
Effect of Cesium and Phosphate Addition to Mo/V/W Mixed Oxide Catalysts for the Gas Phase Oxidation of Methacrolein to Methacrylic Acid

Abstract
:
1. Introduction
2. Results and Discussion
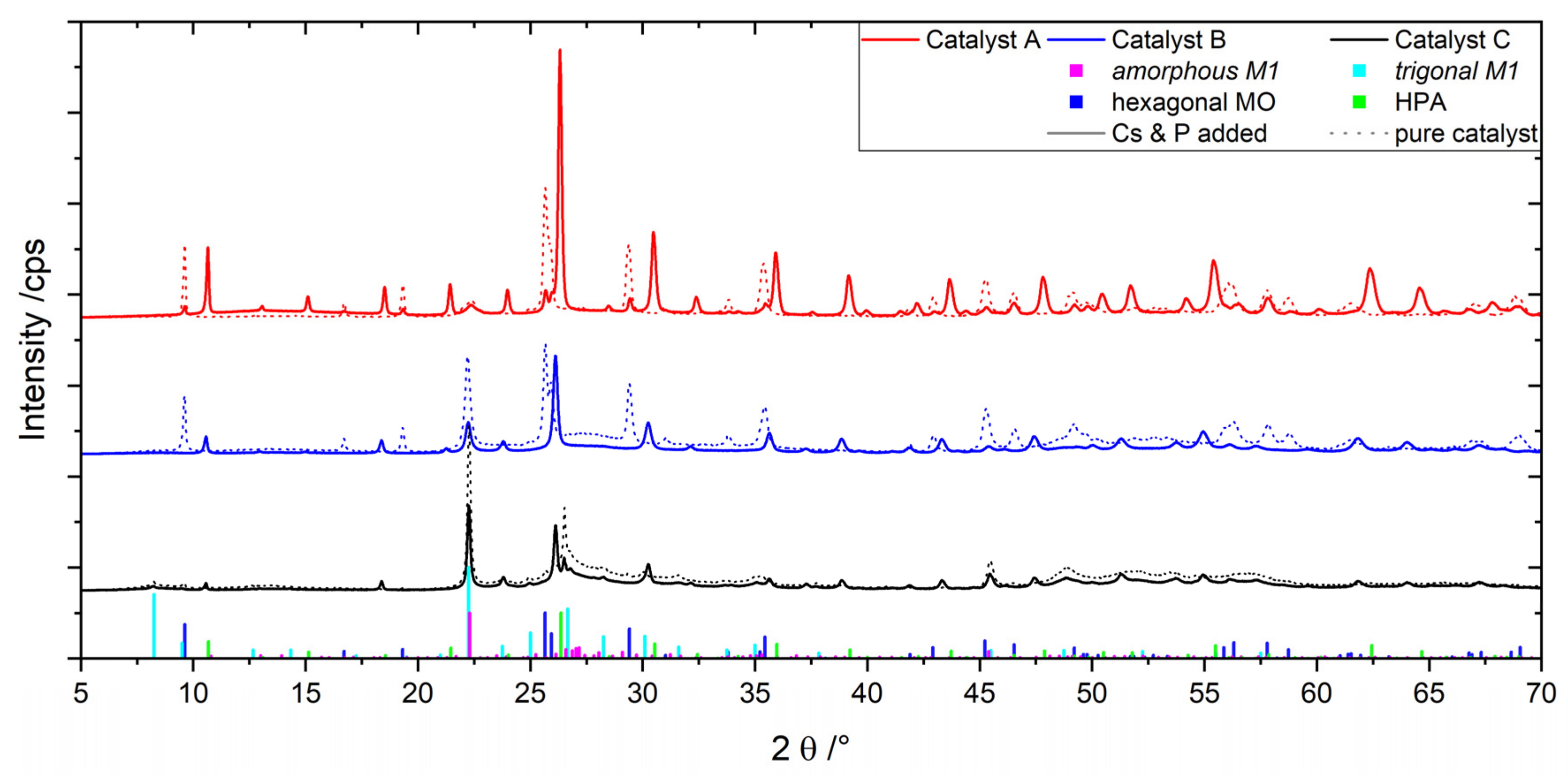
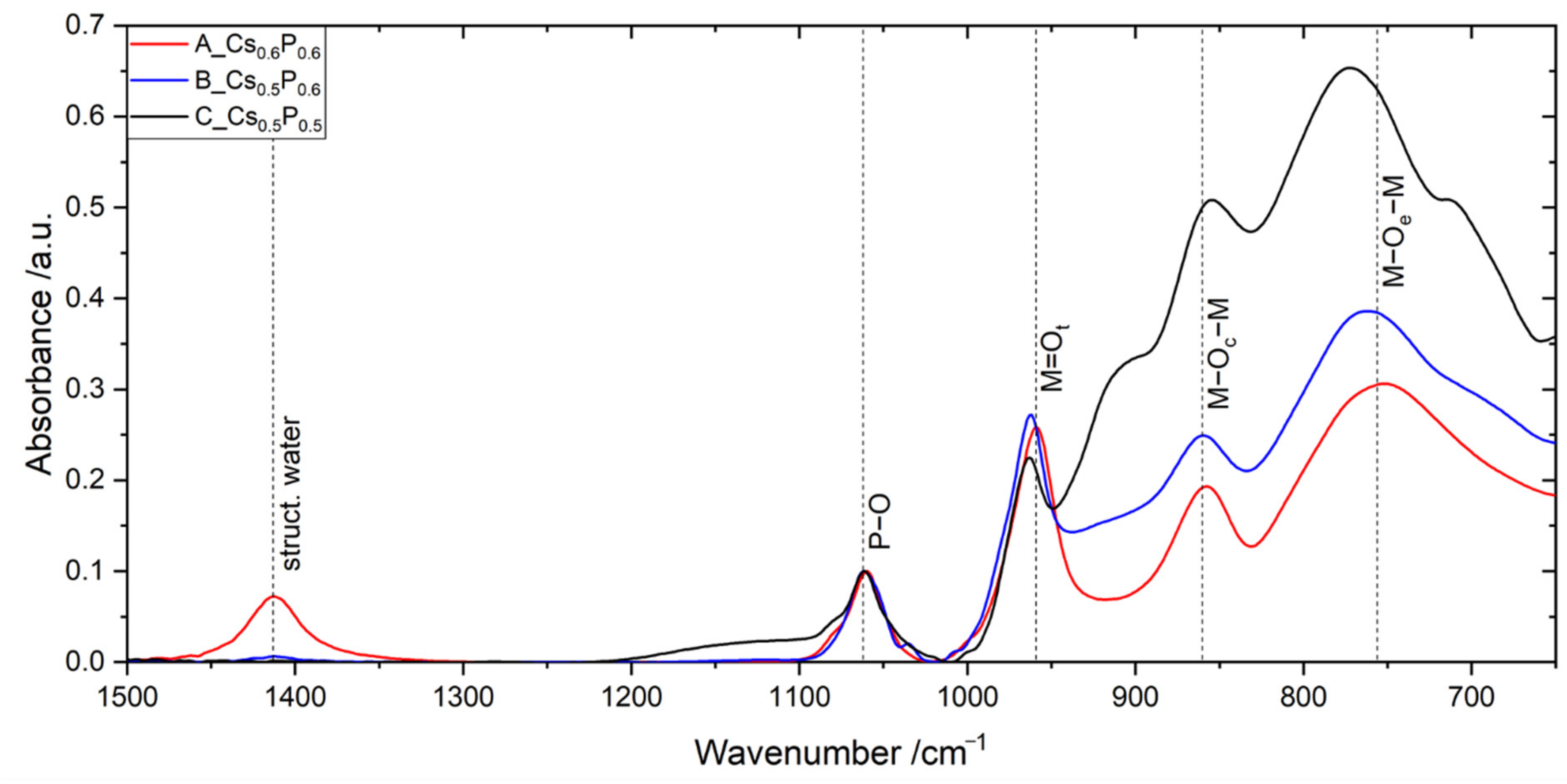
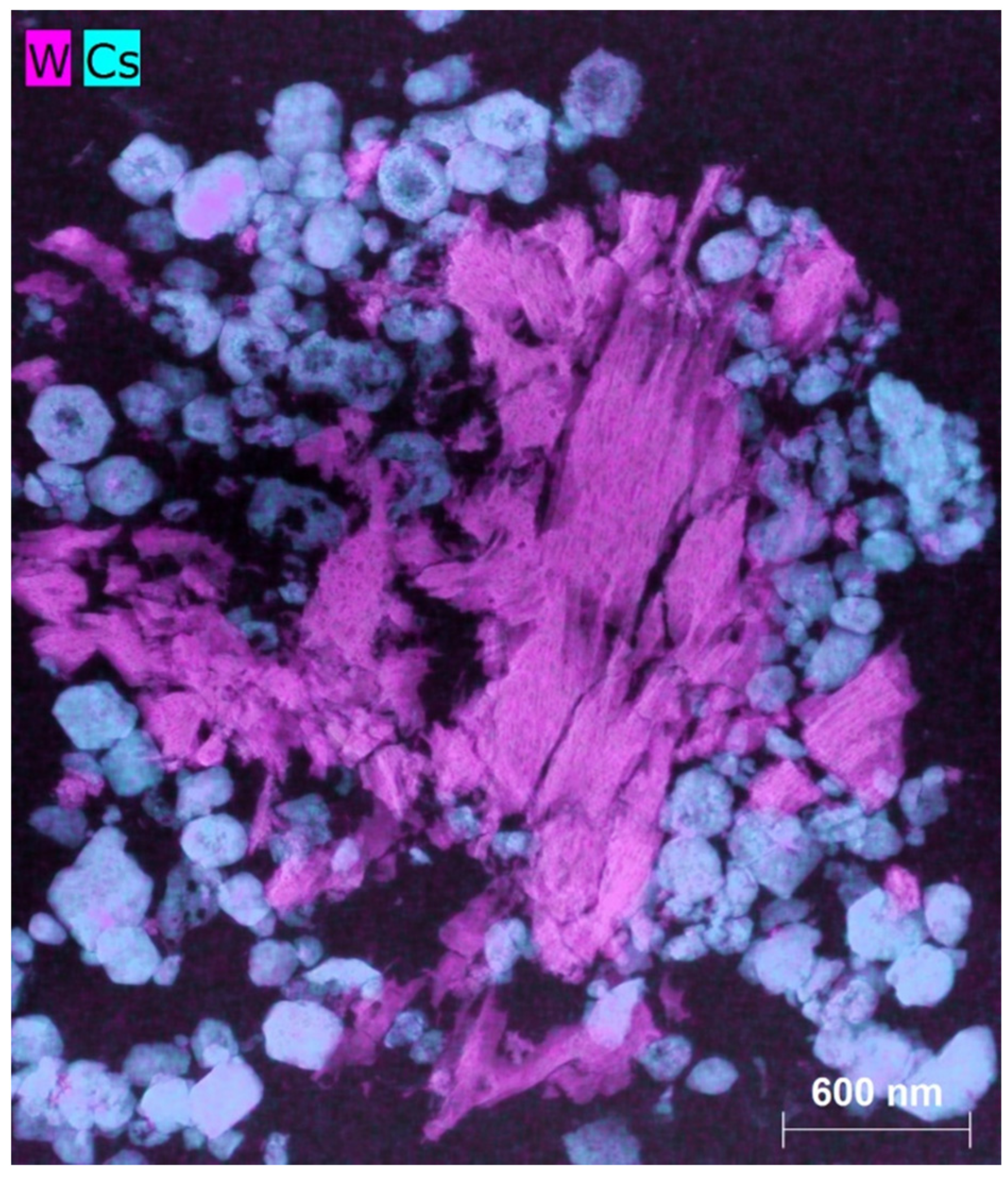
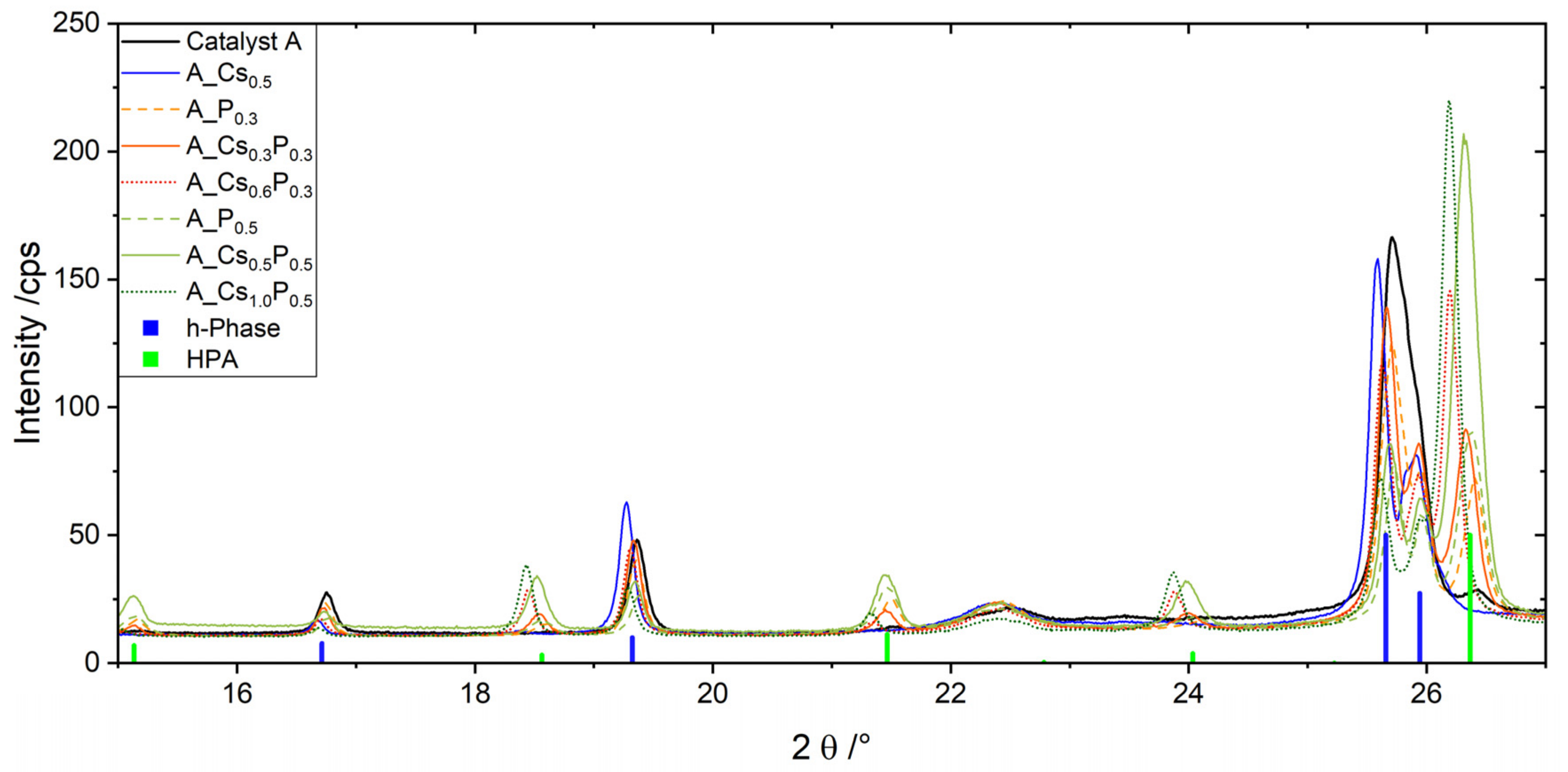
2.1. Catalysts A, B and C Containing Cs/P ≈ 1
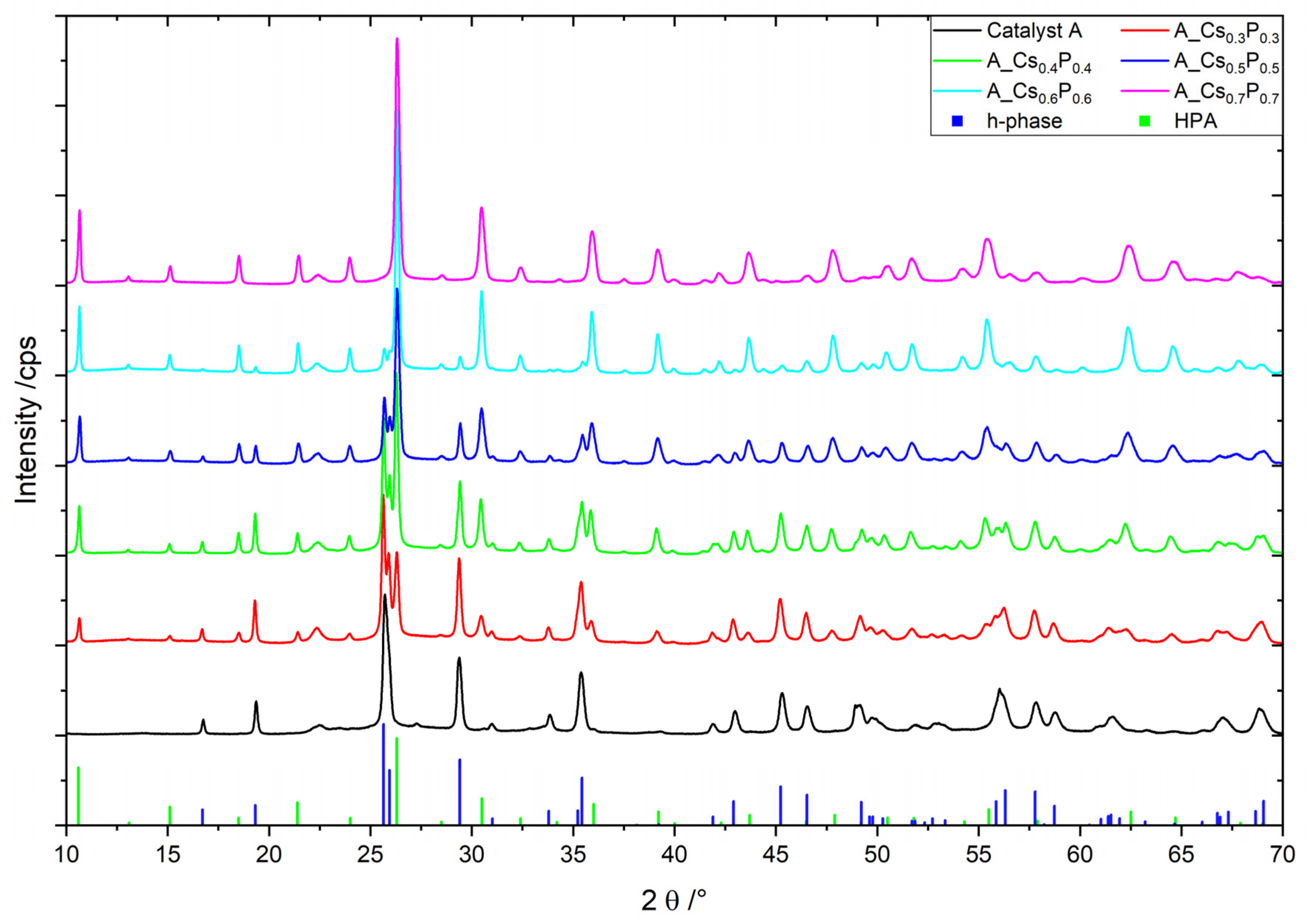
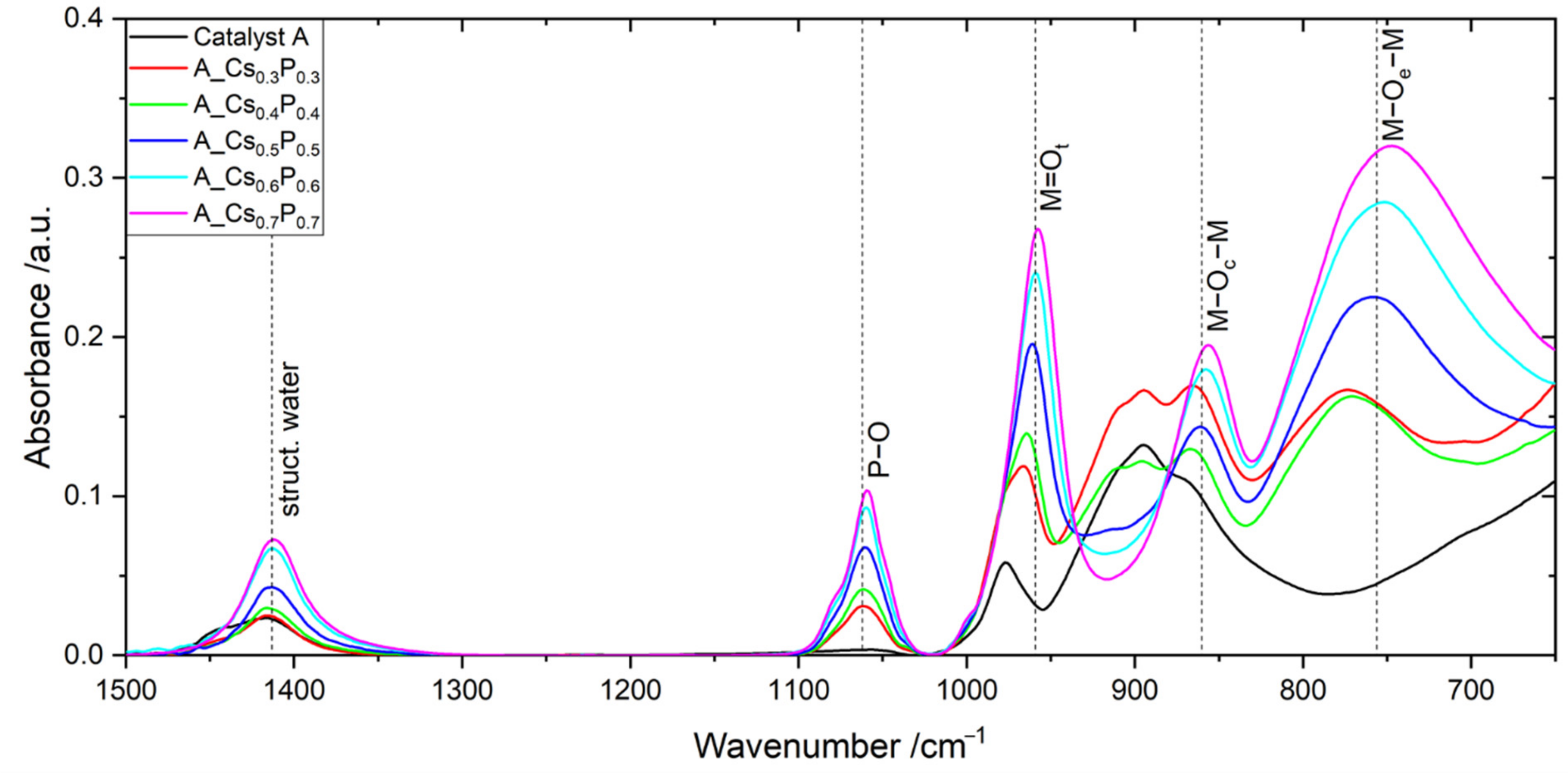
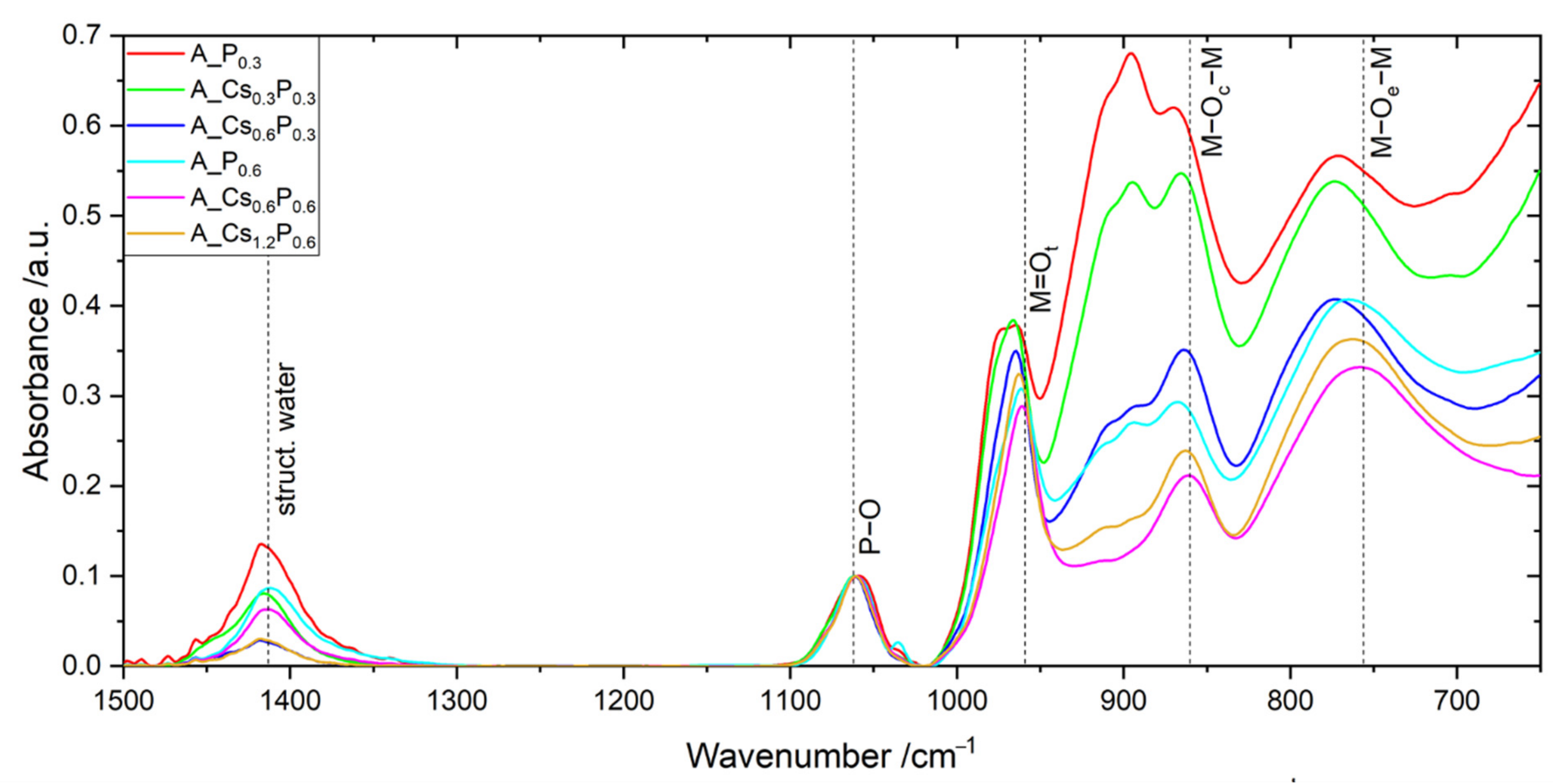
2.2. Catalyst A with Varied Cs/P and (Mo + V + W)/P Ratios
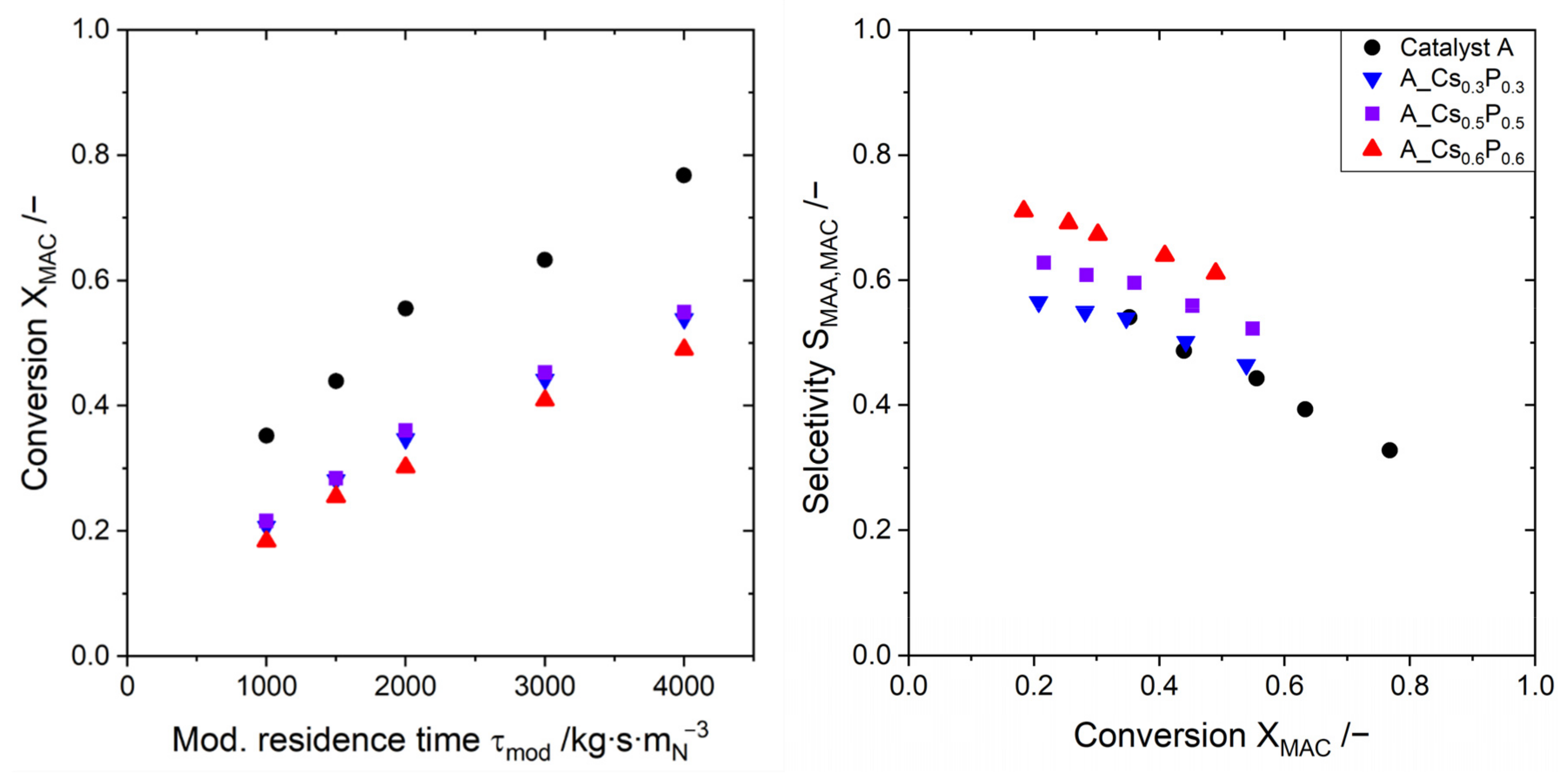
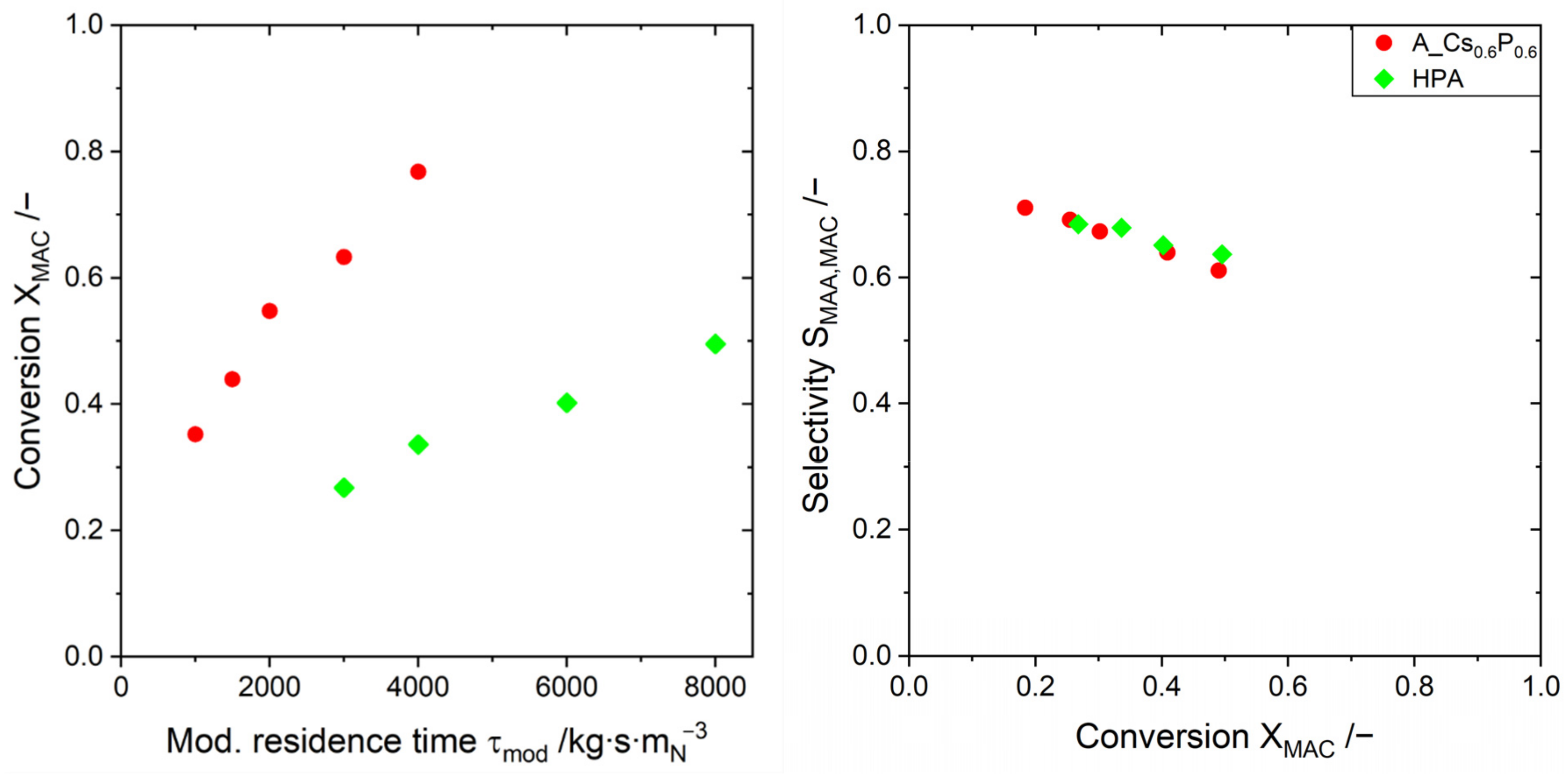
2.3. Evaluating the Catalyst Performance
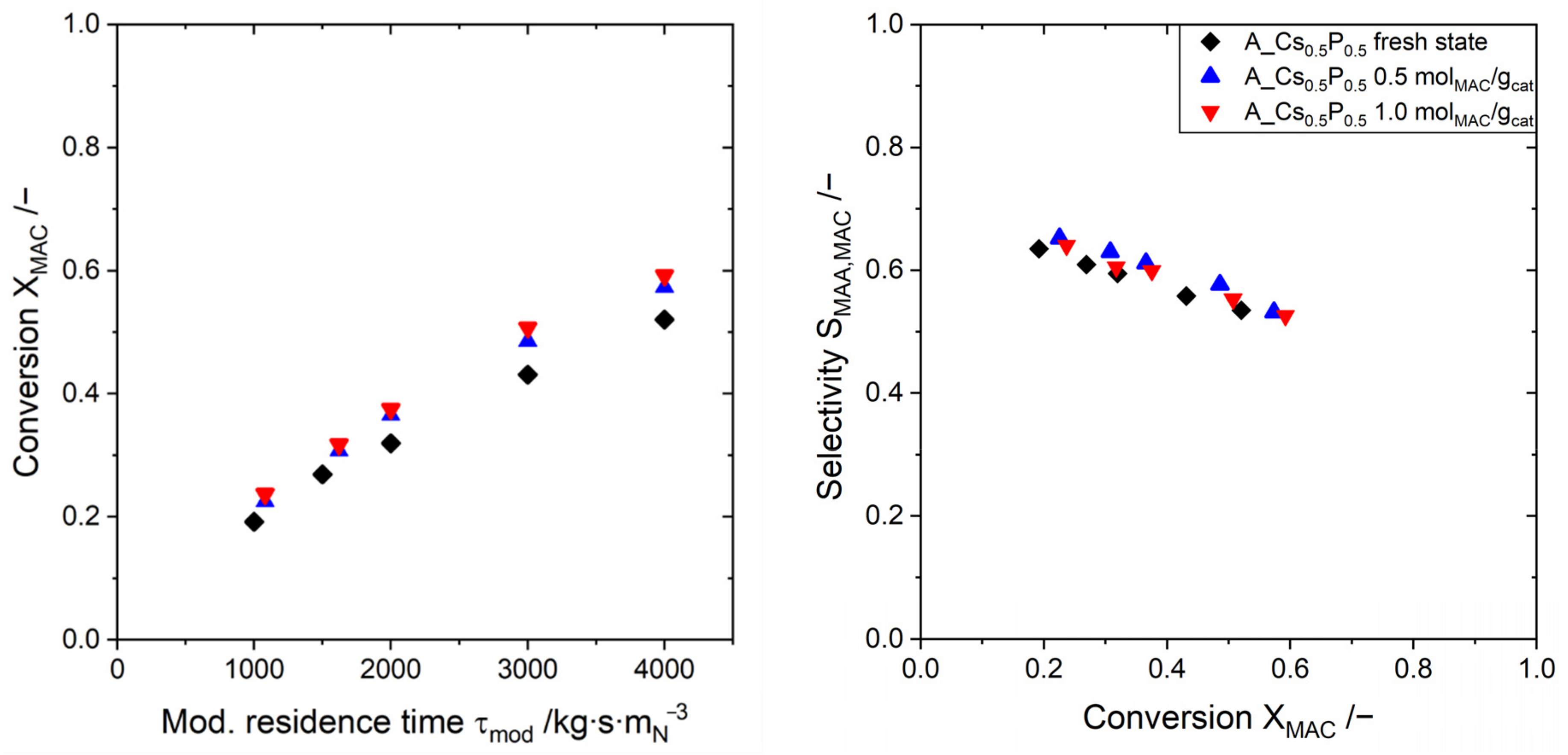
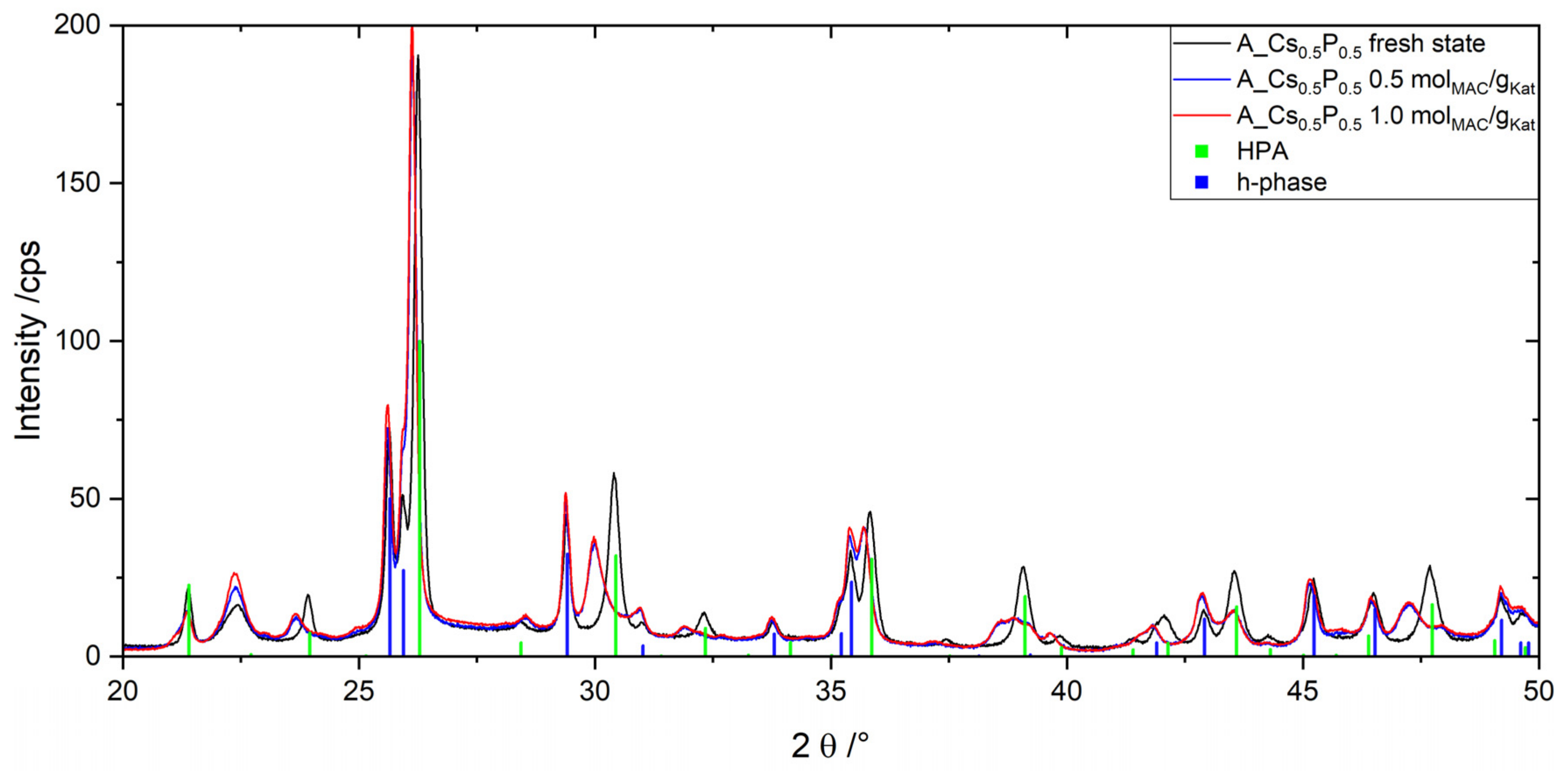
2.4. Catalyst Stability
3. Experimental
3.1. Catalyst Preparation
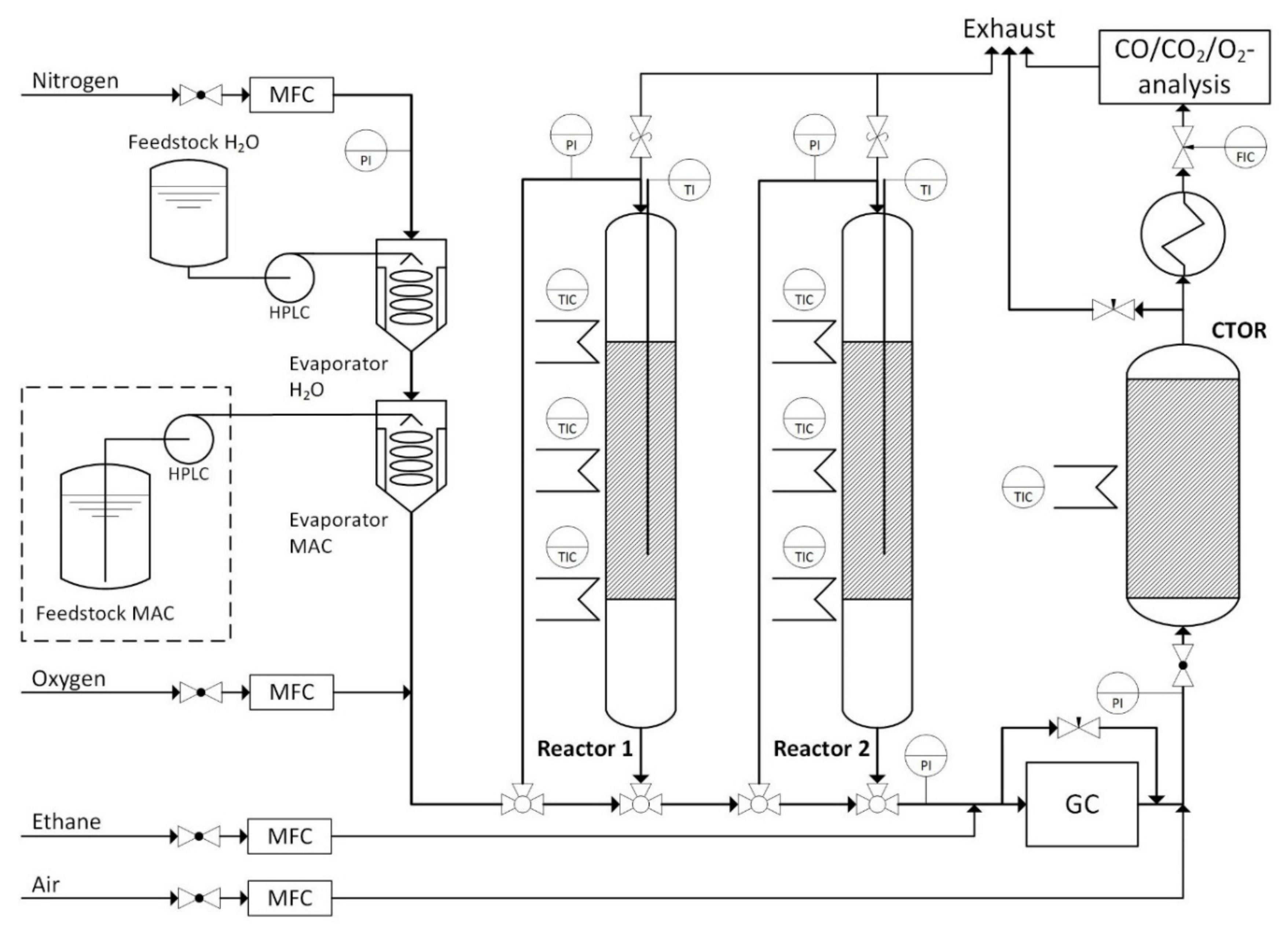
3.2. Reaction Unit
3.3. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Nomenclature
| Abbreviations | |
| FID | flame ionization detector |
| HAc | acetic acid |
| HPA | heteropoly acid |
| MAA | methacrylic acid |
| MAC | methacrolein |
| MO | mixed oxide |
| TCD | thermal conductivity detector |
| wt | weight |
| Symbols | |
| ABET | specific surface area (m2∙g−1) |
| CMAC,s | MAC concentration on the catalyst surface (mol∙m−3) |
| CMAC,g | MAC concentration in the gas phase (mol∙m−3) |
