
Transaminase Catalysis for Enantiopure Saturated Heterocycles as Potential Drug Scaffolds



Abstract
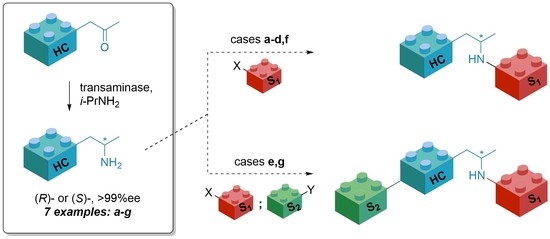
:
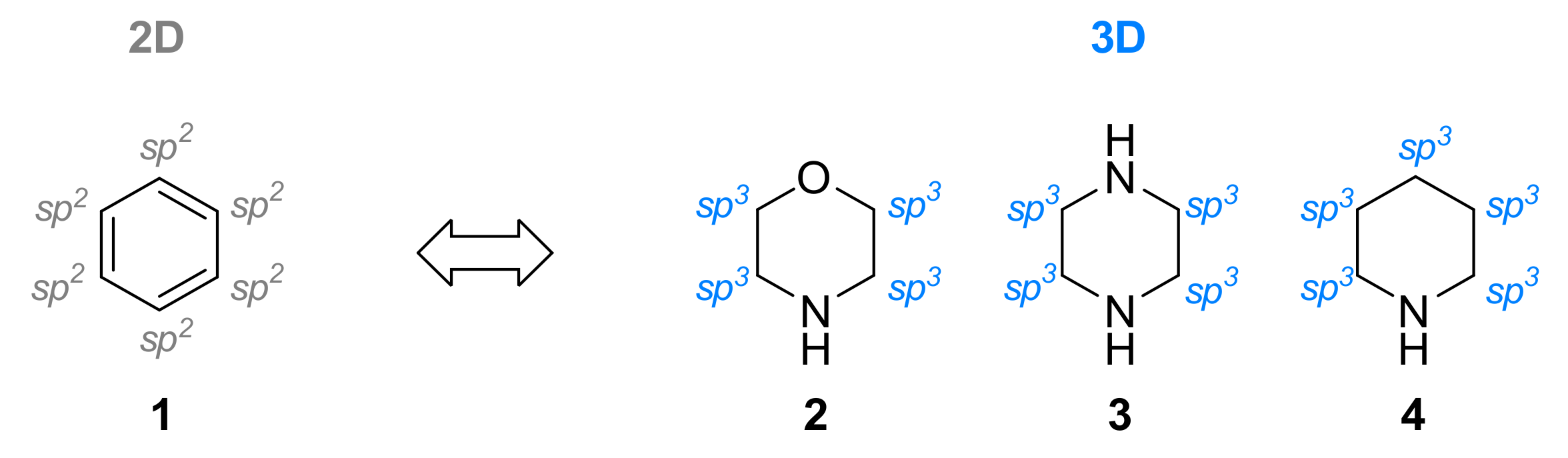
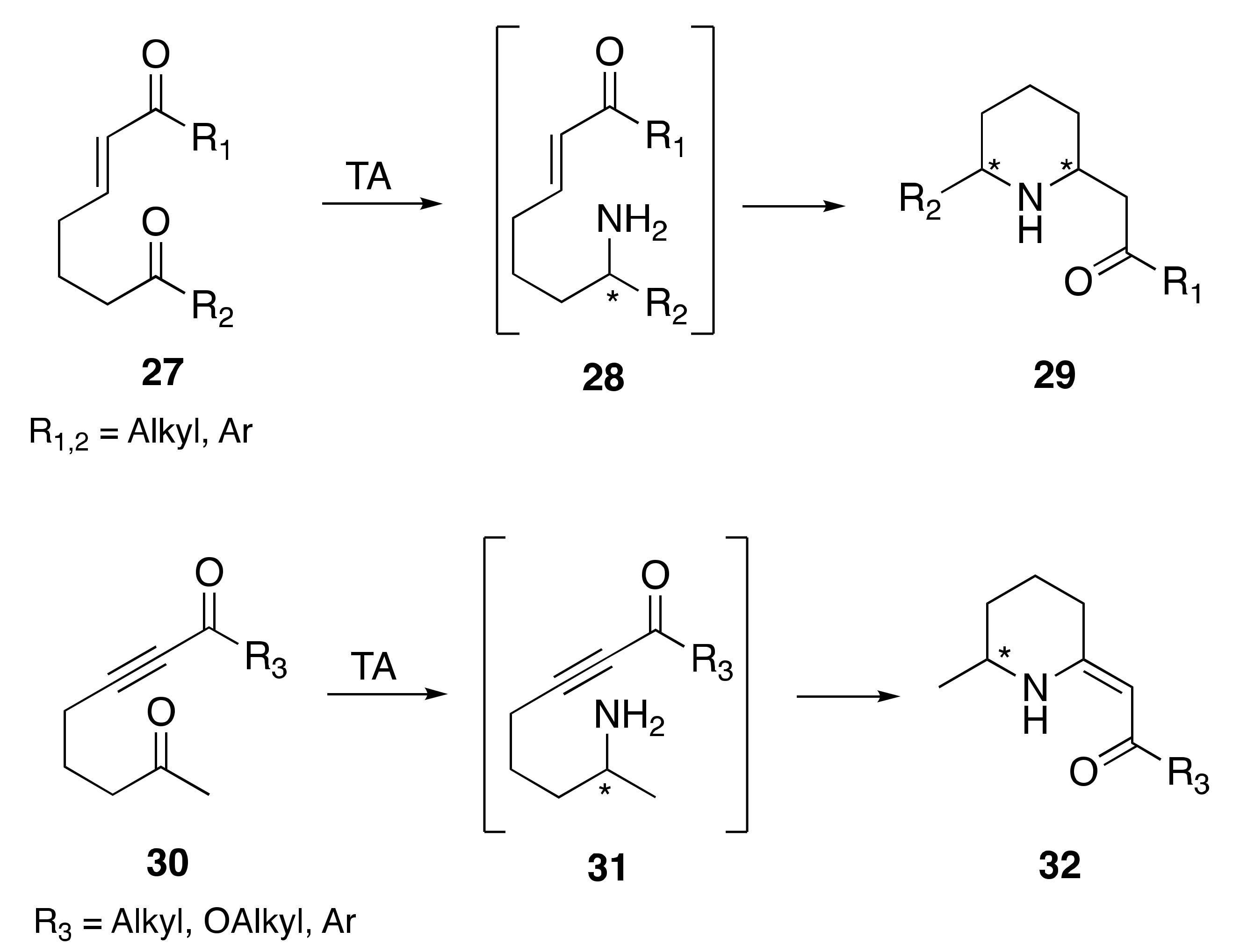
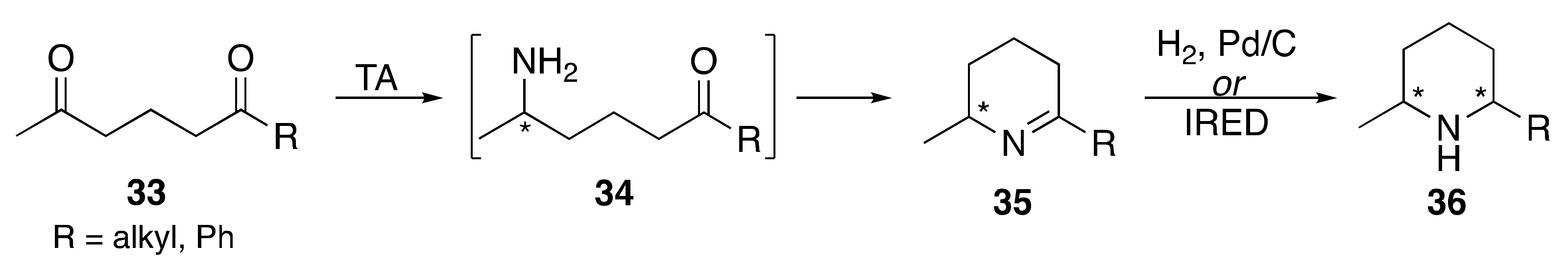
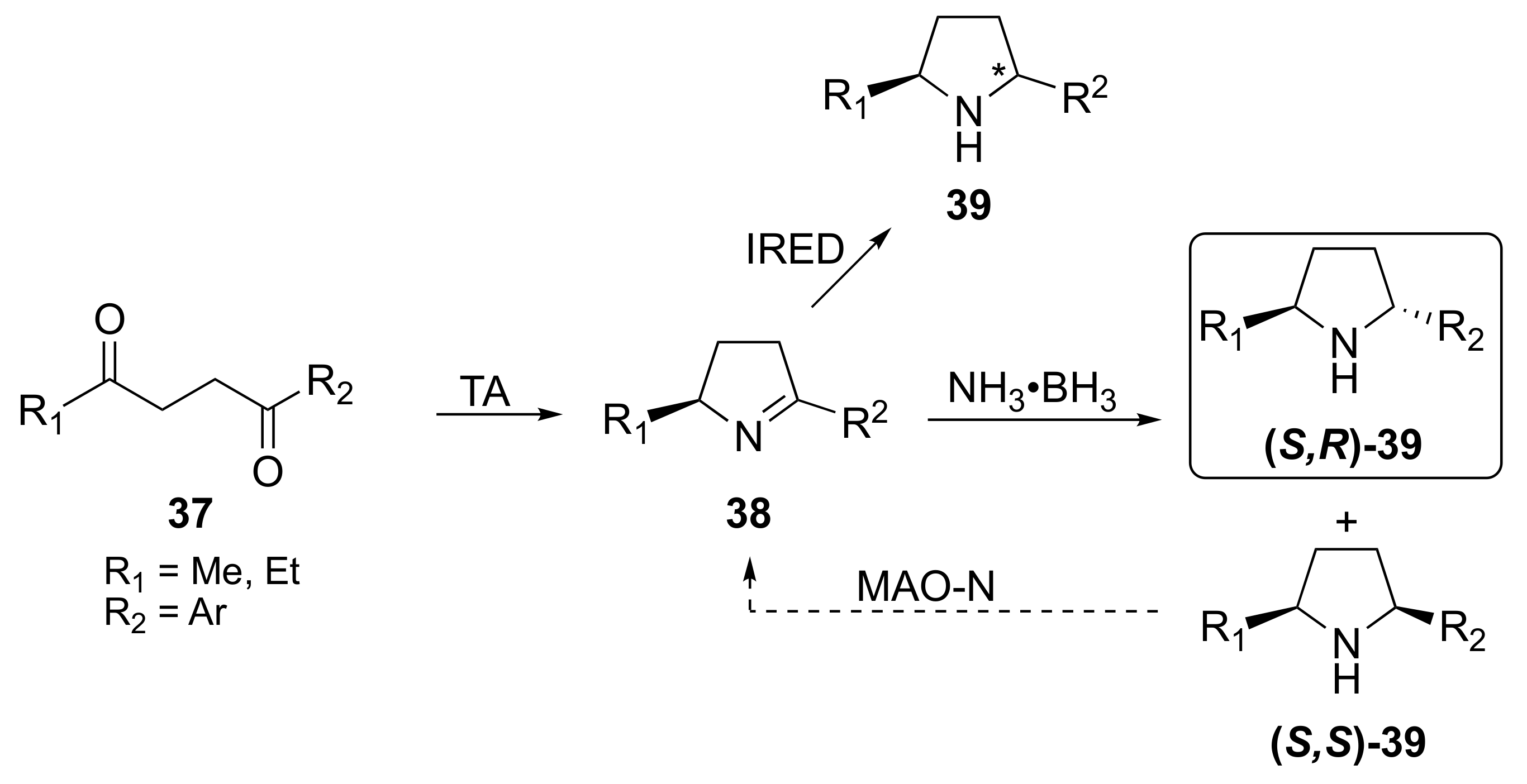
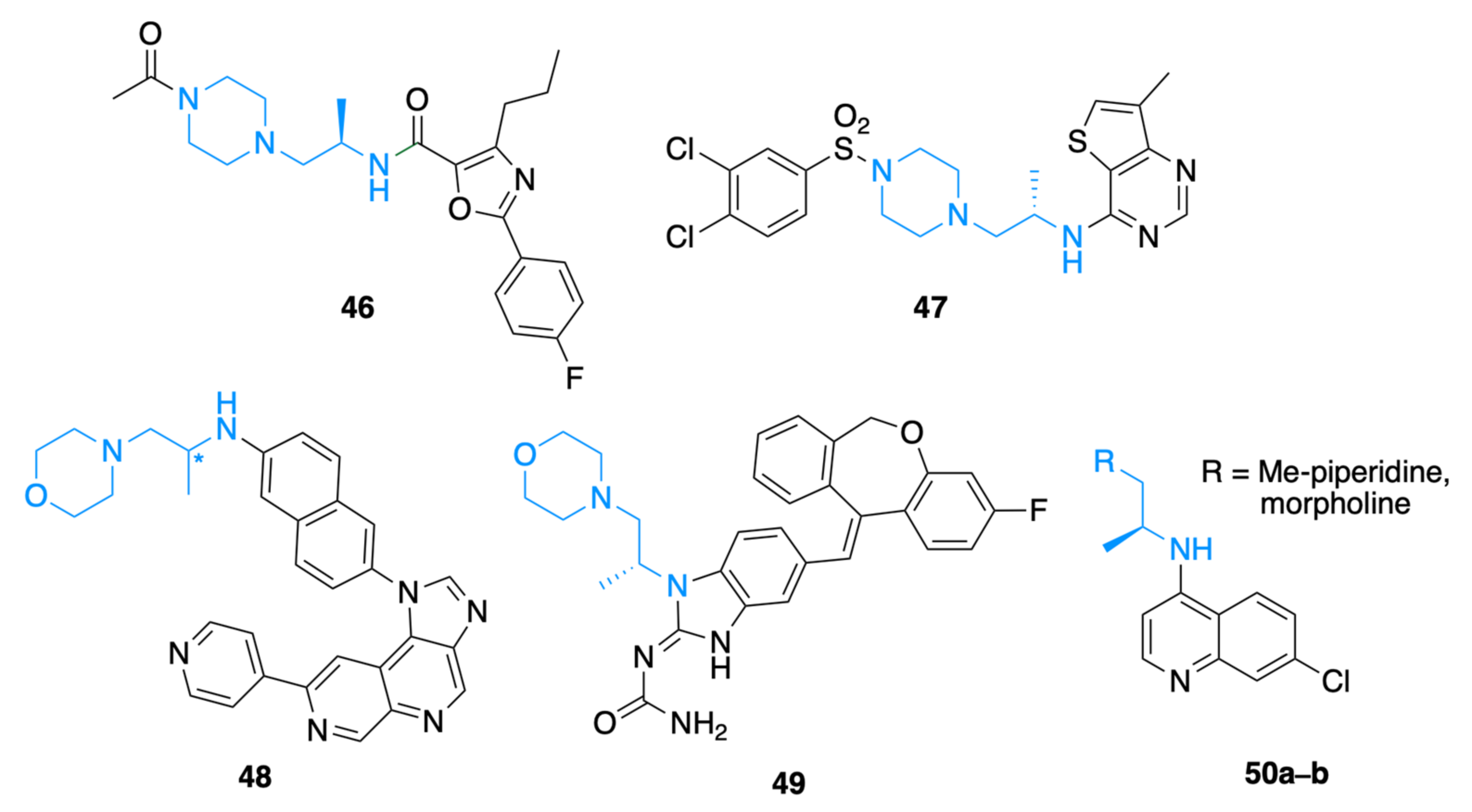
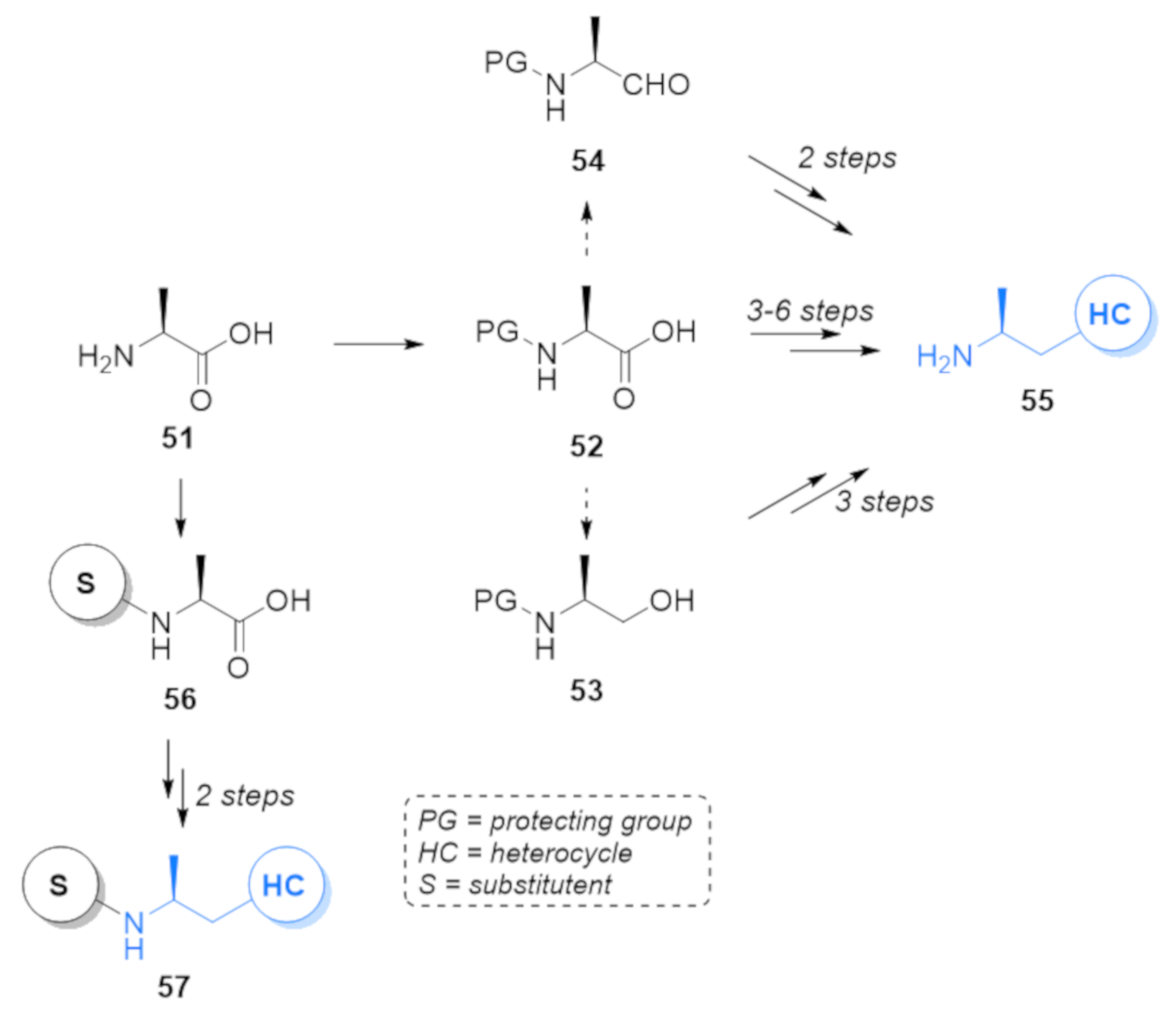
1. Introduction
2. Results and Discussion
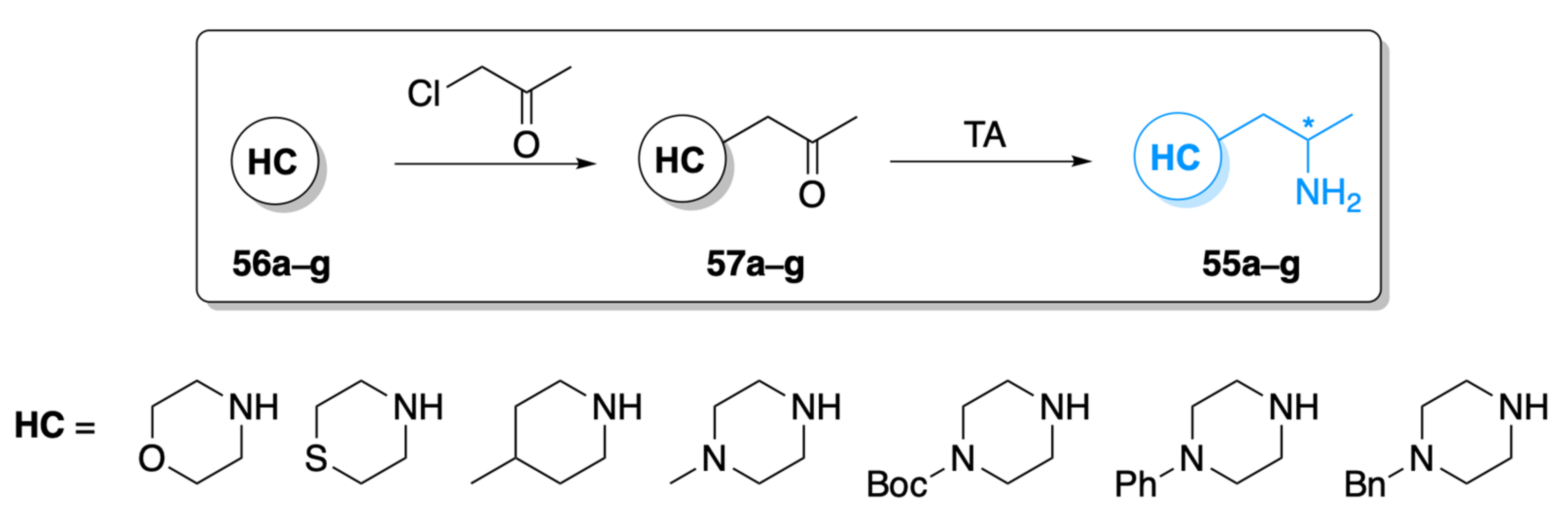
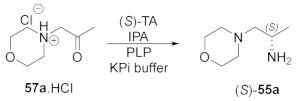
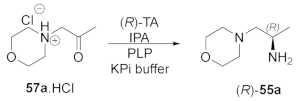
2.1. Screening the Activity of TAs with Morpholine Derivative 57a
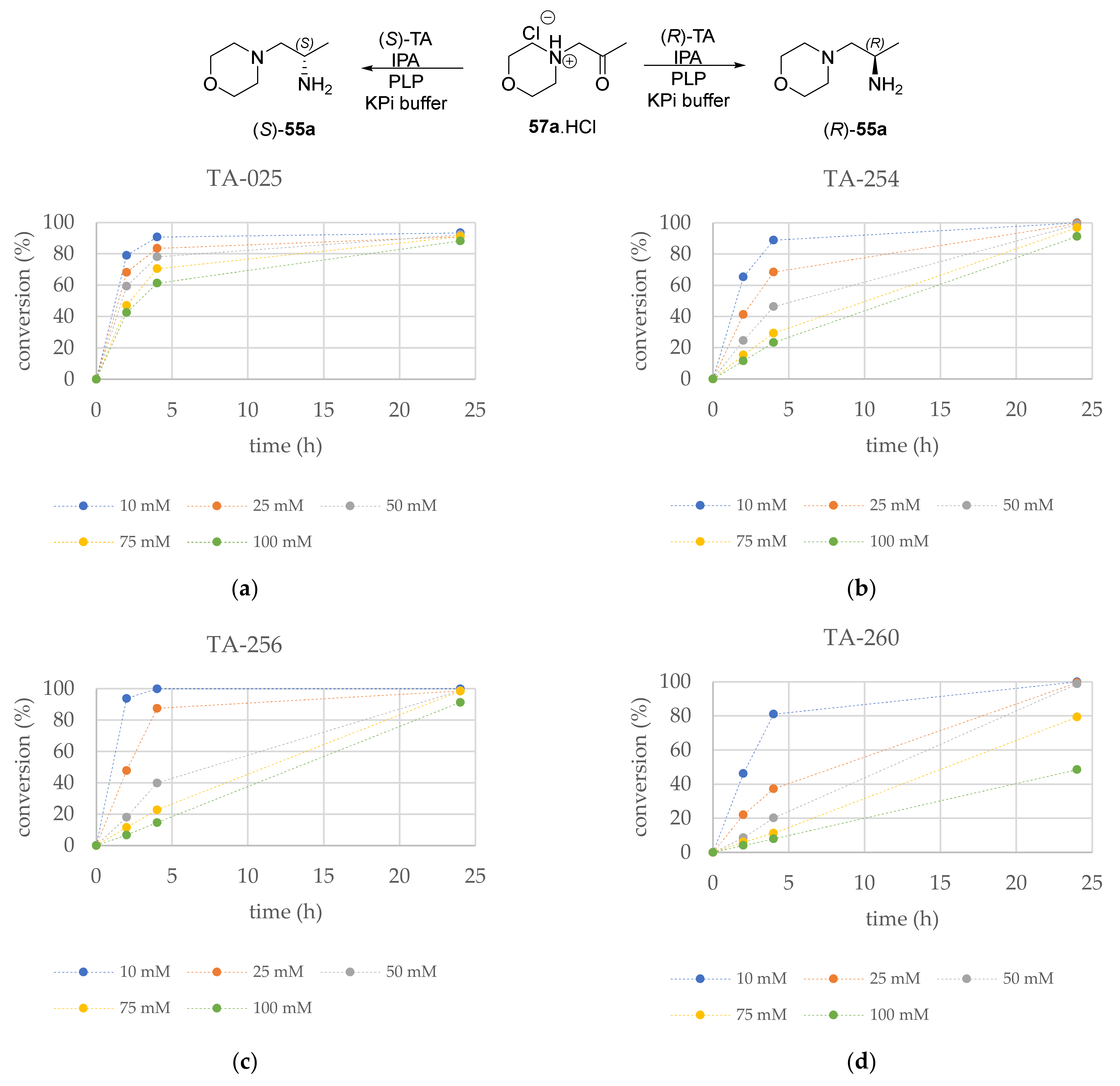
2.2. Screening the Effect of Substrate Concentration with the 1-Morpholinopropan-2-one (57a)
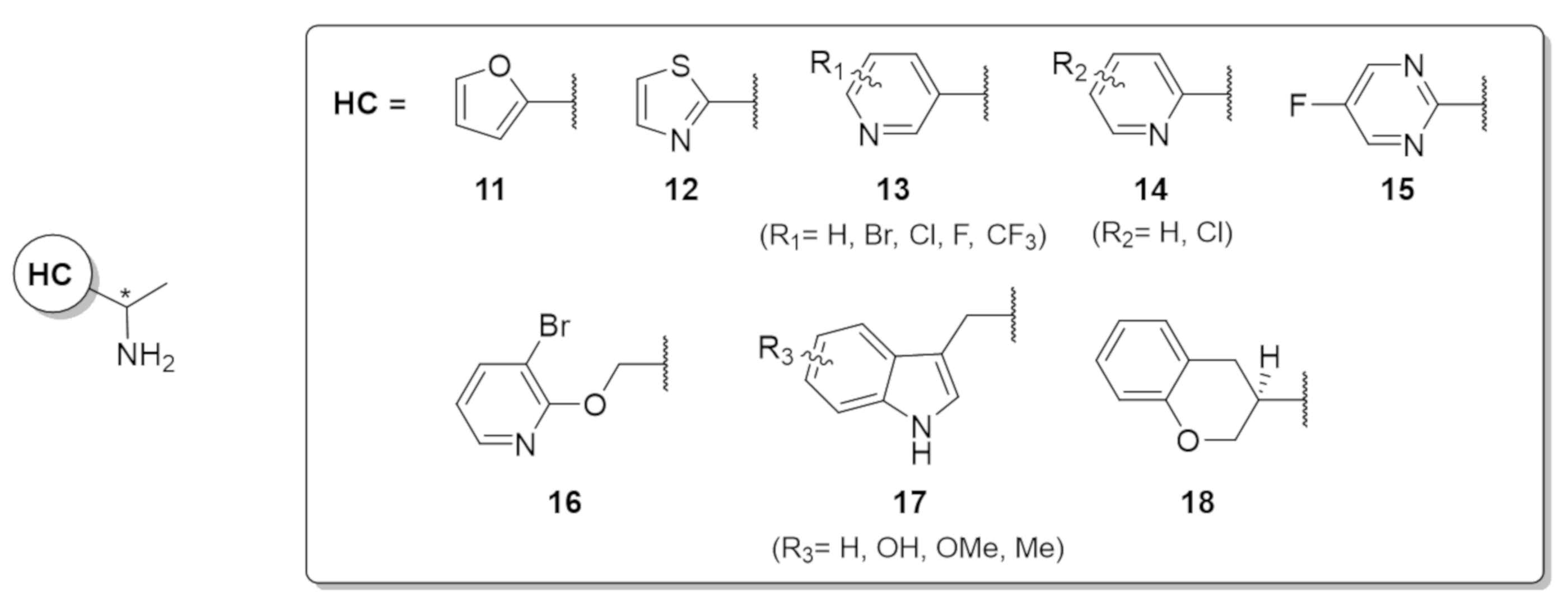
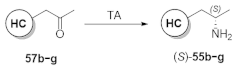
2.3. Screening the Best Transaminases with Further Heterocycle-Containing Ketones (57b–g)
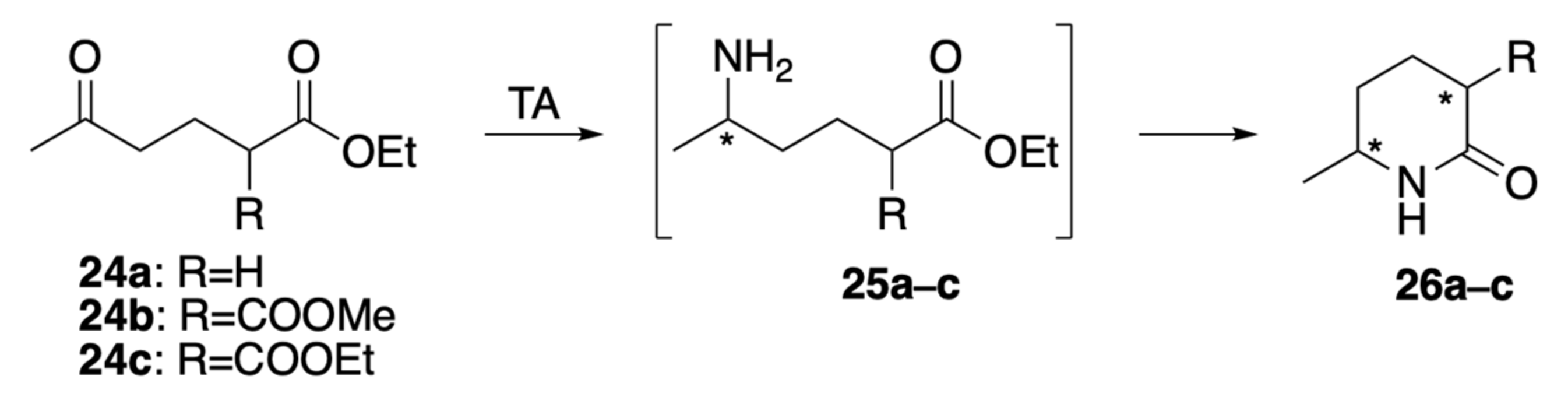
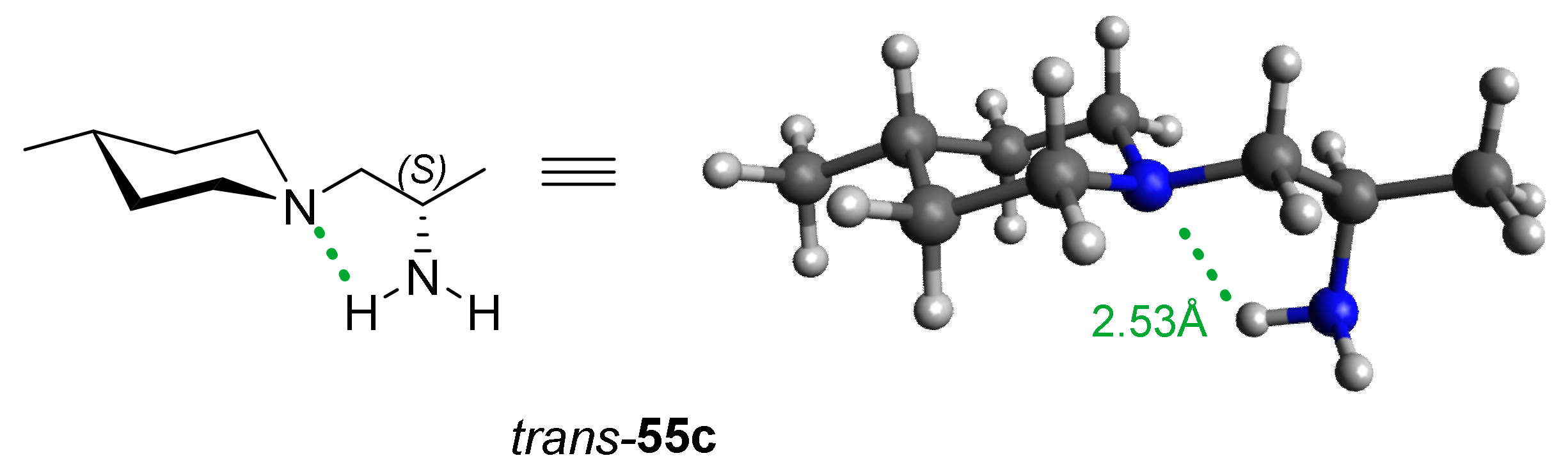
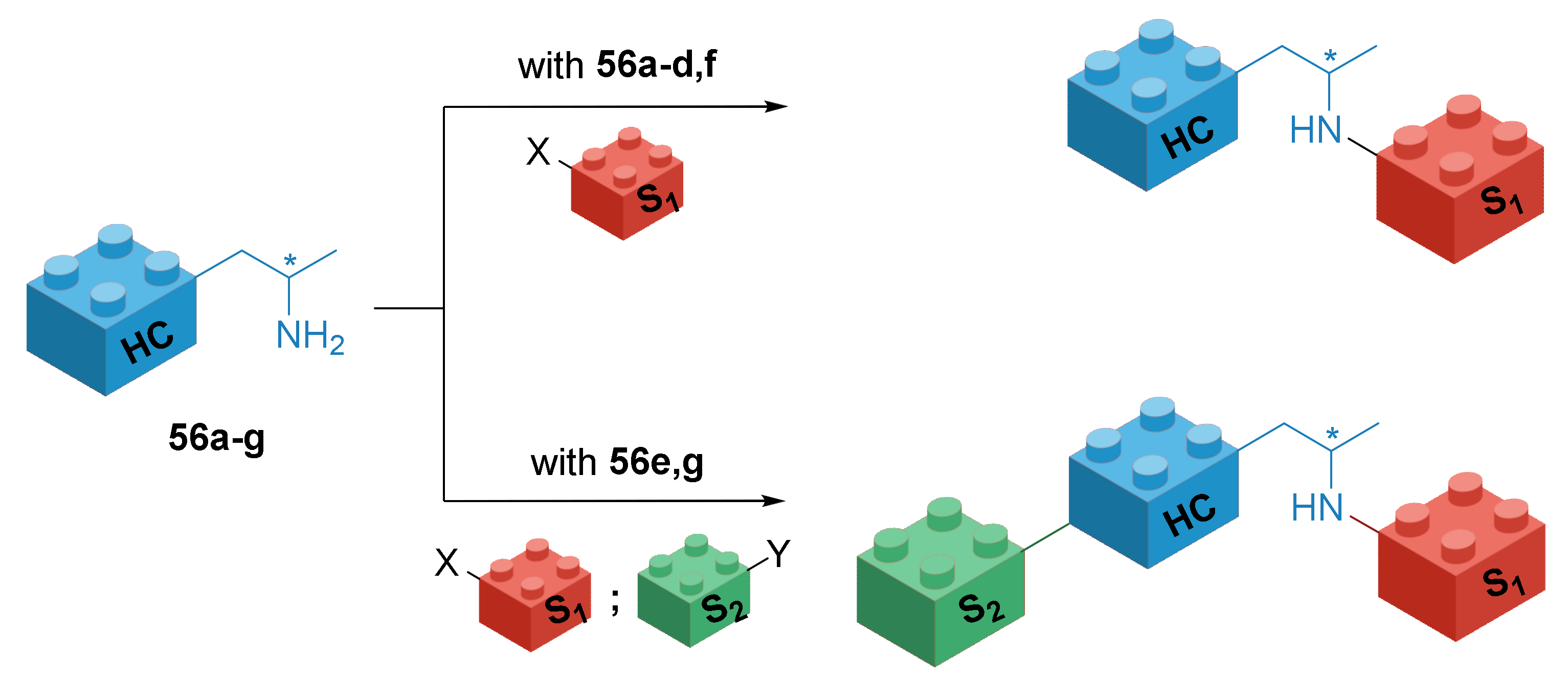
2.4. Structural Stabilization of the Tertiary Amine Bearing the Asymmetric Amine Side Chain
3. Materials and Methods
3.1. Materials and Biocatalysts
3.2. Analytical and Separation Methods
3.3. Synthesis of the Heterocyclic Ketones (57a–g)
3.4. Screening the Asymmetric Transamination of the Ketone 57a with Codexis® TAs
3.5. Screening the Effect of Substrate Concentration on the Optimal TAs with Ketones 57a–g
3.6. Scaling-Up the Transamination of 57c
3.7. Isolation of the Enantiopure Amines (S)-55a–g
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.F. The Impact of Aromatic Ring Count on Compound Developability—Are Too Many Aromatic Rings a Liability in Drug Design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-Oriented Synthesis as a Tool for the Discovery of Novel Biologically Active Small Molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [Green Version]
- Aldeghi, M.; Malhotra, S.; Selwood, D.L.; Chan, A.W.E. Two- and Three-dimensional Rings in Drugs. Chem. Biol. Drug Des. 2014, 83, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; MacDonald, S.J.F.; Young, R.J.; Pickett, S.D. The Impact of Aromatic Ring Count on Compound Developability: Further Insights by Examining Carbo- and Hetero-Aromatic and -Aliphatic Ring Types. Drug Discov. Today 2011, 16, 164–171. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Vardanyan, R. Piperidine-Based Drug Discovery, 1st ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; ISBN 978-0-12-805157-3. [Google Scholar]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine Scaffold: A Remarkable Tool in Generation of Diverse Pharmacological Agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Kourounakis, A.P.; Xanthopoulos, D.; Tzara, A. Morpholine as a Privileged Structure: A Review on the Medicinal Chemistry and Pharmacological Activity of Morpholine Containing Bioactive Molecules. Med. Res. Rev. 2020, 40, 709–752. [Google Scholar] [CrossRef] [PubMed]
- Caron, S. Practical Synthetic Organic Chemistry, 2nd ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2020; ISBN 9781119448853. [Google Scholar]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S.; Gilmore, B.F. Application of ω-Transaminases in the Pharmaceutical Industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef]
- Ghislieri, D.; Turner, N.J. Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Frodsham, L.; Golden, M.; Hard, S.; Kenworthy, M.N.; Klauber, D.J.; Leslie, K.; Macleod, C.; Meadows, R.E.; Mulholland, K.R.; Reilly, J.; et al. Use of ω-Transaminase Enzyme Chemistry in the Synthesis of a JAK2 Kinase Inhibitor. Org. Process Res. Dev. 2013, 17, 1123–1130. [Google Scholar] [CrossRef]
- Meadows, R.E.; Mulholland, K.R.; Schürmann, M.; Golden, M.; Kierkels, H.; Meulenbroeks, E.; Mink, D.; May, O.; Squire, C.; Straatman, H.; et al. Efficient Synthesis of (S)-1-(5-Fluoropyrimidin-2-Yl)Ethylamine Using an ω-Transaminase Biocatalyst in a Two-Phase System. Org. Process Res. Dev. 2013, 17, 1117–1122. [Google Scholar] [CrossRef]
- Molinaro, C.; Bulger, P.G.; Lee, E.E.; Kosjek, B.; Lau, S.; Gauvreau, D.; Howard, M.E.; Wallace, D.J.; O’Shea, P.D. CRTH2 Antagonist MK-7246: A Synthetic Evolution from Discovery through Development. J. Org. Chem. 2012, 77, 2299–2309. [Google Scholar] [CrossRef]
- Peng, Z.; Wong, J.W.; Hansen, E.C.; Puchlopek-Dermenci, A.L.A.; Clarke, H.J. Development of a Concise, Asymmetric Synthesis of a Smoothened Receptor (Smo) Inhibitor: Enzymatic Transamination of a 4-Piperidinone with Dynamic Kinetic Resolution. Org. Lett. 2014, 16, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Mangion, I.K.; Sherry, B.D.; Yin, J.; Fleitz, F.J. Enantioselective Synthesis of a Dual Orexin Receptor Antagonist. Org. Lett. 2012, 14, 3458–3461. [Google Scholar] [CrossRef]
- Chung, C.K.; Bulger, P.G.; Kosjek, B.; Belyk, K.M.; Rivera, N.; Scott, M.E.; Humphrey, G.R.; Limanto, J.; Bachert, D.C.; Emerson, K.M. Process Development of C–N Cross-Coupling and Enantioselective Biocatalytic Reactions for the Asymmetric Synthesis of Niraparib. Org. Process Res. Dev. 2014, 18, 215–227. [Google Scholar] [CrossRef]
