
Preparation and High-Throughput Testing of TiO2-Supported Co Catalysts for Fischer‒Tropsch Synthesis


Abstract
:1. Introduction
2. Results and Discussion
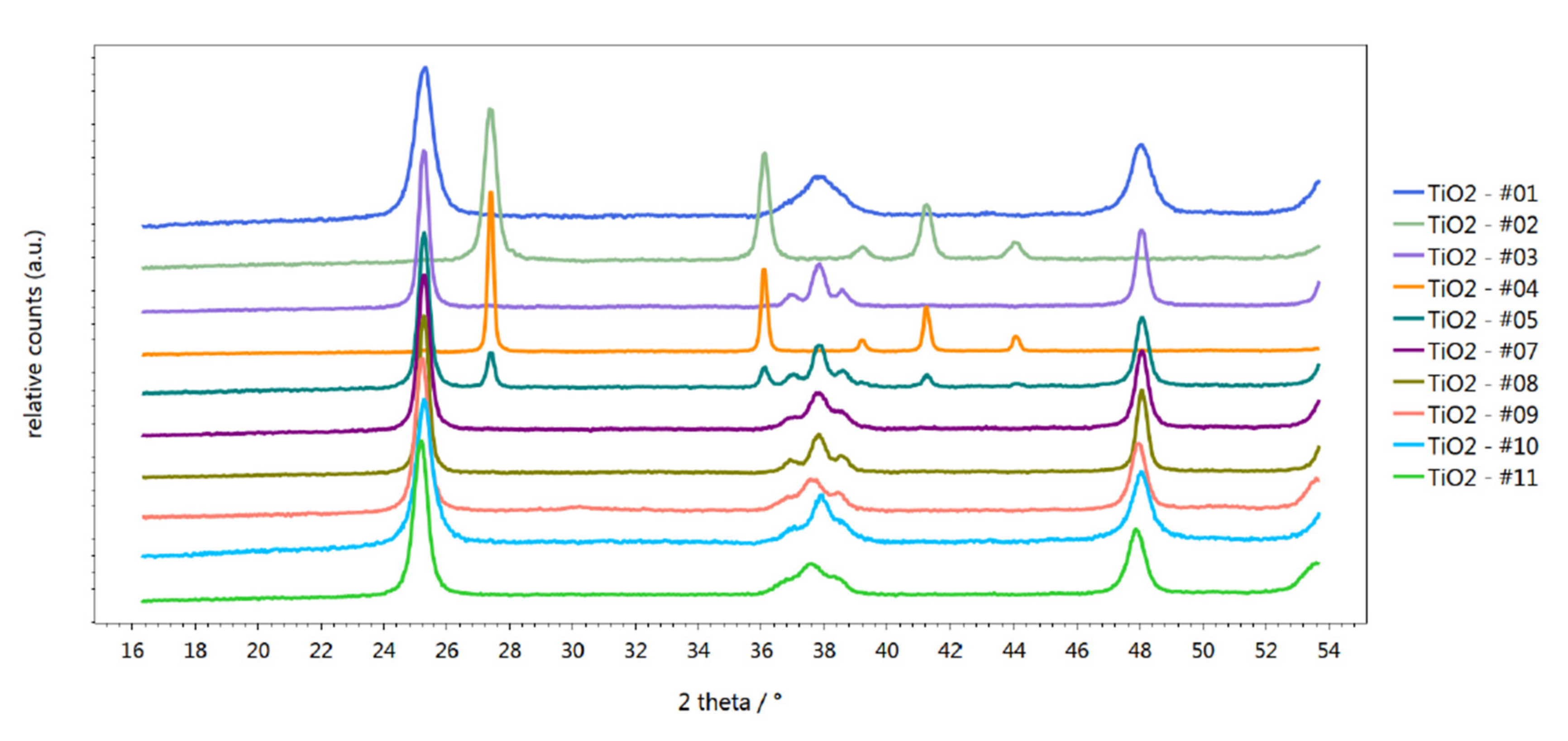
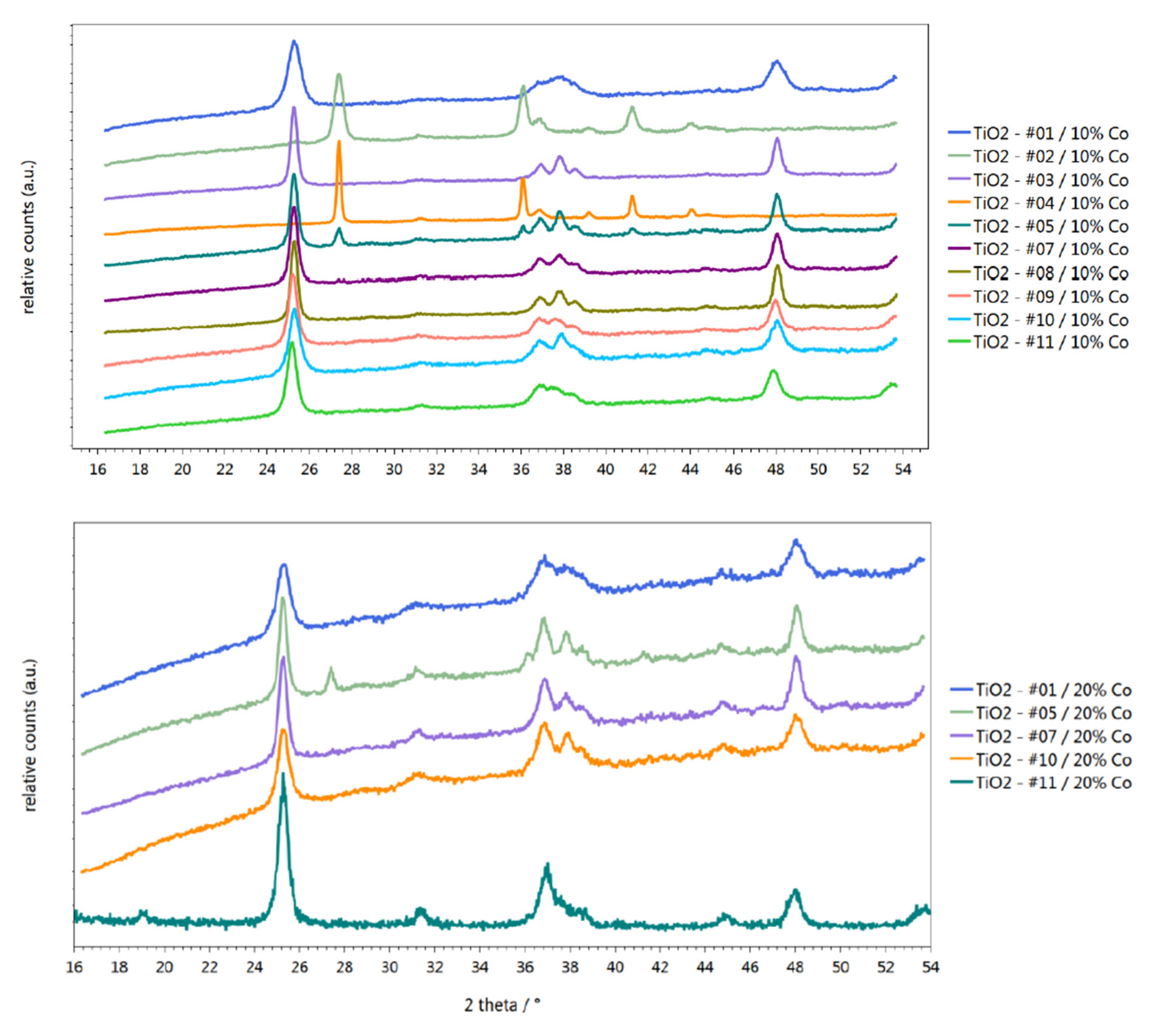
2.1. Characterization of Materials
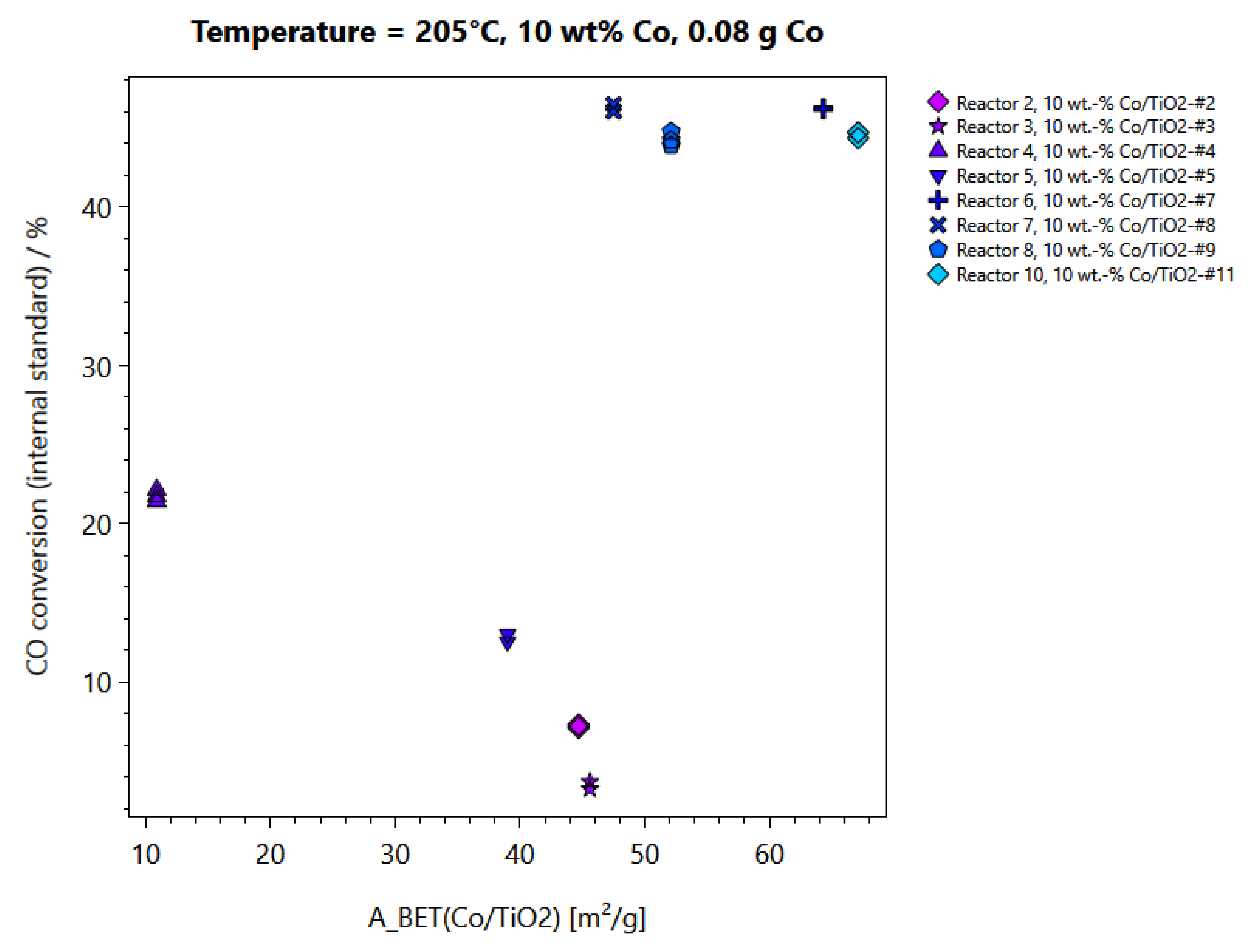
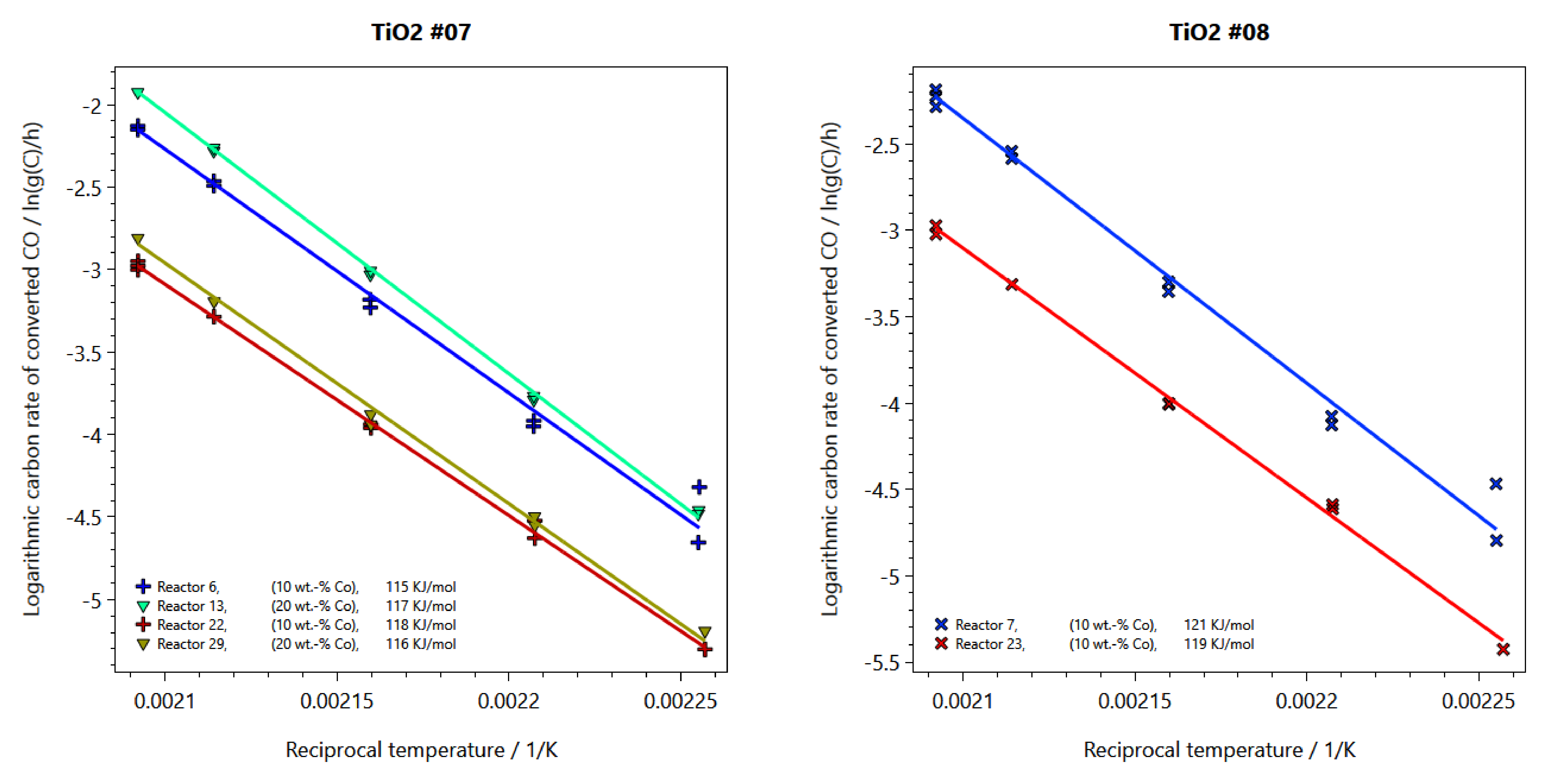
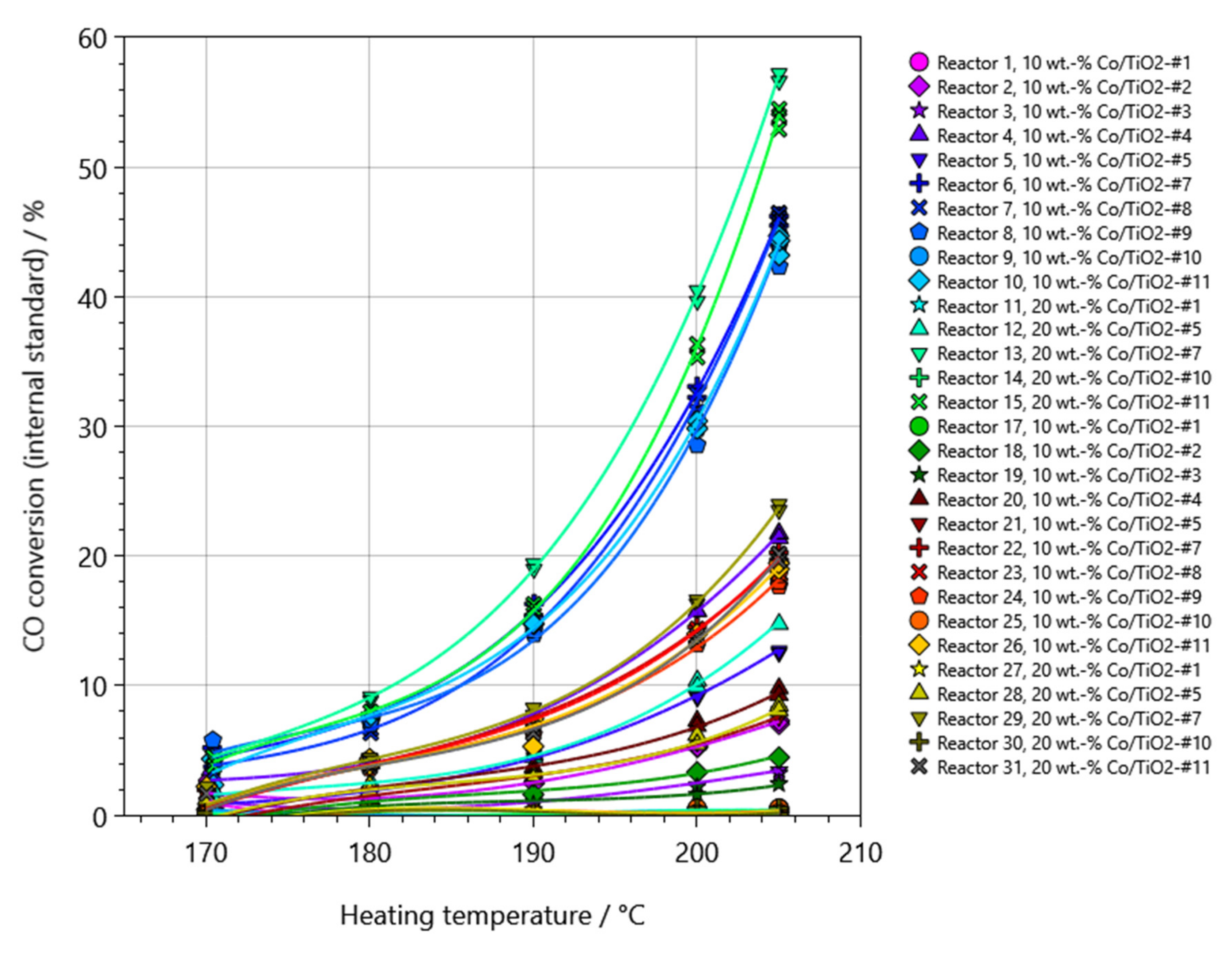
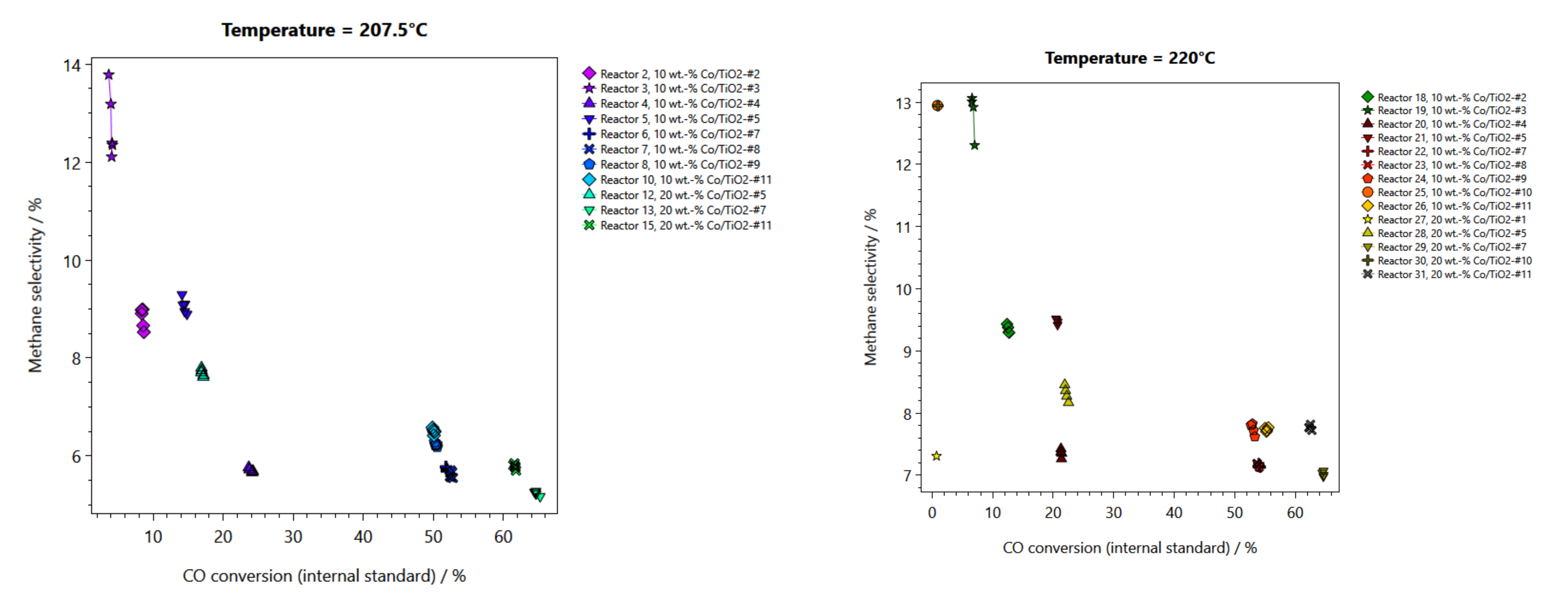
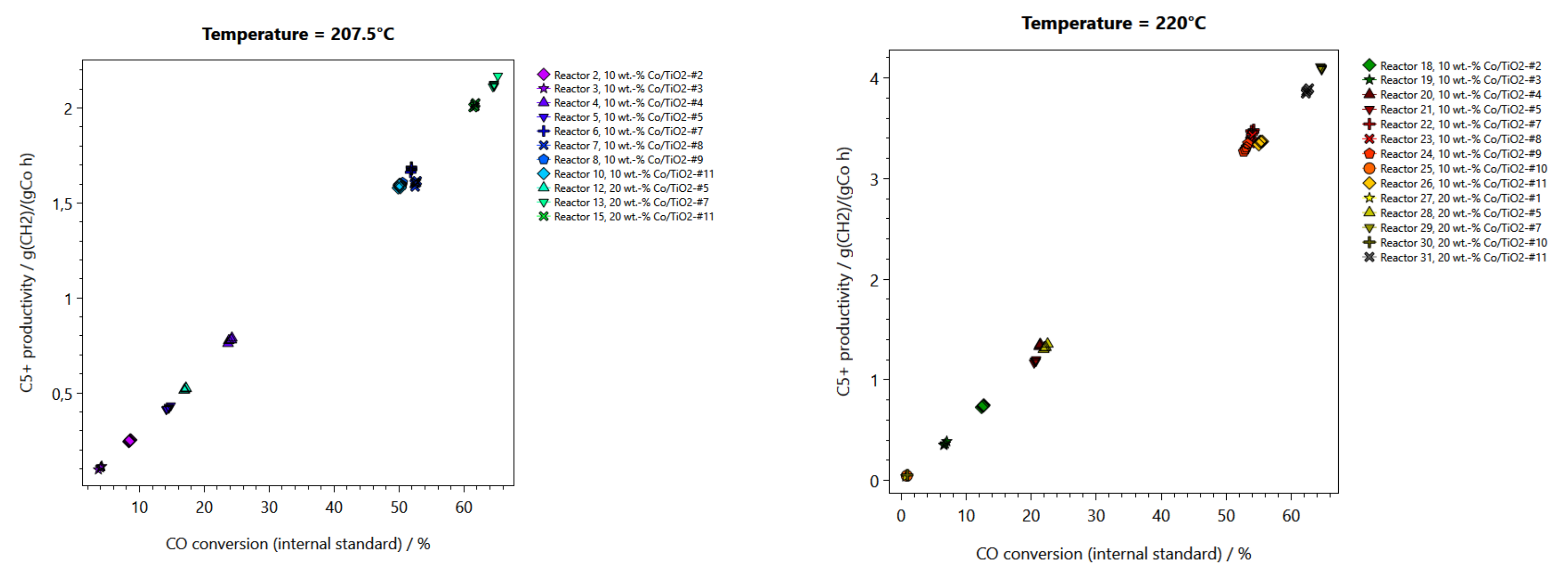
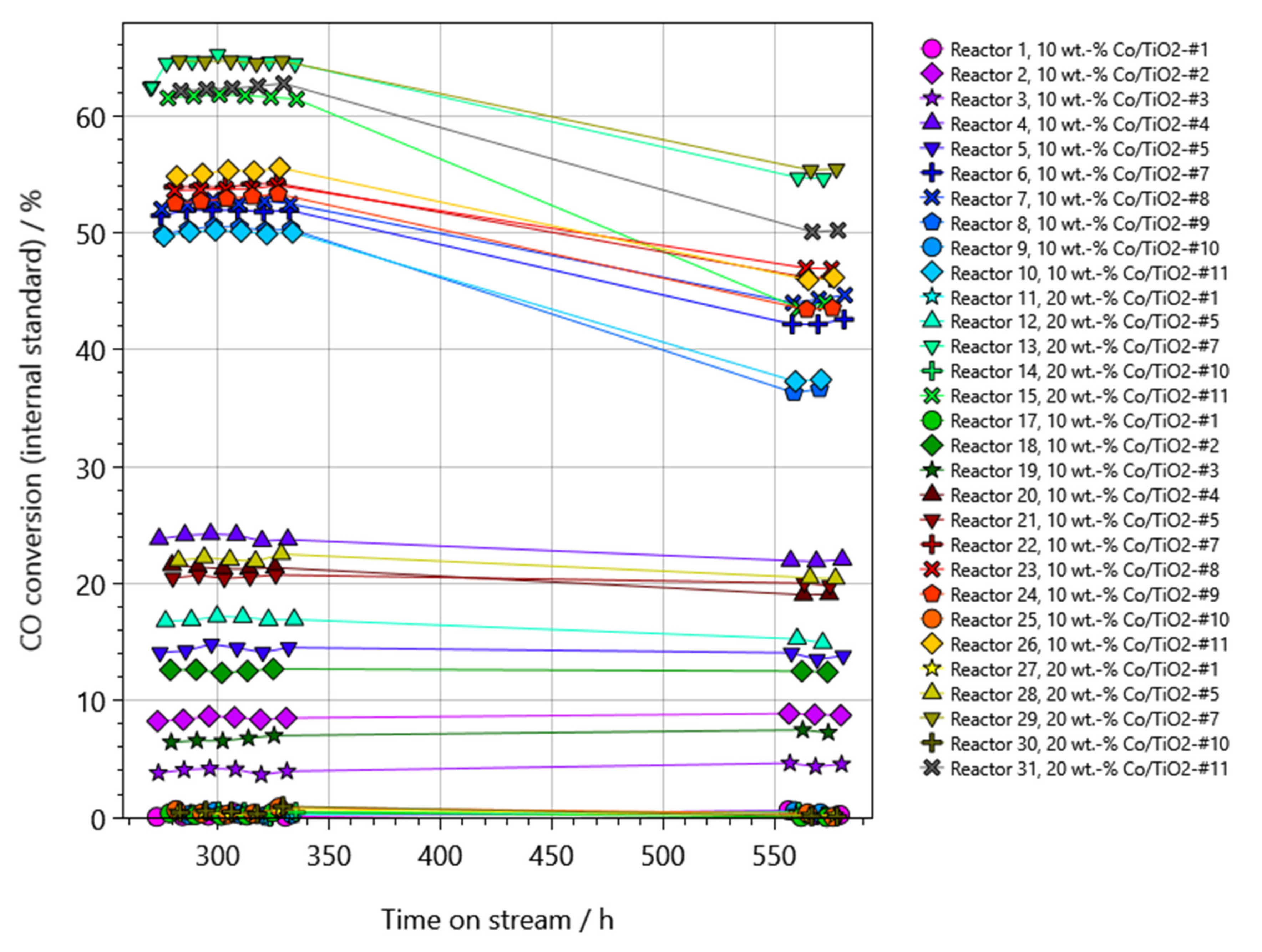
2.2. Performance Testing in FTS
3. Experimental Section
3.1. Catalyst Preparation
- Impregnation of TiO2 with Co(NO3)2 (targeting 10 wt % Co)
- Drying and calcination as described above
- Impregnation of TiO2 with Co(NO3)2 (targeting 15 wt % Co including the previous impregnation step)
- Drying and calcination as described above
- Impregnation of TiO2 with Co(NO3)2 (targeting 20 wt % Co including the previous two impregnation steps)
- Drying and calcination as described above
3.2. Catalyst Characterization
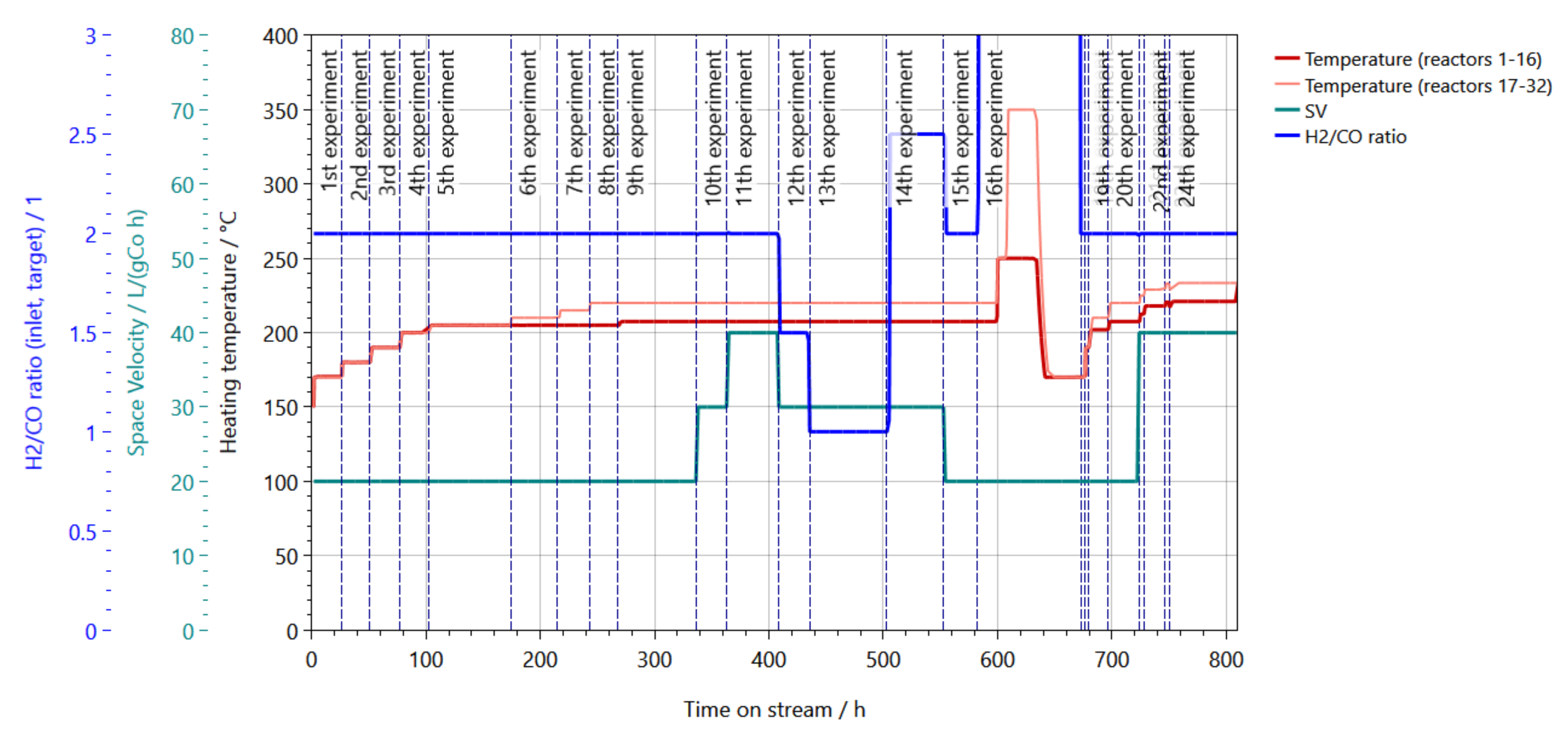
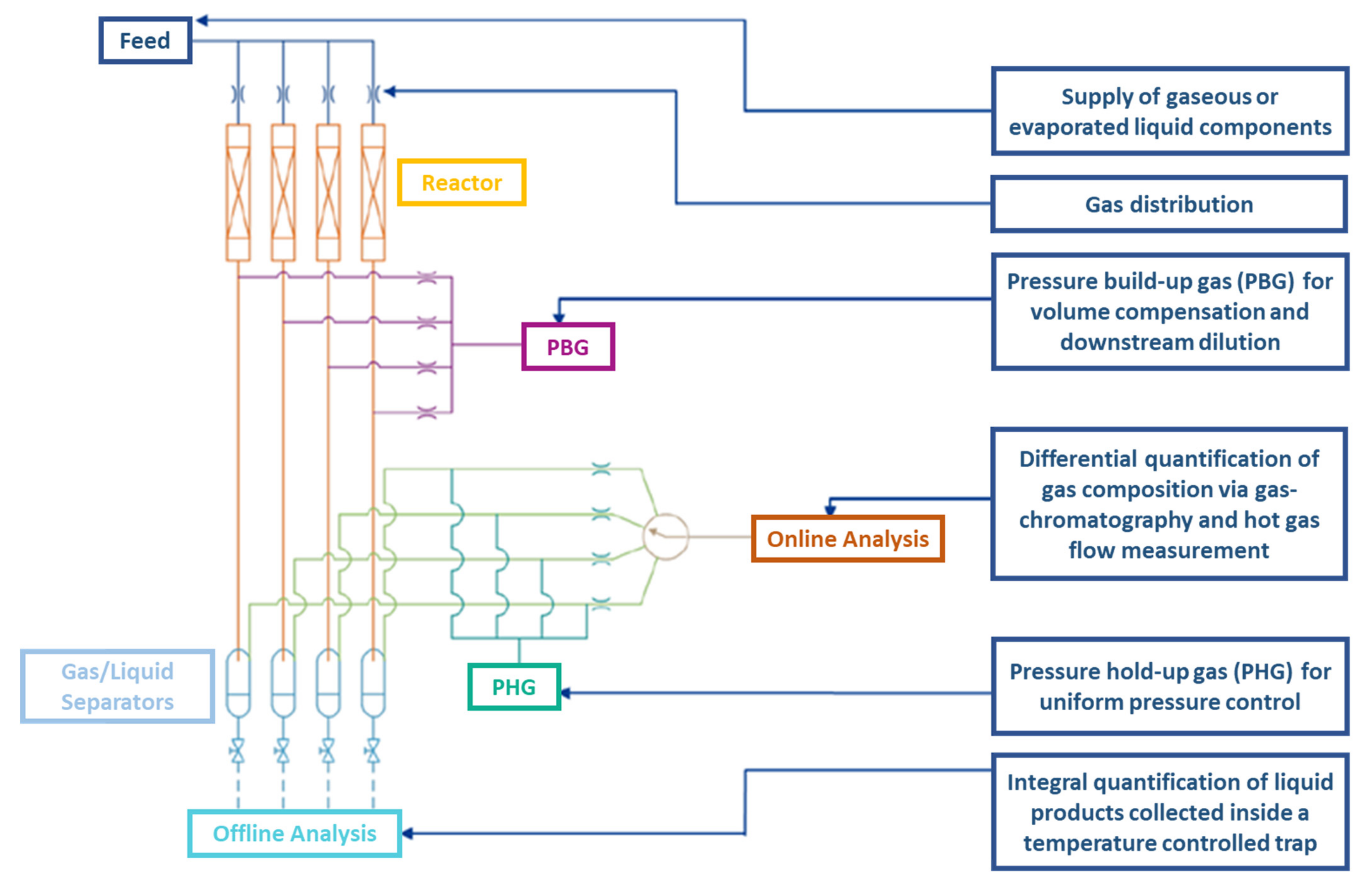
3.3. High-Throughput Testing of Catalysts
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Day | Experiment Number | Temperature (Reactor 1 to 16)/°C | Temperature (Reactor 17 to 32)/°C | Pressure/bar(g) | GHSV (Reactor 1 to 16)/mL/(gCo h) | GHSV (Reactor 17 to 32)/mL/(gCo h) | H2/CO Ratio |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 170 | 170 | 20 | 20,000 | 40,000 | 2.0 |
