen storage properties of Prussian blue analogues. Catal. Today 2007, 120, 311–316. [Google Scholar] [CrossRef]
- Reguera, L.; Balmaseda, J.; Krap, C.P.; Reguera, E. Hydrogen storage in porous transition metal nitroprussides. J. Phys. Chem. C 2008, 112, 10490–10501. [Google Scholar] [CrossRef]
- Zamora, B.; Roque, J.; Balmaseda, J.; Reguera, E. Methane storage in Prussian blue analogues and related porous solids: Nature of the involver adsorption forces. Z. Anorg. Allg. Chem. 2010, 636, 2574–2578. [Google Scholar] [CrossRef]
- Chapman, K.W.; Southon, P.D.; Weeks, C.L.; Kepert, C.J. Reversible hydrogen gas uptake in nanoporous Prussian blue analogues. Chem. Commun. 2005, 26, 3322–3324. [Google Scholar] [CrossRef]
- Barton, G.B.; Hepworth, J.L.; McClanahan, E.D.; Moore, R.L.; Van Tuyl, H.H. Chemical processing wastes: Recovering fission products. Ind. Eng. Chem. 1958, 50, 212–216. [Google Scholar] [CrossRef]
- Valvekens, P.; De Vos, D. Double metal cyanides as heterogeneous catalysts for organic reactions. In New Materials for Catalytic Applications; Parvulescu, V.I., Kemnitz, E., Eds.; Elvesier: San Diego, CA, USA, 2016; pp. 1–12. [Google Scholar]
- Satyarthi, J.K.; Srinivas, D.; Ratnasamy, P. Influence of surface hydrophobicity on the esterification of fatty acids over solid catalysts. Energy Fuels 2010, 24, 2154–2161. [Google Scholar] [CrossRef]
- Srinivas, D.; Satyarthi, J.K. Biodiesel production from vegetable oils and animal fat over solid acid double-metal cyanide catalysts. Catal. Surv. Asia 2011, 15, 145–160. [Google Scholar] [CrossRef]
- Srivastava, R.; Srinivas, D.; Ratnasamy, P. Fe-Zn double-metal cyanide complexes as novel, solid transesterification catalysts. J. Catal. 2006, 241, 34–44. [Google Scholar] [CrossRef]
- Yan, F.; Yuan, Z.; Lu, P.; Luo, W.; Yang, L.; Deng, L. Fe-Zn double-metal cyanide complexes catalyzed biodiesel production from high-acid-value oil. Renew. Energy 2011, 36, 2026–2031. [Google Scholar] [CrossRef]
- Peeters, A.; Valvekens, P.; Vermoortele, F.; Ameloot, R.; Kirschhock, C.; De Vos, D. Lewis acid double metal cyanide catalysts for hydroamination of phenylacetylene. Chem. Commun. 2011, 47, 4114–4116. [Google Scholar] [CrossRef] [Green Version]
- Patil, M.V.; Yadav, M.K.; Jasra, R.V. Prins condensation for synthesis of nopol from β-pinene and paraformaldehyde on novel Fe-Zn double metal cyanide solid acid catalyst. J. Mol. Catal. A Chem. 2007, 273, 39–47. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Grirrane, A.; Reguera, E.; García, H. Mixed (Fe2+ and Cu2+) double metal hexacyanocobaltates as solid catalyst for the aerobic oxidation of oximes to carbonyl compounds. J. Catal. 2014, 311, 386–392. [Google Scholar] [CrossRef]
- Belner, R.J. Method of Making a Polyether Using a Double Metal Cyanide Complex Compound. U.S. Patent 3,278,458A, 11 October 1996. [Google Scholar]
- Herold, R.J.; Livigni, R.A. Hexacyanometalate salt complexes as catalysts for epoxide polymerization. In Polymerization Kinetics and Technology; Platzer, N.A.J., Ed.; American Chemical Society: Washington, DC, USA, 1973; pp. 208–229. [Google Scholar]