- Girardin, M.; Ouellet, S.G.; Gauvreau, D.; Moore, J.C.; Hughes, G.; Devine, P.N.; O’Shea, P.D.; Campeau, L.-C. Convergent Kilogram-Scale Synthesis of Dual Orexin Receptor Antagonist. Org. Process Res. Dev. 2013, 17, 61–68. [Google Scholar] [CrossRef]
- Chung, J.Y.L.; Zhong, Y.-L.; Maloney, K.M.; Reamer, R.A.; Moore, J.C.; Strotman, H.; Kalinin, A.; Feng, R.; Strotman, N.A.; Xiang, B.; et al. Unusual Pyrimidine Participation: Efficient Stereoselective Synthesis of Potent Dual Orexin Receptor Antagonist MK-6096. Org. Lett. 2014, 16, 5890–5893. [Google Scholar] [CrossRef]
- Chung, J.Y.L.; Marcune, B.; Strotman, H.R.; Petrova, R.I.; Moore, J.C.; Dormer, P.G. Synthesis of ((3 R, 6 R)-6-Methylpiperidin-3-Yl)Methanol via Biocatalytic Transamination and Crystallization-Induced Dynamic Resolution. Org. Process Res. Dev. 2015, 19, 1418–1423. [Google Scholar] [CrossRef]
- Dunbabin, A.; Subrizi, F.; Ward, J.M.; Sheppard, T.D.; Hailes, H.C. Furfurylamines from Biomass: Transaminase Catalysed Upgrading of Furfurals. Green Chem. 2017, 19, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.E.; Rodríguez-Mata, M.; Busto, E.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; García-Cerrada, S.; Mendiola, J.; de Frutos, Ó.; Collado, I. Transaminases Applied to the Synthesis of High Added-Value Enantiopure Amines. Org. Process Res. Dev. 2014, 18, 788–792. [Google Scholar] [CrossRef]
- López-Iglesias, M.; González-Martínez, D.; Gotor, V.; Busto, E.; Kroutil, W.; Gotor-Fernández, V. Biocatalytic Transamination for the Asymmetric Synthesis of Pyridylalkylamines. Structural and Activity Features in the Reactivity of Transaminases. ACS Catal. 2016, 6, 4003–4009. [Google Scholar] [CrossRef]
- Galman, J.L.; Slabu, I.; Weise, N.J.; Iglesias, C.; Parmeggiani, F.; Lloyd, R.C.; Turner, N.J. Biocatalytic Transamination with Near-Stoichiometric Inexpensive Amine Donors Mediated by Bifunctional Mono- and Di-Amine Transaminases. Green Chem. 2017, 19, 361–366. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. Stereoselective Amination of Racemic Sec-Alcohols through Sequential Application of Laccases and Transaminases. Green Chem. 2017, 19, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. But-2-Ene-1,4-Diamine and But-2-Ene-1,4-Diol as Donors for Thermodynamically Favored Transaminase- and Alcohol Dehydrogenase-Catalyzed Processes. Adv. Synth. Catal. 2016, 358, 1618–1624. [Google Scholar] [CrossRef] [Green Version]
- Dawood, A.W.H.; de Souza, R.O.M.A.; Bornscheuer, U.T. Asymmetric Synthesis of Chiral Halogenated Amines Using Amine Transaminases. ChemCatChem 2018, 10, 951–955. [Google Scholar] [CrossRef]