| 2 | 2 | 180 | 180 | 20 | 20,000 | 40,000 | 2.0 |
| 3 | 3 | 190 | 190 | 20 | 20,000 | 40,000 | 2.0 |
| 4 | 4 | 200 | 200 | 20 | 20,000 | 40,000 | 2.0 |
| 5 | 5 | 205 | 205 | 20 | 20,000 | 40,000 | 2.0 |
| 8 | 6 | 205 | 210 | 20 | 20,000 | 40,000 | 2.0 |
| 10 | 7 | 205 | 215 | 20 | 20,000 | 40,000 | 2.0 |
| 11 | 8 | 205 | 220 | 20 | 20,000 | 40,000 | 2.0 |
| 12 | 9 | 207.5 | 220 | 20 | 20,000 | 40,000 | 2.0 |
| 15 | 10 | 207.5 | 220 | 20 | 20,000 | 40,000 | 2.0 |
| 16 | 11 | 207.5 | 220 | 20 | 40,000 | 80,000 | 2.0 |
| 18 | 12 | 207.5 | 220 | 20 | 30,000 | 60,000 | 1.5 |
| 19 | 13 | 207.5 | 220 | 20 | 30,000 | 60,000 | 1.0 |
| 22 | 14 | 207.5 | 220 | 20 | 30,000 | 60,000 | 2.5 |
| 24 | 15 | 207.5 | 220 | 20 | 20,000 | 40,000 | 2.0 |
| 25 | 16 | 250 | 350 | 20 | 20,000 | 40,000 | n.a. |
| 29 | 17 | 170 | 170 | 20 | 20,000 | 40,000 | 2.0 |
| 29 | 18 | 190 | 190 | 20 | 20,000 | 40,000 | 2.0 |
| 29 | 19 | 202 | 210 | 20 | 20,000 | 40,000 | 2.0 |
| 30 | 20 | 207.5 | 220 | 20 | 20,000 | 40,000 | 2.0 |
| 31 | 21 | 212.5 | 225 | 20 | 40,000 | 80,000 | 2.0 |
| 32 | 22 | 218 | 229 | 20 | 40,000 | 80,000 | 2.0 |
| 32 | 23 | 220.5 | 233 | 20 | 40,000 | 80,000 | 2.0 |
| 32 | 24 | 221 | 233.5 | 20 | 40,000/ | 80,000 | 2.0 |
References
- Comm/dg/unit. A European Green Deal. Available online: https://ec.europa.eu/info/strategy/priorities-2019-2024/european-green-deal_en (accessed on 7 December 2020).