- Kruper, W.J.; Swart, D.J. Carbon Dioxide Oxirane Copolymers Prepared Using Double Metal Cyanide Complexes. U.S. Patent 4,500,704A, 19 February 1985. [Google Scholar]
- Dharman, M.M.; Yu, J.; Ahn, J.; Park, D. Selective production of cyclic carbonate over polycarbonate using a double metal cyanide-quaternary ammonium salt catalyst system. Green Chem. 2009, 11, 1754–1757. [Google Scholar] [CrossRef]
- Saikia, L.; Satyarthi, J.K.; Gonnade, R.; Srinivas, D.; Ratnasamy, P. Double metal cyanides as efficient solid acid catalysts for synthesis of β-amino alcohols under solvent-free conditions. Catal. Lett. 2008, 123, 24–31. [Google Scholar] [CrossRef]
- Peeters, A.; Valvekens, P.; Ameloot, R.; Sankar, G.; Kirschhock, C.E.A.; De Vos, D.E. Zn-Co double metal cyanides as heterogeneous catalysts for hydroamination: A structure-activity relationship. ACS Catal. 2013, 3, 597–607. [Google Scholar] [CrossRef]
- Zhang, X.; Hua, Z.; Chen, S.; Liu, F.; Sun, X.; Qi, G. Role of zinc chloride and complexing agents in highly active double metal cyanide catalysts for ring-opening polymerization of propylene oxide. Appl. Catal. A 2007, 325, 91–98. [Google Scholar] [CrossRef]
- Lee, I.K.; Ha, J.Y.; Cao, C.; Park, D.; Ha, C.; Kim, I. Effect of complexing agents of double metal cyanide catalyst on the copolymerization of cyclohexene oxide and carbon dioxide. Catal. Today 2009, 148, 389–397. [Google Scholar] [CrossRef]
- Fernández-Dacosta, C.; Van der Spek, M.; Hung, C.R.; Oregionni, G.D.; Skagestad, R.; Parihar, P.; Gokak, D.T.; Strømman, A.H.; Ramirez, A. Prospective techno-economic and environmental assessment of carbon capture at a refinery and CO2 utilisation in polyol synthesis. J. CO2 Util. 2017, 21, 405–422. [Google Scholar] [CrossRef]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–364. [Google Scholar] [CrossRef]
- Von der Assen, N.; Bardow, A. Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: Insight from an industrial case study. Green Chem. 2014, 16, 3272–3280. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Lu, L.; Cheng, Y.; Xu, N.; Pan, L.; Lin, Q.; Wang, Y. Clean and rapid synthesis of double metal cyanide complexes using mechanochemistry. Green Chem. 2011, 12, 2701–2703. [Google Scholar] [CrossRef]
- Dai, C.; Zhu, Q.; Pang, H.; Zhu, L.; Lin, Q. Rapid copolymerization of carbon dioxide and propylene oxide catalyzed by double metal cyanide complexes in an ultrasonic field. Mater. Lett. 2016, 180, 89–92. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, Q. Coupling reaction of CO2 and propylene oxide catalyzed by DMC with co-complexing agents incorporated via ball milling. J. Mol. Catal. A Chem. 2014, 390, 63–68. [Google Scholar] [CrossRef]
- Zhang, X.H.; Chen, S.; Wu, X.M.; Sun, X.K.; Liu, F.; Qi, G.R. Highly active double metal cyanide complexes. Effect of central metal and ligand on reaction of epoxide/CO2. Chin. Chem. Lett. 2007, 18, 887–890. [Google Scholar] [CrossRef]
- Qiang, L.; Zhifang, G.; Lisha, P.; Xue, X. Zn-Cr double metal cyanide catalysts synthesized by ball milling for the copolymerization of CO2/propylene oxide, phtalic anhydride/propylene oxide, and CO2/propylene oxide/phtalic anhydride. Catal. Commun. 2015, 64, 114–118. [Google Scholar] [CrossRef]
