
ω-Transaminase-Mediated Asymmetric Synthesis of (S)-1-(4-Trifluoromethylphenyl)Ethylamine


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
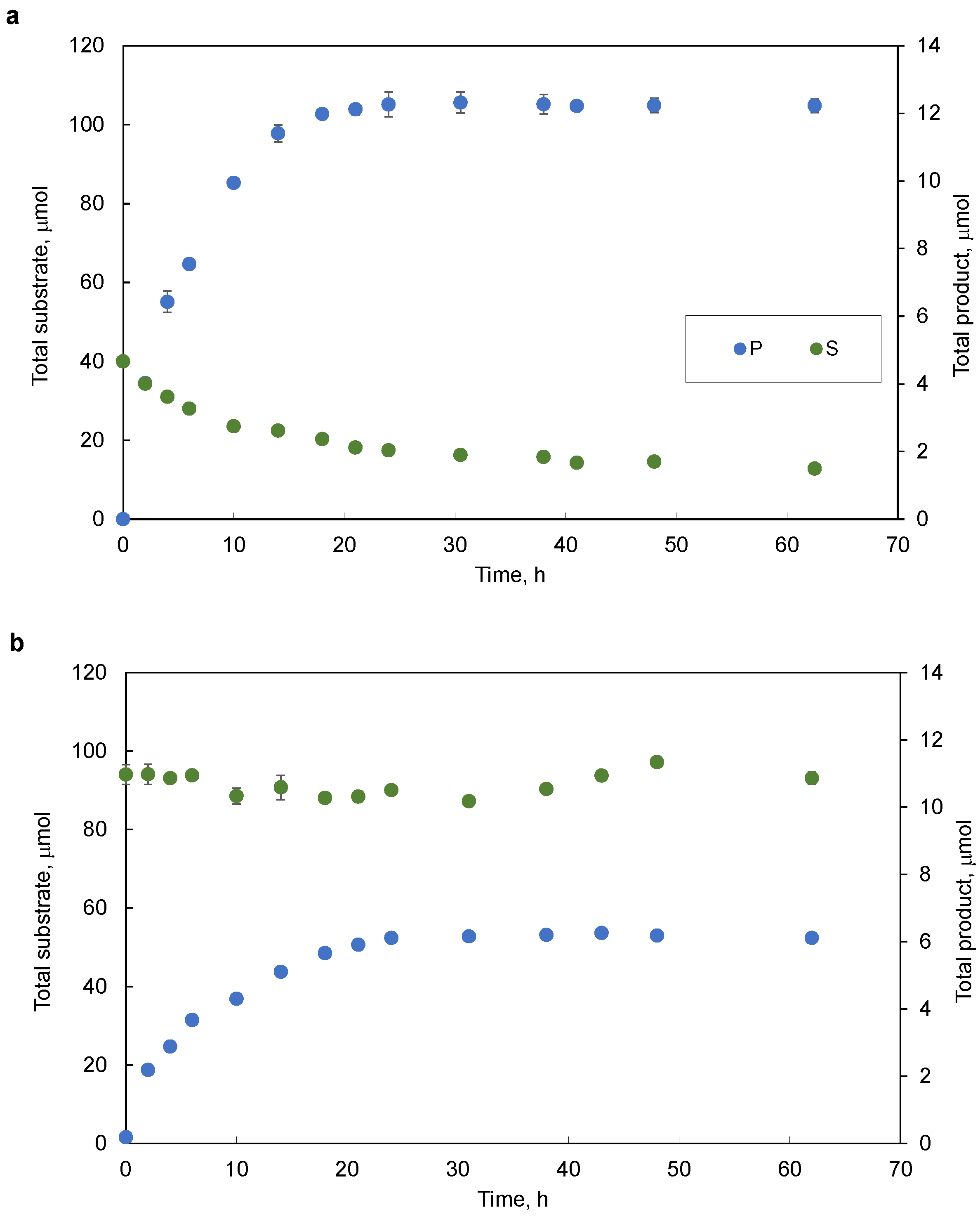
2.1. Bioconversion Conditions
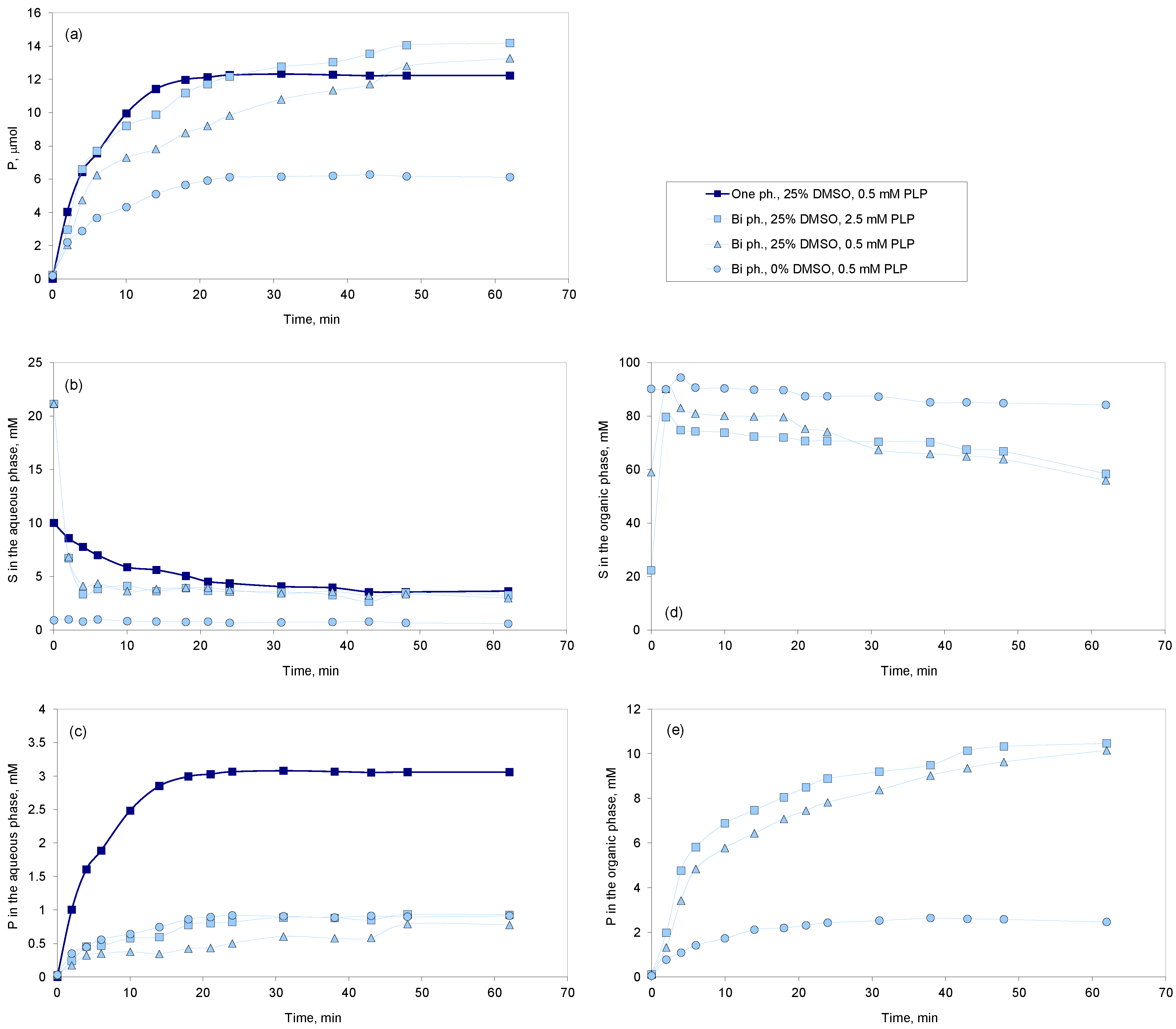
2.2. Two-Phase System
3. Materials and Methods
3.1. Biocatalyst Preparation
3.2. Enzyme Activity Experiments for Optimization of Transaminase Bioconversion
3.2.1. Determination of the Effect of Substrate Concentration
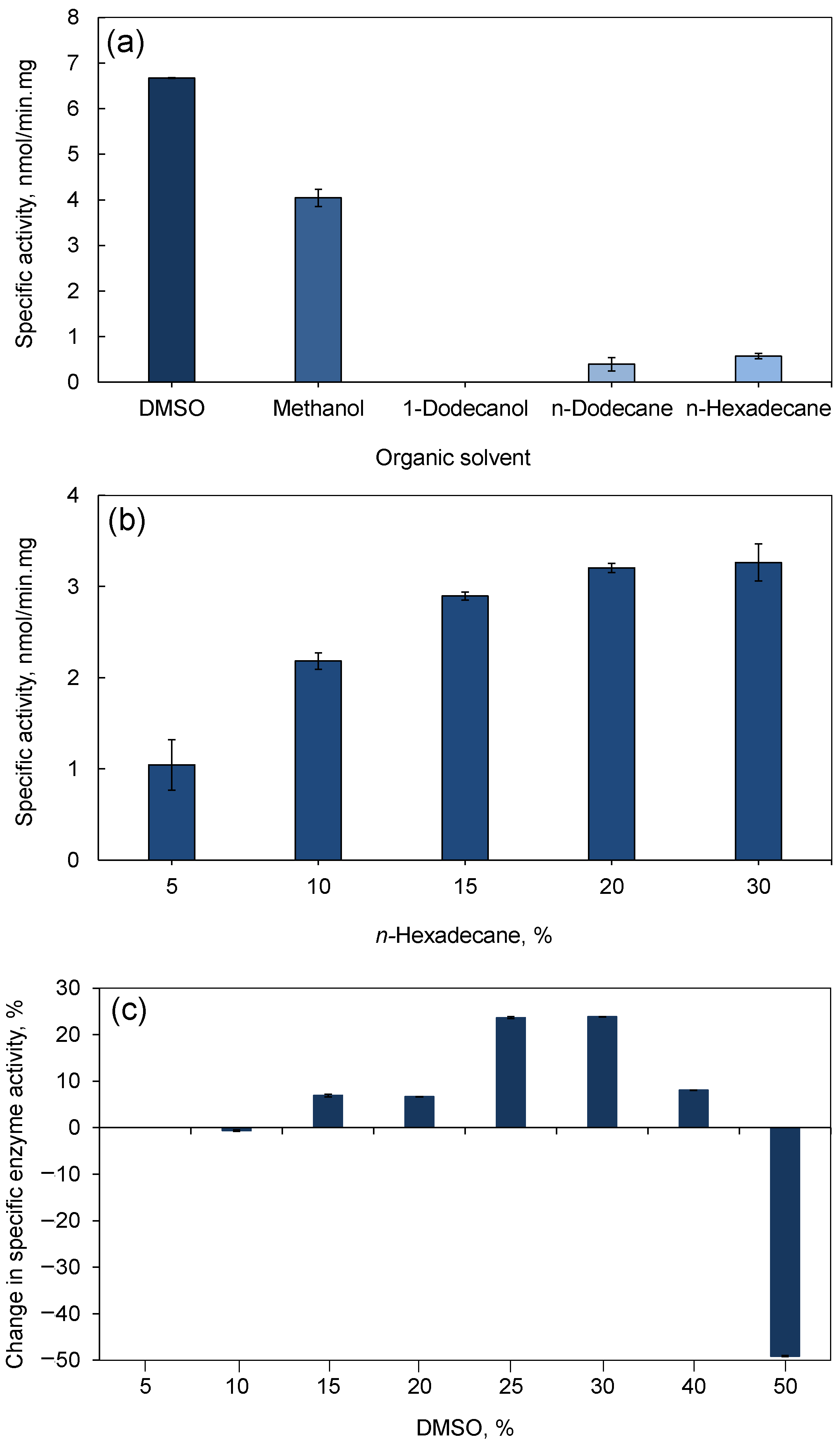
3.2.2. Organic Solvent Screening and Optimization
3.3. Bioconversion Assays in Single- and Two-Phase Systems
3.4. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.A.A. Green chemistry in United States science policy. Green Chem. Lett. Rev. 2019, 12, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Woodley, J.M. New frontiers in biocatalysis for sustainable synthesis. Curr. Opin. Green Sustain. Chem. 2020, 21, 22–26. [Google Scholar] [CrossRef]
- de Carvalho, C.C.C.R.; da Fonseca, M.M.R. 2.40-Biotransformations. In Comprehensive Biotechnology, 3rd ed.; Moo-Young, M., Ed.; Oxford: Pergamon, Turkey, 2017; pp. 574–585. [Google Scholar]
- de Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from Nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-M.; Han, S.-S.; Kim, H.-S. Industrial applications of enzyme biocatalysis: Current status and future aspects. Biotechnol. Adv. 2015, 33, 1443–1454. [Google Scholar] [CrossRef]
- Abdelraheem, E.M.M.; Busch, H.; Hanefeld, U.; Tonin, F. Biocatalysis explained: From pharmaceutical to bulk chemical production. React. Chem. Eng. 2019, 4, 1878–1894. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.J.C.; Pereira, R.F.S.; Fernandes, P.; Cabral, J.M.S.; de Carvalho, C.C.C.R. Cultivation-based strategies to find efficient marine biocatalysts. Biotechnol. J. 2017, 12, 1700036. [Google Scholar] [CrossRef] [PubMed]