| dHPA | Unit cell parameter of HPA phase (nm) |
| Fi | integrated area in XRD diffractogram (−) |
| kF,i | surface specific reaction rate coefficient (m∙s−1) |
| km,I | mass specific reaction rate coefficient (m3∙kg−1∙s−1) |
| molar flow of component i (mol∙s−1) | |
| molar flow of component i at the inlet (mol∙s−1) | |
| p | pressure (bar) |
| RSi | carbon-based reactor selectivity to component i (−) |
| T | temperature (°C) |
| TOS | time on stream (h) |
| total volumetric inlet flow (m3∙s−1) | |
| Wz | Weisz number for inner mass transfer limitation (−) |
| Xi | conversion of component i (−) |
| zi | carbon number of component i (−) |
| τmod | modified residence time at standard conditions (kg∙s∙m−3) |
| Subscripts | |
| 0 | inlet condition |
| c | corner sharing |
| cat | catalyst |
| e | edge sharing |
| i | component i |
| max | maximum |
| N | Norm conditions (0 °C, 1.01325 bar) |
| t | terminal |
| Superscripts | |
| R | reactor |
References
- Védrine, J.C. Metal Oxides in Heterogeneous Oxidation Catalysis: State of the Art and Challenges for a More Sustainable World. ChemSusChem 2019, 12, 577–588. [Google Scholar] [CrossRef]
- Darabi Mahboub, M.J.; Dubois, J.-L.; Cavani, F.; Rostamizadeh, M.; Patience, G.S. Catalysis for the synthesis of methacrylic acid and methyl methacrylate. Chem. Soc. Rev. 2018, 47, 7703–7738. [Google Scholar] [CrossRef]