- Holzer, A.K.; Hiebler, K.; Mutti, F.G.; Simon, R.C.; Lauterbach, L.; Lenz, O.; Kroutil, W. Asymmetric Biocatalytic Amination of Ketones at the Expense of NH 3 and Molecular Hydrogen. Org. Lett. 2015, 17, 2431–2433. [Google Scholar] [CrossRef]
- López-Iglesias, M.; González-Martínez, D.; Rodríguez-Mata, M.; Gotor, V.; Busto, E.; Kroutil, W.; Gotor-Fernández, V. Asymmetric Biocatalytic Synthesis of Fluorinated Pyridines through Transesterification or Transamination: Computational Insights into the Reactivity of Transaminases. Adv. Synth. Catal. 2017, 359, 279–291. [Google Scholar] [CrossRef]
- Mourelle-Insua, Á.; López-Iglesias, M.; Gotor, V.; Gotor-Fernández, V. Stereoselective Access to 1-[2-Bromo(Het)Aryloxy]Propan-2-Amines Using Transaminases and Lipases; Development of a Chemoenzymatic Strategy Toward a Levofloxacin Precursor. J. Org. Chem. 2016, 81, 9765–9774. [Google Scholar] [CrossRef]
- Fischereder, E.-M.M.; Pressnitz, D.; Kroutil, W. Stereoselective Cascade to C3-Methylated Strictosidine Derivatives Employing Transaminases and Strictosidine Synthases. ACS Catal. 2016, 6, 23–30. [Google Scholar] [CrossRef]
- Monti, D.; Forchin, M.C.; Crotti, M.; Parmeggiani, F.; Gatti, F.G.; Brenna, E.; Riva, S. Cascade Coupling of Ene-Reductases and ω-Transaminases for the Stereoselective Synthesis of Diastereomerically Enriched Amines. ChemCatChem 2015, 7, 3106–3109. [Google Scholar] [CrossRef]
- Höhne, M.; Kühl, S.; Robins, K.; Bornscheuer, U.T. Efficient Asymmetric Synthesis of Chiral Amines by Combining Transaminase and Pyruvate Decarboxylase. ChemBioChem 2008, 9, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, E.; Paradisi, F. Implementation of Biocatalysis in Continuous Flow for the Synthesis of Small Cyclic Amines. Chimia 2020, 74, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Luo, Z.; Sun, G.; Chen, M.; Lai, J.; Lin, W.; Goldmann, S.; Zhang, L.; Wang, Z. Development of an Efficient and Scalable Biocatalytic Route to (3R)-3-Aminoazepane: A Pharmaceutically Important Intermediate. Org. Process Res. Dev. 2017, 21, 648–654. [Google Scholar] [CrossRef]
- Lerchner, A.; Achatz, S.; Rausch, C.; Haas, T.; Skerra, A. Coupled Enzymatic Alcohol-to-Amine Conversion of Isosorbide Using Engineered Transaminases and Dehydrogenases. ChemCatChem 2013, 5, 3374–3383. [Google Scholar] [CrossRef]
- Weiß, M.S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Protein-Engineering of an Amine Transaminase for the Stereoselective Synthesis of a Pharmaceutically Relevant Bicyclic Amine. Org. Biomol. Chem. 2016, 14, 10249–10254. [Google Scholar] [CrossRef] [Green Version]