- Coscolín, C.; Katzke, N.; García-Moyano, A.; Navarro-Fernández, J.; Almendral, D.; Martínez-Martínez, M.; Bollinger, A.; Bargiela, R.; Gertler, C.; Chernikova, T.N.; et al. Bioprospecting Reveals Class III ω-Transaminases Converting Bulky Ketones and Environmentally Relevant Polyamines. Appl. Environ. Microbiol. 2019, 85, e02404–e02418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, M.; Méndez-García, C.; Bargiela, R.; Chow, J.; Alonso, S.; García-Moyano, A.; Bjerga, G.E.K.; Steen, I.H.; Schwabe, T.; Blom, C.; et al. Decoding the ocean’s microbiological secrets for marine enzyme biodiscovery. FEMS Microbiol. Lett. 2018, 366. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Coppola, D.; di Prisco, G.; Giordano, D.; Verde, C. Enzymes from Marine Polar Regions and Their Biotechnological Applications. Mar. Drugs 2019, 17, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S.; Gilmore, B.F. Application of ω-Transaminases in the Pharmaceutical Industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef]
- Devine, P.N.; Howard, R.M.; Kumar, R.; Thompson, M.P.; Truppo, M.D.; Turner, N.J. Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem. 2018, 2, 409–421. [Google Scholar] [CrossRef]
- Ferrandi, E.E.; Monti, D. Amine transaminases in chiral amines synthesis: Recent advances and challenges. World J. Microbiol. Biotechnol. 2017, 34, 13. [Google Scholar] [CrossRef]
- Tang, X.-L.; Zhang, N.-N.; Ye, G.-Y.; Zheng, Y.-G. Efficient biosynthesis of (R)-3-amino-1-butanol by a novel (R)-selective transaminase from Actinobacteria sp. J. Biotechnol. 2019, 295, 49–54. [Google Scholar] [CrossRef]
- Trowbridge, A.; Walton, S.M.; Gaunt, M.J. New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev. 2020, 120, 2613–2692. [Google Scholar] [CrossRef] [Green Version]