- Nagai, K.; Ui, T. Trends and Future of Monomer–MMA Technologies. Sumitomo Chem. 2004, 2, 4–13. [Google Scholar]
- Drochner, A.; Ohlig, D.; Knoche, S.; Gora, N.; Heid, M.; Menning, N.; Petzold, T.; Vogel, H. Mechanistic Studies on the Transition Metal Oxide Catalysed Partial Oxidation of (Meth)Acrolein to the Corresponding Carboxylic Acids. Top. Catal. 2016, 59, 1518–1532. [Google Scholar] [CrossRef]
- Wagner, J.B.; Su, D.S.; Schunk, S.A.; Hibst, H.; Petzoldt, J.; Schlögl, R. Structural characterization of high-performance catalysts for partial oxidation—the high-resolution and analytical electron microscopy approach. J. Catal. 2004, 224, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Böhnke, H.; Gaube, J.; Petzoldt, J. Selective Oxidation of Methacrolein towards Methacrylic Acid on Mixed Oxide (Mo, V, W) Catalysts. Part 1. Studies on Kinetics. Ind. Eng. Chem. Res. 2006, 45, 8794–8800. [Google Scholar] [CrossRef]
- Böhnke, H.; Gaube, J.; Petzoldt, J. Selective Oxidation of Methacrolein towards Methacrylic Acid on Mixed Oxide (Mo, V, W) Catalysts. Part 2. Variation of Catalyst Composition and Comparison with Acrolein Oxidation. Ind. Eng. Chem. Res. 2006, 45, 8801–8806. [Google Scholar] [CrossRef]
- Schunk, S.; Sundermann, A.; Hibst, H. Structure oriented library design in gas phase oxidation catalysis. Catal. Today 2008, 137, 36–43. [Google Scholar] [CrossRef]