- Chen, S.; Hua, Z.; Fang, Z.; Qi, G. Copolymerization of carbon dioxide and propylene oxide with highly effective hexacyanocobaltate(III)-based coordination catalyst. Polymer 2004, 45, 6519–6524. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, X. Carbon dioxide-based copolymers: Environmental benefits of PPC, and industrially viable catalyst. Biotechnol. J. 2010, 5, 1164–1180. [Google Scholar] [CrossRef] [PubMed]
- Kember, M.R.; Buchard, A.; Williams, C.K. Catalysts for CO2/epoxide copolymerization. Chem. Commun. 2011, 47, 141–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Qin, Y.; Zhao, X.; Wang, F.; Wang, X. Selective synthesis of oligo(carbonate-ether) diols from copolymerization of CO2 and propylene oxide under zinc-cobalt double metal cyanide complex. J. Polym. Res. 2012, 19, 9878–9887. [Google Scholar] [CrossRef]
- Robertson, N.J.; Qin, Z.; Dallinger, G.C.; Lobkovsky, E.B.; Lee, S.; Coates, G.W. Two-dimensional double metal cyanide complexes: Highly active catalysts for the homopolymerization of propylene oxide and copolymerization of propylene oxide and carbon dioxide. Dalton Trans. 2006, 45, 5390–5395. [Google Scholar] [CrossRef]
- Alferov, K.; Wang, S.; Li, T.; Xiao, M.; Guan, S.; Meng, Y. Co-Ni cyanide bi-metal catalysts: Copolymerization of carbon dioxide with propylene oxide and chain transfer agents. Catalysts 2019, 9, 632. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, S.H.; Duyker, S.G.; Southon, P.D.; Peterson, V.K.; Kepert, C.J. Identification of bridged CO2 binding in a Prussian blue analogue using neutron powder diffraction. Chem. Commun. 2013, 49, 9404–9406. [Google Scholar] [CrossRef]
- Ludi, A.; Güedel, H.U.; Rüegg, M. The structural chemistry of Prussian blue analogs. A single-crystal study of manganese(II) hexacyanocobaltate(III), Mn3[Co(CN)6]2·xH2O. Inorg. Chem. 1970, 9, 2224–22278. [Google Scholar] [CrossRef]
- Kuyper, J.; Boxhoorn, G. Hexacyanometallate salts used as alkene-oxide polymerization catalysts and molecular sieves. J. Catal. 1987, 105, 163–174. [Google Scholar] [CrossRef]
- Kaye, S.S.; Long, J.R. Hydrogen storage in the dehydrated Prussian blue analogues M3[Co(CN)6]2 (M = Mn, Fe, Co, Ni, Cu, Zn). J. Am. Chem. Soc. 2005, 127, 6506–6507. [Google Scholar] [CrossRef]
- Autie-Castro, G.; Autie, M.; Reguera, E.; Santamaría-González, J.; Moreno-Tost, R.; Rodríguez-Castellón, E.; Jiménez-López, A. Adsorption and separation of light alkane hydrocarbons by porous hexacyanocobaltates(III). Surf. Interface Anal. 2009, 41, 730–734. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Dunbar, K.R.; Heintz, R.A. Chemistry of transition metal cyanide compounds: Modern perspectives. In Progress in Inorganic Chemistry; Karlin, K.D., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1997; Volume 45, pp. 283–391. [Google Scholar] [CrossRef]
- Ghosh, S.N. Infrared spectra of the Prussian blue analogs. J. Inorg. Nucl. Chem. 1974, 36, 2465–2466. [Google Scholar] [CrossRef]