- Ghislieri, D.; Turner, N.J. Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Höhne, M.; Bornscheuer, U.T. Biocatalytic Routes to Optically Active Amines. ChemCatChem 2009, 1, 42–51. [Google Scholar] [CrossRef]
- Mathew, S.; Yun, H. ω-Transaminases for the Production of Optically Pure Amines and Unnatural Amino Acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Tufvesson, P.; Lima-Ramos, J.; Jensen, J.S.; Al-Haque, N.; Neto, W.; Woodley, J.M. Process considerations for the asymmetric synthesis of chiral amines using transaminases. Biotechnol. Bioeng. 2011, 108, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Kelefiotis-Stratidakis, P.; Tyrikos-Ergas, T.; Pavlidis, I.V. The challenge of using isopropylamine as an amine donor in transaminase catalysed reactions. Org. Biomol. Chem. 2019, 17, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Lye, G.J.; Woodley, J.M. Application of in situ product-removal techniques to biocatalytic processes. Trends Biotechnol. 1999, 17, 395–402. [Google Scholar] [CrossRef]
- de Carvalho, C.C.R.; van Keulen, F.; Manuela, M.; da Fonseca, R. Production and Recovery of Limonene-1,2-Diol and Simultaneous Resolution of a Diastereomeric Mixture of Limonene-1,2-Epoxide with whole Cells of Rhodococcus erythropolis DCL14. Biocatal. Biotransform. 2000, 18, 223–235. [Google Scholar] [CrossRef]
- Rehn, G.; Adlercreutz, P.; Grey, C. Supported liquid membrane as a novel tool for driving the equilibrium of ω-transaminase catalyzed asymmetric synthesis. J. Biotechnol. 2014, 179, 50–55. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Wang, J.; Sanchez-Rosello, M.; Acena, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [Green Version]