- Grasselli, R.K.; Volpe, A.F. Catalytic Consequences of a Revised Distribution of Key Elements at the Active Centers of the M1 Phase of the MoVNbTeOx System. Top. Catal. 2014, 57, 1124–1137. [Google Scholar] [CrossRef]
- Deusser, L.M.; Petzoldt, J.C.; Gaube, J.W.; Hibst, H. Kinetic Model for Methacrolein Oxidation. Influence of Cesium and Vanadium on Heteropolyacid Catalysts CsxH3-x+y[PMo12-yVyO40]. Ind. Eng. Chem. Res. 1998, 37, 3230–3236. [Google Scholar] [CrossRef]
- Illies, S.; Kraushaar-Czarnetzki, B. Processing Study on the Stability of Heteropoly Acid Catalyst in the Oxidation of Methacrolein to Methacrylic Acid. Ind. Eng. Chem. Res. 2016, 55, 8509–8518. [Google Scholar] [CrossRef]
- Keggin, J.F. Structure of the Crystals of 12-Phosphotungstic Acid. Nature 1933, 132, 351. [Google Scholar] [CrossRef]
- Marosi, L. Catalytic performance of Csx(NH4)yHzPMo12O40 and related heteropolyacids in the methacrolein to methacrylic acid conversion: In situ structural study of the formation and stability of the catalytically active species. J. Catal. 2003, 213, 235–240. [Google Scholar] [CrossRef]
- Chamack, M.; Mahjoub, A.R.; Aghayan, H. Cesium salts of tungsten-substituted molybdophosphoric acid immobilized onto platelet mesoporous silica: Efficient catalysts for oxidative desulfurization of dibenzothiophene. Chem. Eng. J. 2014, 255, 686–694. [Google Scholar] [CrossRef]