- The Paris Agreement|UNFCCC. Available online: https://unfccc.int/process-and-meetings/the-paris-agreement/the-paris-agreement (accessed on 7 December 2020).
- Kätelhön, A.; Meys, R.; Deutz, S.; Suh, S.; Bardow, A. Climate change mitigation potential of carbon capture and utilization in the chemical industry. Proc. Natl. Acad. Sci. USA 2019, 11187–11194. [Google Scholar] [CrossRef] [Green Version]
- Vogel, G.H. Change in Raw Material Base in the Chemical Industry. Chem. Eng. Technol. 2008, 31, 730–735. [Google Scholar] [CrossRef]
- Pagliaro, M. An Industry in Transition: The Chemical Industry and the Megatrends Driving Its Forthcoming Transformation. Angew. Chem. Int. Ed. 2019, 11154–11159. [Google Scholar] [CrossRef]
- bp Global. Oil|Energy Economics|Home. Available online: https://www.bp.com/en/global/corporate/energy-economics/energy-outlook/demand-by-fuel/oil.html (accessed on 7 December 2020).
- Shell’s Ambition to Be a Net-Zero Emissions Energy Business. Available online: https://www.shell.com/energy-and-innovation/the-energy-future/shells-ambition-to-be-a-net-zero-emissions-energy-business.html (accessed on 10 January 2020).
- Melero, J.A.; Iglesias, J.; Garcia, A. Biomass as renewable feedstock in standard refinery units. Feasibility, opportunities and challenges. Energy Environ. Sci. 2012, 5, 7393. [Google Scholar] [CrossRef]