- Hunter, L. The C–F bond as a conformational tool in organic and biological chemistry. Beilstein J. Org. Chem. 2010, 6, 38. [Google Scholar] [CrossRef] [Green Version]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.A. Sitagliptin Manufacture: A Compelling Tale of Green Chemistry, Process Intensification, and Industrial Asymmetric Catalysis. Angew. Chem. Int. Ed. 2011, 50, 1974–1976. [Google Scholar] [CrossRef] [PubMed]
- Frodsham, L.; Golden, M.; Hard, S.; Kenworthy, M.N.; Klauber, D.J.; Leslie, K.; Macleod, C.; Meadows, R.E.; Mulholland, K.R.; Reilly, J.; et al. Use of ω-Transaminase Enzyme Chemistry in the Synthesis of a JAK2 Kinase Inhibitor. Org. Process Res. Dev. 2013, 17, 1123–1130. [Google Scholar] [CrossRef]
- Meadows, R.E.; Mulholland, K.R.; Schürmann, M.; Golden, M.; Kierkels, H.; Meulenbroeks, E.; Mink, D.; May, O.; Squire, C.; Straatman, H.; et al. Efficient Synthesis of (S)-1-(5-Fluoropyrimidin-2-yl)ethylamine Using an ω-Transaminase Biocatalyst in a Two-Phase System. Org. Process Res. Dev. 2013, 17, 1117–1122. [Google Scholar] [CrossRef]
- Han, X.; Zhang, J.; Zhang, Y.; Liu, J.; Fang, W.; Fang, Z.; Xiao, Y. Amphritea opalescens sp. nov., isolated from sediment adjacent to Fildes Peninsula, Antarctica. Int. J. Syst. Evol. Microbiol. 2019, 69, 1585–1590. [Google Scholar] [CrossRef]
- Jang, H.; Yang, S.-H.; Seo, H.-S.; Lee, J.-H.; Kim, S.-J.; Kwon, K.K. Amphritea spongicola sp. nov., isolated from a marine sponge, and emended description of the genus Amphritea. Int. J. Syst. Evol. Microbiol. 2015, 65, 1866–1870. [Google Scholar] [CrossRef]
- Kim, Y.-O.; Park, S.; Kim, D.N.; Nam, B.-H.; Won, S.-M.; An, D.H.; Yoon, J.-H. Amphritea ceti sp. nov., isolated from faeces of Beluga whale (Delphinapterus leucas). Int. J. Syst. Evol. Microbiol. 2014, 64, 4068–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Solis, L.; Bayir, N.; Halim, M.; Du, C.; Ward, J.M.; Baganz, F.; Lye, G.J. Non-linear kinetic modelling of reversible bioconversions: Application to the transaminase catalyzed synthesis of chiral amino-alcohols. Biochem. Eng. J. 2013, 73, 38–48. [Google Scholar] [CrossRef]