- Xue, J.; Yin, H.; Li, H.; Zhang, D.; Jiang, T.; Yu, L.; Shen, Y. Oxidation of cyclopentene catalyzed by tungsten-substituted molybdophosphoric acids. Korean J. Chem. Eng. 2009, 26, 654–659. [Google Scholar] [CrossRef]
- Krauß, K. Entwicklung und Einsatz einer DRIFTS-Meßzelle zur In-Situ-Spektroskopie Heterogen Katalysierter Gasphasenoxidationen; Technische Universität Darmstadt: Darmstadt, Germany, 2000. [Google Scholar]
- Dürr, N. Partialoxidation von Methacrolein zu Methacrylsäure an Heteropolysäurekatalysatoren; Technische Universität Darmstadt: Darmstadt, Germany, 2014. [Google Scholar]
- Mestl, G.; Verbruggen, N.F.D.; Bosch, E.; Knözinger, H. Mechanically Activated MoO3. 5. Redox Behavior. Langmuir 1996, 12, 2961–2968. [Google Scholar] [CrossRef]
- Akimoto, M. 12-Heteropolymolybdates as catalysts for vapor-phase oxidative dehydrogenation of isobutyric acid 3. Molybdotungstophosphoric and molybdovanadophosphoric acids. J. Catal. 1984, 89, 196–208. [Google Scholar] [CrossRef]
- Essayem, N.; Coudurier, G.; Fournier, M.; Védrine, J.C. Acidic and catalytic properties of CsxH3−xPW12O40 heteropolyacid compounds. Catal. Lett. 1995, 34, 223–235. [Google Scholar] [CrossRef]
- Xian-e, C.; Daichun, D.; Jianping, N.; Youming, J.; Jing, Z.; Yixiang, Q. Enthalpies of formation and DSC and TG results of heteropoly acids containing tungsten and molybdenum. Thermochim. Acta 1997, 292, 45–50. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Heterogeneous acid catalysis by heteropoly acids: Approaches to catalyst deactivation. J. Mol. Catal. A Chem. 2009, 305, 104–111. [Google Scholar] [CrossRef]