- Pressnitz, D.; Fuchs, C.S.; Sattler, J.H.; Knaus, T.; Macheroux, P.; Mutti, F.G.; Kroutil, W. Asymmetric Amination of Tetralone and Chromanone Derivatives Employing ω-Transaminases. ACS Catal. 2013, 3, 555–559. [Google Scholar] [CrossRef]
- Truppo, M.D.; Rozzell, J.D.; Turner, N.J. Efficient Production of Enantiomerically Pure Chiral Amine at Conc 50 g/L Using Transaminase. Org. Process Res. Dev. 2010, 14, 234–237. [Google Scholar] [CrossRef]
- Ryan, J.; Šiaučiulis, M.; Gomm, A.; Maciá, B.; O’Reilly, E.; Caprio, V. Transaminase Triggered Aza-Michael Approach for the Enantioselective Synthesis of Piperidine Scaffolds. J. Am. Chem. Soc. 2016, 138, 15798–15800. [Google Scholar] [CrossRef] [PubMed]
- Taday, F.; Ryan, J.; Argent, S.P.; Caprio, V.; Maciá, B.; O’Reilly, E. Asymmetric Construction of Alkaloids by Employing a Key ω-Transaminase Cascade. Chem. A Eur. J. 2020, 26, 3729–3732. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.C.; Grischek, B.; Zepeck, F.; Steinreiber, A.; Belaj, F.; Kroutil, W. Regio- and Stereoselective Monoamination of Diketones without Protecting Groups. Angew. Chemie Int. Ed. 2012, 51, 6713–6716. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.C.; Zepeck, F.; Kroutil, W. Chemoenzymatic Synthesis of All Four Diastereomers of 2,6-Disubstituted Piperidines through Stereoselective Monoamination of 1,5-Diketones. Chem. A Eur. J. 2013, 19, 2859–2865. [Google Scholar] [CrossRef]
- Simon, R.C.; Fuchs, C.S.; Lechner, H.; Zepeck, F.; Kroutil, W. Concise Chemoenzymatic Three-Step Total Synthesis of Isosolenopsin through Medium Engineering. Eur. J. Org. Chem. 2013, 2013, 3397–3402. [Google Scholar] [CrossRef] [PubMed]
- France, S.P.; Hussain, S.; Hill, A.M.; Hepworth, L.J.; Howard, R.M.; Mulholland, K.R.; Flitsch, S.L.; Turner, N.J. One-Pot Cascade Synthesis of Mono- and Disubstituted Piperidines and Pyrrolidines Using Carboxylic Acid Reductase (CAR), ω-Transaminase (ω-TA), and Imine Reductase (IRED) Biocatalysts. ACS Catal. 2016, 6, 3753–3759. [Google Scholar] [CrossRef]
- O’Reilly, E.; Iglesias, C.; Ghislieri, D.; Hopwood, J.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. A Regio- and Stereoselective ω-Transaminase/Monoamine Oxidase Cascade for the Synthesis of Chiral 2,5-Disubstituted Pyrrolidines. Angew. Chemie Int. Ed. 2014, 53, 2447–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepworth, L.J.; France, S.P.; Hussain, S.; Both, P.; Turner, N.J.; Flitsch, S.L. Enzyme Cascades in Whole Cells for the Synthesis of Chiral Cyclic Amines. ACS Catal. 2017, 7, 2920–2925. [Google Scholar] [CrossRef] [Green Version]