- Chen, B.H.; Hibbert, E.G.; Dalby, P.A.; Woodley, J.M. A new approach to bioconversion reaction kinetic parameter identification. AICHE J. 2008, 54, 2155–2163. [Google Scholar] [CrossRef]
- Chen, S.; Campillo-Brocal, J.C.; Berglund, P.; Humble, M.S. Characterization of the stability of Vibrio fluvialis JS17 amine transaminase. J. Biotechnol. 2018, 282, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Börner, T.; Rämisch, S.; Reddem, E.R.; Bartsch, S.; Vogel, A.; Thunnissen, A.-M.W.H.; Adlercreutz, P.; Grey, C. Explaining Operational Instability of Amine Transaminases: Substrate-Induced Inactivation Mechanism and Influence of Quaternary Structure on Enzyme–Cofactor Intermediate Stability. ACS Catal. 2017, 7, 1259–1269. [Google Scholar] [CrossRef]
- Skalden, L.; Peters, C.; Dickerhoff, J.; Nobili, A.; Joosten, H.-J.; Weisz, K.; Höhne, M.; Bornscheuer, U.T. Two Subtle Amino Acid Changes in a Transaminase Substantially Enhance or Invert Enantiopreference in Cascade Syntheses. ChemBioChem 2015, 16, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, C.C.C.R.; da Fonseca, M.M.R. Maintenance of cell viability in the biotransformation of (−)-carveol with whole cells of Rhodococcus erythropolis. J. Mol. Catal. B Enzym. 2002, 19, 389–398. [Google Scholar] [CrossRef]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef]
- Feng, Y.; Luo, Z.; Sun, G.; Chen, M.; Lai, J.; Lin, W.; Goldmann, S.; Zhang, L.; Wang, Z. Development of an Efficient and Scalable Biocatalytic Route to (3R)-3-Aminoazepane: A Pharmaceutically Important Intermediate. Org. Process Res. Dev. 2017, 21, 648–654. [Google Scholar] [CrossRef]