- Micek-Ilnicka, A. The role of water in the catalysis on solid heteropolyacids. J. Mol. Catal. A Chem. 2009, 308, 1–14. [Google Scholar] [CrossRef]
- Sennerich, M.; Weidler, P.; Kraushaar-Czarnetzki, B. Hexagonal Mo/V/W mixed oxide as a catalyst for the partial oxidation of methacrolein to methacrylic acid. Catal. Commun. 2020, 141, 106016. [Google Scholar] [CrossRef]
- Hibst, H.; Tenten, A.; Marosi, L. Multimetalloxide. German Patent DE19542755A1, 22 May 1997. [Google Scholar]
- Rosowski, F.; Müller-Engel, K.J.; Klaus, J.; Blickhan, N.; Dürr, N.; Jekewitz, T.; Menning, N.; Petzold, T.; Schmidt, S. Verfahren der heterogen katalysierten Gasphasenpartialoxidation von (Meth)acrolein zu (Meth)acrylsäure. German Patent DE102012207811A1, 12 July 2012. [Google Scholar]
- Bruker AXS. TOPAS V6: General Profile and Structure Analysis Software for Powder Diffraction Data—User’s Manual; Bruker: Billerica, MA, USA, 2017. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Chemical Composition | ABET (m2/g) | Cryst. (wt.%) | h-Phase (wt.%) * | M1 (wt.%) * | HPA (wt.%) * |
|---|---|---|---|---|---|---|
| A | Mo8.0V2.0W1.0 | 13.9 | ≈79 | >95 | <5 | 0 |
| B | Mo8.0V2.0W0.5 | 20.6 | ≈68 | ≈55 | ≈45 | 0 |