- Fernando, S.; Adhikari, S.; Chandrapal, C.; Murali, N. Biorefineries: Current Status, Challenges, and Future Direction. Energy Fuels 2006, 20, 1727–1737. [Google Scholar] [CrossRef]
- Rauch, R.; Kiennemann, A.; Sauciuc, A. Fischer-Tropsch Synthesis to Biofuels (BtL Process). In The Role of Catalysis for the Sustainable Production of Bio-Fuels and Bio-Chemicals; Elsevier: Amsterdam, The Netherlands, 2013; pp. 397–443. [Google Scholar] [CrossRef]
- Luo, S.; Zhou, Y.; Yi, C. Syngas production by catalytic steam gasification of municipal solid waste in fixed-bed reactor. Energy 2012, 44, 391–395. [Google Scholar] [CrossRef]
- Arena, U. Process and technological aspects of municipal solid waste gasification. A review. Waste Manag. 2012, 32, 625–639. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Syngas in perspective. Catal. Today 2002, 71, 243–247. [Google Scholar] [CrossRef]
- Saeidi, S.; Talebi Amiri, M.; Saidina Amin, N.A.; Rahimpour, M.R. Progress in Reactors for High-Temperature Fischer–Tropsch Process: Determination Place of Intensifier Reactor Perspective. Int. J. Chem. React. Eng. 2014, 12, 639–664. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, H.; Mahmoudi, M.; Doustdar, O.; Jahangiri, H.; Tsolakis, A.; Gu, S.; LechWyszynski, M. A review of Fischer Tropsch synthesis process, mechanism, surface chemistry and catalyst formulation. Biofuels Eng. 2017, 2, 11–31. [Google Scholar] [CrossRef]