- Land, H.; Ruggieri, F.; Szekrenyi, A.; Fessner, W.-D.; Berglund, P. Engineering the Active Site of an (S)-Selective Amine Transaminase for Acceptance of Doubly Bulky Primary Amines. Adv. Synth. Catal. 2020, 362, 812–821. [Google Scholar] [CrossRef] [Green Version]
- Heintz, S.; Börner, T.; Ringborg, R.H.; Rehn, G.; Grey, C.; Nordblad, M.; Krühne, U.; Gernaey, K.V.; Adlercreutz, P.; Woodley, J.M. Development of in situ product removal strategies in biocatalysis applying scaled-down unit operations. Biotechnol. Bioeng. 2017, 114, 600–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellechner, O.; Blatkiewicz, M.; Smirnova, I. Reactive Separations for In Situ Product Removal of Enzymatic Reactions: A Review. Chem. Ing. Tech. 2019, 91, 1522–1543. [Google Scholar] [CrossRef]
- Moldoveanu, S.; David, V. Derivatization Methods in GC and GC/MS. In Gas Chromatography—Derivatization, Sample Preparation, Application; Kusch, P., Ed.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Orata, F. Derivatization reactions and reagents for gas chromatography analysis. In Advanced Gas Chromatography—Progress in Agricultural, Biomedical and Industrial Applications; Mohd, M.A., Ed.; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, C.J.C.; Ferrer, M.; de Carvalho, C.C.C.R. ω-Transaminase-Mediated Asymmetric Synthesis of (S)-1-(4-Trifluoromethylphenyl)Ethylamine. Catalysts 2021, 11, 307. https://doi.org/10.3390/catal11030307
Rodrigues CJC, Ferrer M, de Carvalho CCCR. ω-Transaminase-Mediated Asymmetric Synthesis of (S)-1-(4-Trifluoromethylphenyl)Ethylamine. Catalysts. 2021; 11(3):307. https://doi.org/10.3390/catal11030307
Chicago/Turabian StyleRodrigues, Carlos J. C., Manuel Ferrer, and Carla C. C. R. de Carvalho. 2021. "ω-Transaminase-Mediated Asymmetric Synthesis of (S)-1-(4-Trifluoromethylphenyl)Ethylamine" Catalysts 11, no. 3: 307. https://doi.org/10.3390/catal11030307