- Brotherton-Pleiss, C.E.; Dillon, M.P.; Ford, A.P.D.W.; Gever, J.R.; Carter, D.S.; Gleason, S.K.; Lin, C.J.; Moore, A.G.; Thompson, A.W.; Villa, M.; et al. Discovery and Optimization of RO-85, a Novel Drug-like, Potent, and Selective P2X3 Receptor Antagonist. Bioorganic Med. Chem. Lett. 2010, 20, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Glatthar, R.; Stojanovic, A.; Troxler, T.; Mattes, H.; Möbitz, H.; Beerli, R.; Blanz, J.; Gassmann, E.; Drückes, P.; Fendrich, G.; et al. Discovery of Imidazoquinolines as a Novel Class of Potent, Selective, and in Vivo Efficacious Cancer Osaka Thyroid (COT) Kinase Inhibitors. J. Med. Chem. 2016, 59, 7544–7560. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.M.; Kallman, N.J.; Koenig, T.M.; Linder, R.J.; Richey, R.N.; Rizzo, J.R.; Ward, J.A.; Yu, H.; Zhang, T.Y.; Mitchell, D. Double Heck Route to a Dibenzoxepine and Convergent Suzuki Cross-Coupling Strategy for the Synthesis of an MR Antagonist. Org. Process Res. Dev. 2017, 21, 208–217. [Google Scholar] [CrossRef]
- Kondaparla, S.; Soni, A.; Manhas, A.; Srivastava, K.; Puri, S.K.; Katti, S.B. Synthesis and Antimalarial Activity of New 4-Aminoquinolines Active against Drug Resistant Strains. RSC Adv. 2016, 6, 105676–105689. [Google Scholar] [CrossRef]
- Dola, V.R.; Soni, A.; Agarwal, P.; Ahmad, H.; Raju, K.S.R.; Rashid, M.; Wahajuddin, M.; Srivastava, K.; Haq, W.; Dwivedi, A.K.; et al. Synthesis and Evaluation of Chirally Defined Side Chain Variants of 7-Chloro-4-Aminoquinoline To Overcome Drug Resistance in Malaria Chemotherapy. Antimicrob. Agents Chemother. 2017, 61, e01152-16. [Google Scholar] [CrossRef] [Green Version]
- Beck, H.P.; Kohn, T.; Rubenstein, S.; Hedberg, C.; Schwandner, R.; Hasslinger, K.; Dai, K.; Li, C.; Liang, L.; Wesche, H.; et al. Discovery of Potent LPA2 (EDG4) Antagonists as Potential Anticancer Agents. Bioorganic Med. Chem. Lett. 2008, 18, 1037–1041. [Google Scholar] [CrossRef]
- Abou-Gharbia, M.A.; Childers, W.E.; Fletcher, H.; McGaughey, G.; Patel, U.; Webb, M.B.; Yardley, J.; Andree, T.; Boast, C.; Kucharik, R.J.; et al. Synthesis and SAR of Adatanserin: Novel Adamantyl Aryl- and Heteroarylpiperazines with Dual Serotonin 5-HT(1A) and 5-HT2 Activity as Potential Anxiolytic and Antidepressant Agents. J. Med. Chem. 1999, 42, 5077–5094. [Google Scholar] [CrossRef]