- Nakagawa, I.; Shimanouchi, T. Far infra-red spectra of some hexacyano-complex salts. Spectrochim. Acta A Mol. Spectrosc. 1970, 26, 131–141. [Google Scholar] [CrossRef]
- Shriver, D.F.; Sheriver, S.A.; Anderson, S.E. Ligand field strength of the nitrogen end of cyanide and structures of cubic cyanide polymers. Inorg. Chem. 1965, 4, 725–730. [Google Scholar] [CrossRef]
- Ng, C.W.; Ding, J.; Shi, Y.; Gan, L.M. Structure and magnetic properties of copper(II) hexacyanoferrate(III) compound. J. Phys. Chem. Solids 2001, 62, 767–775. [Google Scholar] [CrossRef]
- Ojwang, D.O.; Grins, J.; Wardecki, D.; Valvo, M.; Renman, V.; Häggström, L.; Ericsson, T.; Gustafsson, T.; Mahmoud, A.; Hermann, R.P.; et al. Structure characterization and properties of K-containing copper hexacyanoferrate. Inorg. Chem. 2016, 55, 5924–5934. [Google Scholar] [CrossRef]
- Avila, M.; Reguera, L.; Rodríguez-Hernández, J.; Balmaseda, J.; Reguera, E. Porous framework of T2[Fe(CN)6]·xH2O with T = Co, Ni, Cu, Zn and H2 storage. J. Solid State Chem. 2008, 181, 2899–2907. [Google Scholar] [CrossRef]
- Sebastian, J.; Srinivas, D. Effects of method of preparation on catalytic activity of Co-Zn double-metal cyanide catalysts for copolymerization of CO2 and epoxide. Appl. Catal. A Gen. 2014, 482, 300–308. [Google Scholar] [CrossRef]
- Reguera, E.; Fernández-Bertrán, J.; Balmaseda, J. The existence of ferrous ferricyanide. Transit. Met. Chem. 1999, 24, 648–654. [Google Scholar] [CrossRef]
- Fluck, E.; Kerler, W.; Neuwirth, W. The Mössbauer effect and its significance in chemistry. Angew. Chem. Int. Ed. 1963, 2, 277–287. [Google Scholar] [CrossRef]
- Ito, A.; Suenaga, M.; Ono, K. Mössbauer study of soluble Prussian blue, insoluble Prussian blue and Turnbull’s blue. J. Chem. Phys. 1968, 48, 3597–3599. [Google Scholar] [CrossRef]
- Amr El-Sayed, M.F.; Sheline, R.K. The position of the C≡N stretching frequency in organic and inorganic molecules. J. Inorg. Nucl. Chem. 1958, 6, 187–193. [Google Scholar] [CrossRef]
- Li, K.; Xue, D. Estimation of electronegativity values of elements in different valence states. J. Phys. Chem. A 2006, 110, 11332–11337. [Google Scholar] [CrossRef]
- Mullica, D.F.; Milligan, W.O.; Beall, G.W.; Reeves, W.L. Crystal structure of Zn3[Co(CN)6]2·12H2O. Acta Cryst. B 1978, 34, 3558–3561. [Google Scholar] [CrossRef] [Green Version]
- Scherrer, P. Bestimmung der grösse und der inneren struktur von kolloidteilchen mittels röntgenstrahlen. Nachr. Ges. Wiss. Gottingen Math. Phys. Kl. 1918, 2, 98–100. [Google Scholar]
- Williamson, G.K.; Hall, W.H. X-ray line broadening from filed aluminium and wolfram. Acta Metall. 1953, 1, 22–31. [Google Scholar] [CrossRef]
- Roque, J.; Reguera, E.; Balmaseda, J.; Rodríguez-Hernández, J.; Reguera, L.; del Castillo, L.F. Porous hexacyanocobaltates(III): Role of the metal on the framework properties. Microporous Mesoporous Mater. 2007, 103, 57–71. [Google Scholar] [CrossRef]
- Huang, Y.J.; Qi, G.R.; Chen, L.S. Effects of morphology and compositions on catalytic performance of double metal cyanide complex catalyst. Appl. Catal. A 2003, 240, 263–271. [Google Scholar] [CrossRef]