- Ponec, V. Some Aspects of the Mechanism of Methanation and Fischer-Tropsch Synthesis. Catal. Rev. 1978, 18, 151–171. [Google Scholar] [CrossRef]
- Jager, B.; Espinoza, R. Advances in low temperature Fischer-Tropsch synthesis. Catal. Today 1995, 23, 17–28. [Google Scholar] [CrossRef]
- Yang, J.; Ma, W.; Chen, D.; Holmen, A.; Davis, B.H. Fischer–Tropsch synthesis: A review of the effect of CO conversion on methane selectivity. Appl. Catal. A Gen. 2014, 470, 250–260. [Google Scholar] [CrossRef]
- Van der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and Selectivity of the Fischer–Tropsch Synthesis: A Literature Review. Catal. Rev. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Viswanathan, B.; Gopalakrishnan, R. Effect of support and promoter in Fischer-Tropsch cobalt catalysts. J. Catal. 1986, 99, 342–348. [Google Scholar] [CrossRef]
- Borg, Ø.; Eri, S.; Blekkan, E.A.; Storsæter, S.; Wigum, H.; Rytter, E.; Holmen, A. Fischer–Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables. J. Catal. 2007, 248, 89–100. [Google Scholar] [CrossRef]
- Bessel, S. Support effects in cobalt-based Fischer-Tropsch catalysis. Appl. Catal. A Gen. 1993, 96, 253–268. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer–Tropsch synthesis: Support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- Sakurai, K.; Mizusawa, M. X-ray diffraction imaging of anatase and rutile. Anal. Chem. 2010, 82, 3519–3522. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Meng, X.; Tang, F.; Ren, X.; Chen, D.; Ren, J. General Synthesis and Optical Properties of Monodisperse Multifunctional Metal-Ion-Doped TiO 2 Hollow Particles. J. Phys. Chem. C 2009, 113, 20240–20245. [Google Scholar] [CrossRef]
- Yan, Z.; Wu, H.; Han, A.; Yu, X.; Du, P. Noble metal-free cobalt oxide (CoO) nanoparticles loaded on titanium dioxide/cadmium sulfide composite for enhanced photocatalytic hydrogen production from water. Int. J. Hydrogen Energy 2014, 39, 13353–13360. [Google Scholar] [CrossRef]
- Reuel, R. Effects of support and dispersion on the CO hydrogenation activity/selectivity properties of cobalt. J. Catal. 1984, 85, 78–88. [Google Scholar] [CrossRef]
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Fischer–Tropsch synthesis over TiO2 supported cobalt catalyst: Effect of TiO2 crystal phase and metal ion loading. Appl. Catal. A Gen. 2013, 460–461, 8–14. [Google Scholar] [CrossRef]
- Post, M.F.M.; Van’t Hoog, A.C.; Minderhoud, J.K.; Sie, S.T. Diffusion limitations in fischer-tropsch catalysts. AICHE J. 1989, 35, 1107–1114. [Google Scholar] [CrossRef]
- Lyu, S.; Wang, L.; Zhang, J.; Liu, C.; Sun, J.; Peng, B.; Wang, Y.; Rappé, K.G.; Zhang, Y.; Li, J.; et al. Role of Active Phase in Fischer–Tropsch Synthesis: Experimental Evidence of CO Activation over Single-Phase Cobalt Catalysts. ACS Catal. 2018, 8, 7787–7798. [Google Scholar] [CrossRef]