- Li, F.F.; Stubbing, L.A.; Kavianinia, I.; Abbattista, M.R.; Harris, P.W.R.; Smaill, J.B.; Patterson, A.V.; Brimble, M.A. Synthesis and Antiproliferative Activity of C- and N-Terminal Analogues of Culicinin D. Bioorganic Med. Chem. Lett. 2020, 30, 127331. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Land, H.; Berglund, P. An Efficient Single-Enzymatic Cascade for Asymmetric Synthesis of Chiral Amines Catalyzed by ω-Transaminase. Chem. Commun. 2013, 49, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Mourelle-Insua, Á.; Zampieri, L.A.; Lavandera, I.; Gotor-Fernández, V. Conversion of γ- and δ-Keto Esters into Optically Active Lactams. Transaminases in Cascade Processes. Adv. Synth. Catal. 2018, 360, 686–695. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric Synthesis of Optically Pure Pharmacologically Relevant Amines Employing ω-Transaminases. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakó, Á.; Poppe, L.; Mendonça, R. Efficient Synthesis of Pharmaceutically Relevant Prochiral Heterocyclic Aminoketones. Period. Polytech. Chem. Eng. 2021, 65, 177–182. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | TA | Substrate Conc. (mM) | IPA Equiv. (—) | c (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | 113 | 10 | 100 | 93 | >99 |
| 2 | 10 | 10 | 92 | >99 | |
| 3 | 25 | 100 | 0 | >99 | |
| 4 | 200 | 10 | 100 | 24 | >99 |
| 5 | 217 | 10 | 100 | 76 | >99 |
| 6 | 234 | 10 | 100 | 24 | >99 |
| 7 | 237 | 10 | 100 | 98 | >99 |
| 8 | 10 | 10 | 48 | >99 | |
| 9 | 25 | 100 | 94 | >99 | |
| 10 | 50 | 100 | 0 | >99 | |
| 11 | 238 | 10 | 100 | 41 | >99 |
| 12 | 251 | 10 | 100 | 99 | >99 |
| 13 | 10 | 10 | 92 | >99 | |
| 14 | 25 | 100 | 99 | >99 | |
| 15 | 50 | 100 | 23 | >99 | |
| 16 | 254 | 10 | 100 | 99 | >99 |
| 17 | 10 | 10 | 86 | >99 | |
| 18 | 25 | 100 | 99 | >99 | |
| 19 | 50 | 100 | 99 | >99 | |
| 20 | 256 | 10 | 100 | 99 | >99 |
| 21 | 10 | 10 | 85 | >99 | |
| 22 | 25 | 100 | 99 | >99 | |
| 23 | 50 | 100 | 99 | >99 | |
| 24 | 260 | 10 | 100 | 99 | >99 |
| 25 | 10 | 10 | 34 | >99 | |
| 26 | 25 | 100 | 99 | >99 | |
| 27 | 50 | 100 | 99 | >99 | |
| 28 | P1-B04 | 10 | 100 | 17 | >99 |
| 29 | P1-F03 | 10 | 100 | 53 | >99 |
| 30 | P1-G05 | 10 | 100 | 4 | >99 |
| Entry | TA | Substrate Conc. (mM) | IPA Equiv. (—) | c (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | 007 | 10 | 100 | 2 | >99 |
| 2 | 013 | 10 | 100 | 46 | >99 |
| 3 | 025 | 10 | 100 | 93 | >99 |
| 4 | 10 | 10 | 48 | >99 | |
| 5 | 25 | 100 | 93 | >99 | |
| 6 | 50 | 100 | 4 | >99 | |
| 7 | 117 | 10 | 100 | 13 | >99 |
| 8 | 301 | 10 | 100 | 6 | 74 |
| 9 | 303 | 10 | 100 | 11 | 90 |
| 10 | 412 | 10 | 100 | 41 | >99 |
| 11 | 415 | 10 | 100 | 54 | >99 |
| 12 | P2-A01 | 10 | 100 | 40 | >99 |