- Nandan, B.; Venugopal, B.; Amirthapandian, S.; Panigrahi, B.K.; Thangadurai, P. Effect of Pd ion doping in the band gap of SnO2 nanoparticles: Structural and optical studies. J. Nanopart. Res. 2013, 15, 1–11. [Google Scholar] [CrossRef]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the BET equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 2007, 160, 49–56. [Google Scholar]
- Lippens, B.C.; Boer, J.H. Studies on pore systems in catalysts V. The t method. J. Catal. 1965, 4, 319–323. [Google Scholar] [CrossRef]
- Dubinin, M.M. Physical adsorption of gases and vapors in micropores. In Progress in Surface Science and Membrane Science, 1st ed.; Cadenheat, D.A., Ed.; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Balmaseda, J.; Reguera, E.; Rodríguez-Hernández, J.; Reguera, L.; Autie, M. Behavior of transition metals ferricyanides as microporous materials. Microporous Mesoporous Mater. 2006, 26, 222–236. [Google Scholar] [CrossRef]
- Márquez, C.; Rivera-Torrente, M.; Paalanen, P.P.; Weckhuysen, B.M.; Cirujano, F.G.; De Vos, D.; De Baerdemaeker, T. Increasing the abailability of active sites in Zn-Co double metal cyanides by dispersion onto a SiO2 support. J. Catal. 2017, 354, 92–99. [Google Scholar] [CrossRef]
- Sreeprasanth, P.S.; Srivastava, R.; Srinivas, D.; Ratnasamy, P. Hydrophobic, solid acid catalysts for production of biofuels and lubricants. Appl. Catal. A Gen. 2006, 314, 148–159. [Google Scholar] [CrossRef]
- Jadhav, A.R.; Bandal, H.A.; Kim, H. Synthesis of substituted amines: Catalytic reductive amonation of carbonyl compounds using Lewis acid Zn-Co-double metal cyanide/polymethylhydrosiloxane. Chem. Eng. J. 2016, 295, 376–383. [Google Scholar] [CrossRef]
- Chen, C.; Cai, L.X.; Tan, B.; Zhang, Y.J.; Yang, X.D.; Zhang, J. Ammonia detection by using flexible Lewis acidic sites in luminescent porous frameworks constructed from a bipyridinium derivative. Chem. Commun. 2015, 51, 8189–8192. [Google Scholar] [CrossRef]
- Chen, S.; Xiao, Z.; Ma, M. Copolymerization of carbon dioxide and epoxides with a novel effective Zn-Ni double-metal cyanide complex. Appl. Polym. Sci. 2008, 107, 3871–3877. [Google Scholar] [CrossRef]
- Kember, M.; Kabir, R.; Williams, C.K. Method for Preparing Polyols. U.S. Patent 0,148,539A1, 2018. [Google Scholar]
- Huang, Y.J.; Zhang, X.H.; Hua, Z.J.; Chen, S.L.; Qi, G.R. Ring-opening polymerization of propylene oxide catalyzed by a calcium-chloride-modified zinc-cobalt double metal-cyanide complex. Macromol. Chem. Phys. 2010, 211, 1229–1237. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, J.T.; Ha, C.S.; Yang, C.S.; Park, I. Polymerization of propylene oxide by using double metal cyanide catalysts and the application to polyurethane elastomer. Polymer 2003, 44, 3417–3428. [Google Scholar] [CrossRef]
- Coates, G.W.; Moore, D.R. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: Discovery, reactivity, optimization and mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef]
- Kobayashi, M.; Inoue, S.; Tsuruta, T. Copolymerization of carbon dioxide and epoxide by he dialkylzinc-carboxylic acid system. J. Polym. Sci. Polym. Chem. Ed. 1973, 11, 2383–2385. [Google Scholar] [CrossRef]