- Jarosch, K.T.; Tonkovich, A.L.Y.; Perry, S.T.; Kuhlmann, D.; Wang, Y. Microchannel Reactors for Intensifying Gas-to-Liquid Technology. In Microreactor Technology and Process Intensification; ACS Symposium Series; ACS: Washington, DC, USA, 2005; pp. 258–272. [Google Scholar] [CrossRef]
- Wolf, M.; Fischer, N.; Claeys, M. Water-induced deactivation of cobalt-based Fischer–Tropsch catalysts. Nat. Catal. 2020, 3, 962–965. [Google Scholar] [CrossRef]
- Tsakoumis, N.E.; Rønning, M.; Borg, Ø.; Rytter, E.; Holmen, A. Deactivation of cobalt based Fischer–Tropsch catalysts: A review. Catal. Today 2010, 154, 162–182. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Carbon Deposition in Steam Reforming and Methanation. Catal. Rev. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Moodley, D.J. On the deactivation of cobalt-based Fischer-Tropsch synthesis catalysts. Am. Chem. Soc. Div. Petrochem. Chem. 2008, 53, 122–125. [Google Scholar]
- Snyder, R.L.; Fiala, J.; Bunge, H.-J. Defect and Microstructure Analysis by Diffraction; Oxford Science Publications: Oxford, UK; Oxford University Press: Oxford, UK, 1999; Volume 10. [Google Scholar]
- Brenner, A.; Friess, M.; Haas, A.; Köchel, O.; Strehlau, W.; Zech, T. Device and Method for Pressure and Flow Control in Parallel Reactors. Patent PCT/EP2004/014509, 23 12 2003. [Google Scholar]