| C | Mo8.0V2.3W0.8 | 25.8 | ≈58 | <5 | >95 | 0 |
| A_Cs0.6P0.6 | Mo8.0V2.0W0.9Cs0.6P0.6 | 8.8 | ≈71 | <5 | ≈0 | >95 |
| B_Cs0.5P0.6 | Mo8.0V2.5W0.5Cs0.5P0.6 | 15.3 | ≈65 | 0 | ≈25 | ≈75 |
| C_Cs0.5P0.5 | Mo8.0V2.4W0.8Cs0.5P0.5 | 24.9 | ≈62 | 0 | ≈60 | ≈40 |
| Catalyst | Chemical Composition | Cryst. (wt.%) | h-phase (wt.%) * | HPA (wt.%) * | ABET (m2/g) |
|---|---|---|---|---|---|
| A | Mo8.0V2.0W1.0 | ≈78 | 100 | 0 | 11.6 |
| A_Cs0.3P0.3 | Mo8.0V2.0W0.9Cs0.3P0.2 | ≈72 | 55 | 45 | 9.4 |
| A_Cs0.4P0.4 | Mo8.0V2.0W1.0Cs0.4P0.4 | ≈75 | 40 | 60 | 7.4 |
| A_Cs0.5P0.5 | Mo8.0V2.0W1.0Cs0.5P0.5 | ≈72 | 25 | 75 | 9.0 |
| A_Cs0.6P0.6 | Mo8.0V2.0W0.9Cs0.6P0.6 | ≈71 | 5 | 95 | 8.8 |
| A_Cs0.7P0.7 | Mo8.0V2.0W0.9Cs0.7P0.7 | ≈75 | 0 | 100 | 9.5 |
| Catalyst | Chemical Composition | Cryst. (wt.%) | h-Phase (wt.%) * | HPA (wt.%) * | dHPA (nm) | ABET (m2/g) |
|---|---|---|---|---|---|---|
| A_P0.3 | Mo8.0V2.0W1.0P0.3 | ≈77 | 60 | 40 | 1.168 | 6.7 |
| A_Cs0.3P0.3 | Mo8.0V2.0W0.9Cs0.3P0.2 | ≈72 | 65 | 45 | 1.173 | 9.4 |
| A_Cs0.6P0.3 | Mo8.0V2.0W0.9Cs0.7P0.3 | ≈73 | 40 | 60 | 1.179 | 11.4 |
| A_P0.5 | Mo8.0V2.0W1.0P0.5 | ≈73 | 40 | 60 | 1.170 | 5.2 |
| A_Cs0.5P0.5 | Mo8.0V2.0W1.0Cs0.5P0.5 | ≈72 | 25 | 75 | 1.174 | 9.0 |
| A_Cs1.0P0.5 | Mo8.0V2.0W1.0Cs0.9P0.5 | ≈79 | 20 | 80 | 1.178 | 10.8 |
| Catalyst | Ageing State | dHPA (nm) | ABET (m2∙g−1) |
|---|---|---|---|