- Eberhardt, R.; Allmendinger, M.; Zintl, M.; Troll, C.; Luinstra, G.A.; Rieger, B. New zinc dicarboxylate catalysts for the CO2/Propylene oxide copolymerization reaction: Activity enhancement through Zn(II)-ethylsulfinate initiating groups. Macromol. Chem. Phys. 2004, 205, 42–47. [Google Scholar] [CrossRef]
- Kim, J.S.; Ree, M.; Shin, T.J.; Han, O.H.; Cho, S.J.; Hwang, Y.T.; Bae, J.Y.; Lee, J.M.; Ryoo, R.; Kim, H. X-ray absorption and NMR spectroscopic investigations of zinc glutarates prepared from various zinc sources and their catalytic activities in the copolymerization of carbon dioxide and propylene oxide. J. Catal. 2003, 218, 209–219. [Google Scholar] [CrossRef]
- Ree, M.; Bae, J.Y.; Hung, J.H.; Shin, T.J. A new copolymerization process leading to poly(propylene carbonate) with a highly enhanced yield from carbon dioxide and propylene oxide. J. Polym. Sci. A Polym. Chem. 1999, 37, 1863–1876. [Google Scholar] [CrossRef]
- Hsu, T.J.; Tan, C.S. Synthesis of polyethercarbonate from carbon dioxide and cyclohexene oxide by yttrium-metal coordination catalyst. Polymer 2001, 42, 5143–5150. [Google Scholar] [CrossRef]
- Shen, Z.; Chen, X.; Zhang, Y. New catalytic systems for the fixation of carbon dioxide, 2. Synthesis of high molecular weight epichlorohydrin/carbon dioxide copolymer with rare earth phosphonates/triisobutyl-aluminium systems. Macromol. Chem. Phys. 1994, 195, 2003–2011. [Google Scholar] [CrossRef]
- Chen, X.; Shen, Z.; Zhang, Y. New catalytic systems for the fixation of carbon dioxide. 1. Copolymerization of carbon dioxide and propylene oxide with new rare-earth catalysts-RE(P204)3-Al(i-Bu)3-R(OH)n. Macromolecules 1991, 24, 5305–5308. [Google Scholar] [CrossRef]
- Li, Z.; Qin, Y.; Zhao, X.; Wang, F.; Zhang, S.; Wang, X. Synthesis and stabilization of high-molecular-weight poly(propylene carbonate) from Zn-Co-based double metal cyanide catalyst. Eur. Polym. J. 2011, 47, 2152–2157. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, P.; Chen, L. Fe/Zn double metal cyanide (DMC) catalyzed ring-opening polymerization of propylene oxide: Part 3. Synthesis of DMC catalysts. Prog. Org. Coat. 2004, 50, 269–272. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, X.; Chen, S.; Du, B.; Wang, Q.; Fan, Z.; Qi, G. One-pot terpolymerization of CO2, cyclohexene oxide and maleic anhydride using a highly active heterogeneous double metal cyanide complex catalyst. Polymer 2010, 51, 5719–5725. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, R.; Sun, X.; Zhang, J.; Du, B.; Fan, Z.; Qi, G. Selective copolymerization of carbon dioxide with propylene oxide catalyzed by a nanolamellar double metal cyanide complex catalyst at low polymerization temperatures. Polymer 2011, 52, 5494–5502. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | δ(MII-CN) | ν(MII-C) | ν(CN) | δ(HOH) | ν(H2O) |
|---|---|---|---|---|---|
| [Co(CN)6]3− | 416 s | 565 m | 2125 s | - | - |
| [Fe(CN)6]3− | 417 m | 511 m, 583 w | 2118 s | - | - |
| Ni-Co | - | 461 m | 2141 m, 2183 s | 1609; 1634 (sh) | 3404; 3646 (br, s) |
| Co-Co | - | 457 m | 2176 s | 1609; 1634 (sh) | 3409; 3646 (br, s) |