| 13 | P2-A07 | 10 | 100 | 82 | >99 |
| 14 | 10 | 10 | 42 | >99 | |
| 15 | 25 | 100 | 10 | >99 | |
| 16 | P2-B01 | 10 | 100 | 85 | 99 |
| 17 | 10 | 10 | 71 | >99 | |
| 18 | 25 | 100 | 4 | >99 |
| Entry | Substrate | TA | Substrate Concentration (mM) | c (%) | ee (%) |
|---|---|---|---|---|---|
| 1 |  | TA-254 | 10 | 98 | >99 |
| 2 | 25 | 98 | >99 | ||
| 3 | 50 | 97 | >99 | ||
| 4 | 75 | 90 | >99 | ||
| 5 | 100 | 77 | >99 | ||
| 6 |  | TA-256 | 10 | 96 | >99 |
| 7 | 25 | 95 | >99 | ||
| 8 | 50 | 95 | >99 | ||
| 9 | 75 | 91 | >99 | ||
| 10 | 100 | 83 | >99 | ||
| 11 |  | TA-256 | 10 | 97 | >99 |
| 12 | 25 | 95 | >99 | ||
| 13 | 50 | 95 | >99 | ||
| 14 | 75 | 89 | >99 | ||
| 15 | 100 | 88 | >99 | ||
| 16 |  | TA-256 | 10 | 94 | >99 |
| 17 | 25 | 94 | >99 | ||
| 18 | 50 | 94 | >99 | ||
| 19 | 75 | 94 | >99 | ||
| 20 | 100 | 93 | >99 | ||
| 21 | 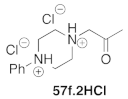 | TA-256 | 10 | 96 | >99 |
| 22 | 25 | 95 | >99 | ||
| 23 | 50 | 95 | >99 | ||
| 24 | 75 | 93 | >99 | ||
| 25 | 100 | 93 | >99 | ||
| 26 | 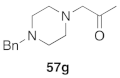 | TA-254 | 10 | 93 | >99 |
| 27 | 25 | 92 | >99 | ||
| 28 | 50 | 92 | >99 | ||
| 29 | 75 | 92 | >99 | ||
| 30 | 100 | 91 | >99 |

| Entry | Substrate a | TA | Yield a (%) | ee b (%) | [α] (c) c |
|---|---|---|---|---|---|
| 1 | 57a | TA-254 | 47 | >99 | +31.2 (1.12) |
| 2 | 57b | TA-254 | 74 | >99 | +44.0 (0.64) |
| 3 | 57c | TA-256 | 83 | >99 | +34.2 (1.20) |
| 4 | 57d | TA-256 | 65 | >99 | +36.6 (1.20) |
| 5 | 57e | TA-256 | 59 | >99 | +29.4 (1.20) |
| 6 | 57f | TA-256 | 81 | >99 | +36.0 (1.20) |
| 7 | 57g | TA-254 | 94 | >99 | +26.9 (1.20) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malta-Lakó, Á.; Zhang, F.; Mendonça, R.; Poppe, L. Transaminase Catalysis for Enantiopure Saturated Heterocycles as Potential Drug Scaffolds. Catalysts 2021, 11, 1501. https://doi.org/10.3390/catal11121501
Malta-Lakó Á, Zhang F, Mendonça R, Poppe L. Transaminase Catalysis for Enantiopure Saturated Heterocycles as Potential Drug Scaffolds. Catalysts. 2021; 11(12):1501. https://doi.org/10.3390/catal11121501
Chicago/Turabian StyleMalta-Lakó, Ágnes, Fangyi Zhang, Ricardo Mendonça, and László Poppe. 2021. "Transaminase Catalysis for Enantiopure Saturated Heterocycles as Potential Drug Scaffolds" Catalysts 11, no. 12: 1501. https://doi.org/10.3390/catal11121501
APA StyleMalta-Lakó, Á., Zhang, F., Mendonça, R., & Poppe, L. (2021). Transaminase Catalysis for Enantiopure Saturated Heterocycles as Potential Drug Scaffolds. Catalysts, 11(12), 1501. https://doi.org/10.3390/catal11121501