| A_Cs0.5P0.5 | Fresh state | 1.175 | 8.21 |
| A_Cs0.5P0.5 | 0.5 molMAC/gKat | 1.186 | 9.52 |
| A_Cs0.5P0.5 | 1.0 molMAC/gKat | 1.189 | 10.76 |
| Parameter | Value |
|---|---|
| isothermal reactor temperature (°C) | 300/340 * |
| methacrolein (MAC) content in the feed (% v/v) | 3.4 |
| molar feed ratio MAC:O2 | 1:2 |
| water vapor content (% v/v) | 21 |
| total pressure (bar(a)) | 1.5 |
| total volumetric inlet flow (mLN/min) | 150–1200 |
| modified residence time (kg·s/m3) | 500–8000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sennerich, M.; Weidler, P.G.; Heißler, S.; Kraushaar-Czarnetzki, B. Effect of Cesium and Phosphate Addition to Mo/V/W Mixed Oxide Catalysts for the Gas Phase Oxidation of Methacrolein to Methacrylic Acid. Catalysts 2021, 11, 231. https://doi.org/10.3390/catal11020231
Sennerich M, Weidler PG, Heißler S, Kraushaar-Czarnetzki B. Effect of Cesium and Phosphate Addition to Mo/V/W Mixed Oxide Catalysts for the Gas Phase Oxidation of Methacrolein to Methacrylic Acid. Catalysts. 2021; 11(2):231. https://doi.org/10.3390/catal11020231
Chicago/Turabian StyleSennerich, Maximilian, Peter G. Weidler, Stefan Heißler, and Bettina Kraushaar-Czarnetzki. 2021. "Effect of Cesium and Phosphate Addition to Mo/V/W Mixed Oxide Catalysts for the Gas Phase Oxidation of Methacrolein to Methacrylic Acid" Catalysts 11, no. 2: 231. https://doi.org/10.3390/catal11020231