| Fe-Co | - | 469 m | 2175 s | 1607; 1639 (sh) | 3415; 3646 (br, s) |
| Ni-Fe | 432 sh, 439 w | 541 w, 594 w | 2098 m, 2167 s | 1609; 1662 (sh) | 3396; 3646 (br, s) |
| Co-Fe | 431 w, 440 sh | 540 w, 593 w | 2116 s, 2159 m | 1609; 1651 (sh) | 3404;3641 (br, s) |
| Fe-Fe | 501 m, 512 sh | 603 w | 2080 s | 1606; 1638 (sh) | 3369; 3595; 3625 (br, br, s) |
| Compound | Cell Edge (Å) | V (Å3) | Crystallite Size (nm) | Strain (ε) | |
|---|---|---|---|---|---|
| D-S | W-H | ||||
| Ni-Co | 10.07 | 1020 | 9.4 | 13.2 | 0.00220 |
| Co-Co | 10.16 | 1049 | 21.5 | 29.4 | 0.00093 |
| Fe-Co | 10.17 | 1051 | 26.2 | 33.5 | 0.00061 |
| Ni-Fe | 10.15 | 1047 | 8.3 | 11.4 | 0.00243 |
| Co-Fe | 10.22 | 1068 | 27.6 | 43.6 | 0.00103 |
| Fe-Fe | 10.16 | 1049 | 34.6 | 44.1 | 0.00049 |
| Compound | H2O, wt % | Dehydration Temp. (°C) * | Decomposition Temp. (°C) * | ||
|---|---|---|---|---|---|
| N2 | Air | N2 | Air | ||
| Ni-Co | 29.30 | 145 | 150 | 490 | 310 |
| Co-Co | 29.71 | 110 | 115 | 515 | 265 |
| Fe-Co | 25.15 | 94 | 96 | 495 | 290 |
| Ni-Fe | 29.73 | 140 | 150 | 540 | 305 |
| Co-Fe | 30.83 | 105 | 108 | 500 | 255 |
| Fe-Fe | 22.46 | 107 | 110 | 525 | 225 |
| Compound | Cell Contraction (%) | Cell Edge after Dehydration (Å) | Cell Volume Reduction (%) | Cell Volume after Dehydration (Å3) |
|---|---|---|---|---|
| Ni-Co | 1.7 | 9.89 | 5.1 | 968 |
| Co-Co | 2.0 | 9.96 | 5.7 | 989 |
| Fe-Co | 1.2 | 10.05 | 3.5 | 1015 |
| Ni-Fe | 1.8 | 9.97 | 5.4 | 991 |
| Co-Fe | 2.4 | 9.98 | 7.0 | 993 |
| Fe-Fe | 1.4 | 10.02 | 4.1 | 1006 |
| Compound | XRF (wt%) | T/M (at./at.) | EA (wt%) | Estimated Catalyst Formulation | ||||
|---|---|---|---|---|---|---|---|---|
| T | M | K | Cl | C | N | |||
| Ni-Co | 24.5 | 12.3 | 0.2 | 1.9 | 2.0 | 14.4 | 17.6 | Ni1.99[Co(CN)6.01]·7.78 H2O *·0.26 Cl−·0.03 K+ |
| Co-Co | MI=MII: 38.3 | 0.4 | 1.8 | - | 14.4 | 16.2 | - | |
| Fe-Co | 21.8 | 12.8 | 0.1 | 1.4 | 1.8 | 17.3 | 20.1 | Fe1.81[Co(CN)6.45]·6.41H2O *·0.19 Cl−·0.01 K+ |
| Ni-Fe | 24.8 | 11.8 | 0.2 | 1.5 | 2.0 | 14.3 | 17.8 | Ni2.01[Fe(CN)6.02]·7.80 H2O *·0.19 Cl−·0.02 K+ |
| Co-Fe | 23.5 | 13.0 | 0.6 | 1.1 | 1.7 | 15.5 | 17.3 | Co1.72[Fe(CN)5.30]·6.95 H2O *·0.14 Cl−·0.06 K+ |
| Fe-Fe | MI=MII: 36.6 | 1.5 | 0.9 | - | 16.8 | 19.6 | - | |
| Compound | SBET a (m2/g) | Sext b (m2/g) | Surface Area Contributed by Micropores (%) |
|---|---|---|---|
| Ni-Co | 373 | - | 100 |
| Co-Co | 801 | 155 | 81 |
| Fe-Co | 757 | 156 | 79 |
| Ni-Fe | 161 | 4 | 98 |
| Co-Fe | 624 | 95 | 85 |
| Fe-Fe | 213 | 75 | 65 |
| Compound | Vt a (cm3/g) | Vmicro (cm3/g) | Pore Volume Contributed by Micropores e (%) | ||
|---|---|---|---|---|---|
| Vm(BET) = Vmicro + Vm(ext) b | DA-Plot c | t-Plot d | |||
| Ni-Co | 0.146 | 0.132 | 0.146 | 0.144 | 97 |
| Co-Co | 0.496 | 0.230 | 0.325 | 0.258 | 55 |
| Fe-Co | 0.680 | 0.214 | 0.309 | 0.263 | 39 |
| Ni-Fe | 0.066 | 0.056 | 0.065 | 0.062 | 92 |
| Co-Fe | 0.590 | 0.188 | 0.267 | 0.216 | 38 |
| Fe-Fe | 0.201 | 0.049 | 0.097 | 0.068 | 36 |
| Compound | Total Acidity a (μmol NH3 g−1) | Acid Sites Density a (μmol NH3 m−2) |
|---|---|---|
| Ni-Co | 1880 | 5.0 |
| Co-Co | 2780 | 3.5 |
| Fe-Co | 2010 | 2.7 |
| Ni-Fe | 1960 | 12.2 |
| Co-Fe | 3780 | 6.1 |
| Fe-Fe | 1550 | 7.3 |
| Compound | Yield b (g) | TOF c | FCU d mol (%) | FCO2 e (wt%) | WPC f (wt%) | SCO2 g (%) | SPO h (%) | RPEC (%) | Mw (g mol−1) | ĐM |
|---|---|---|---|---|---|---|---|---|---|---|
| Ni-Co | 3.2 | 4 | 22.3 | 14.4 | 0.4 | 98.9 | 99.7 | 79.5 | 11,800 | 10.5 |
| Co-Co | 18.9 | 23 | 20.0 | 13.1 | 4.2 | 87.4 | 97.2 | 71.9 | 68,600 | 4.1 |
| Fe-Co | 14.8 | 18 | 16.3 | 11.0 | 8.4 | 73.6 | 94.5 | 75.1 | 85,400 | 6.3 |
| Ni-Fe | 3.0 | 4 | 24.0 | 15.3 | 0.6 | 98.4 | 99.6 | 73.9 | 11,700 | 15.8 |
| Co-Fe | 11.6 | 12 | 33.5 | 20.2 | 13.3 | 75.4 | 90.2 | 66.0 | 50,000 | 5.9 |
| Fe-Fe | 6.0 | 7 | 17.3 | 11.6 | 43.1 | 26.2 | 67.2 | 83.3 | 6000 | 8.4 |
| Catalyst Formulation | Time (h) | PO/Cat. (g/g) | P (bar) | T (°C) | TON a | TOF b | FCU (mol%) | WPC (wt%) | SCO2 (%) | Mw (g mol−1) | ĐM | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ni3[Co(CN)6]2 | 24 | 400 | 20 | 90 | 86 | 4 | 22 | 0.4 | 99 | 11,800 | 10.5 | - |
| Co3[Co(CN)6]2 | 24 | 400 | 20 | 90 | 544 | 23 | 20 | 4.2 | 87 | 68,600 | 4.1 | - |
| Fe3[Co(CN)6]2 | 24 | 400 | 20 | 90 | 428 | 18 | 16 | 8.4 | 74 | 85,400 | 6.3 | - |
| Ni3[Fe(CN)6]2 | 24 | 400 | 20 | 90 | 84 | 4 | 15 | 0.6 | 98 | 11,700 | 15.8 | - |
| Co3[Fe(CN)6]2 | 24 | 400 | 20 | 90 | 296 | 12 | 20 | 13.3 | 75 | 50,000 | 5.9 | - |
| Fe4[Fe(CN)6]3 | 24 | 400 | 20 | 90 | 162 | 7 | 17 | 43.1 | 26 | 6000 | 8.4 | - |
| Zn3[Co(CN)6]2 | 10 | 42,000 | 38 | 90 | 64,414 | 6441 | 13 | 4.4 | 82 | 10,800 | 1.8 | [103] |
| Zn3[Fe(CN)6]2 c | 10 | 4611 | 50 | 110 | 137 | 14 | 15 | 62.5 | 12 | 2160 | n.a. | [50] |
| Zn3[Cr(CN)6]2 c | 10 | 4611 | 50 | 140 | 334 | 33 | 10 | 36.6 | 22 | 1350 | n.a. | [50] |
| Zn[Cd(CN)4] c | 10 | 4611 | 50 | 130 | 35 | 4 | 2 | 6.5 | 33 | n.a. | n.a. | [50] |
| Zn2[Mo(CN)8] c | 10 | 4611 | 50 | 130 | 196 | 20 | 14 | 16.5 | 53 | 1420 | n.a. | [50] |
| Zn2[Fe(CN)6]2 c | 10 | 4611 | 50 | 110 | 233 | 23 | 5 | 77.8 | 2 | n.a. | n.a. | [50] |
| Zn[Ni(CN)4] | 20 | 553 | 50 | 110 | 210 | 11 | 62 | 6.6 | 91 | 2360 | 1.8 | [91] |
| Co[Ni(CN)4]2 | 1 | 664 | 54.4 | 90 | 1510 | 1510 | 27 | - | 100 | 526,400 | 2.8 | [56] |
| Co[Pd(CN)4]2 | 1 | 546 | 54.4 | 90 | 18 | 18 | 43 | - | 100 | 92,160 | 3.6 | [56] |
| Co[Pt(CN)4]2 | 1 | 411 | 54.4 | 90 | 13 | 13 | 44 | - | 100 | 103,230 | 3.7 | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penche, G.; González-Velasco, J.R.; González-Marcos, M.P. Porous Hexacyanometallate(III) Complexes as Catalysts in the Ring-Opening Copolymerization of CO2 and Propylene Oxide. Catalysts 2021, 11, 1450. https://doi.org/10.3390/catal11121450
Penche G, González-Velasco JR, González-Marcos MP. Porous Hexacyanometallate(III) Complexes as Catalysts in the Ring-Opening Copolymerization of CO2 and Propylene Oxide. Catalysts. 2021; 11(12):1450. https://doi.org/10.3390/catal11121450
Chicago/Turabian StylePenche, Guillermo, Juan R. González-Velasco, and M. Pilar González-Marcos. 2021. "Porous Hexacyanometallate(III) Complexes as Catalysts in the Ring-Opening Copolymerization of CO2 and Propylene Oxide" Catalysts 11, no. 12: 1450. https://doi.org/10.3390/catal11121450