- Eschemann, T.O.; Oenema, J.; de Jong, K.P. Effects of noble metal promotion for Co/TiO2 Fischer-Tropsch catalysts. Catal. Today 2016, 261, 60–66. [Google Scholar] [CrossRef]


| Process | Low-Temperature FTS | High-Temperature FTS |
|---|---|---|
| Reaction temperature | 220 to 240 °C | 300 to 350 °C |
| Catalyst | Co-based | Fe-based |
| Product | Paraffins, long chains/waxes | Olefins, short chains, gasoline, chemicals |
| Reactor Type | Fixed bed, slurry bubble column | Fluidized bed reactors |
| Sample Description | Specific Surface Area of Support m2/g | Co Loading/wt % | Specific Surface Area of Impregnated Support/m2/g | Phase Composition of TiO2 |
|---|---|---|---|---|
| 10 wt % Co/TiO2—#01 | 158.9 | 10 | 100.6 | Anatase |
| 10 wt % Co/TiO2—#02 | 56.7 | 10 | 44.7 | Rutile |
| 10 wt % Co/TiO2—#03 | 57.4 | 10 | 45.6 | Anatase |
| 10 wt % Co/TiO2—#04 | 5.4 | 10 | 10.9 | Rutile |
| 10 wt % Co/TiO2—#05 | 48.8 | 10 | 39.0 | Anatase/Rutile |
| 10 wt % Co/TiO2—#07 | 75.6 | 10 | 64.3 | Anatase |
| 10 wt % Co/TiO2—#08 | 53.1 | 10 | 47.5 | Anatase |
| 10 wt % Co/TiO2—#09 | 55.0 | 10 | 52.1 | Anatase |
| 10 wt % Co/TiO2—#10 | 111.4 | 10 | 62.5 | Anatase |
| 10 wt % Co/TiO2—#11 | 76.8 | 10 | 67.1 | Anatase |
| 20 wt % Co/TiO2—#01 | 158.9 | 20 | 59.2 | Anatase |
| 20 wt % Co/TiO2—#05 | 48.8 | 20 | 29.2 | Anatase/Rutile |
| 20 wt % Co/TiO2—#07 | 75.6 | 20 | 52.6 | Anatase |
| 20 wt % Co/TiO2—#10 | 111.4 | 20 | 34.5 | Anatase |
| 20 wt % Co/TiO2—#11 | 76.8 | 20 | 52.5 | Anatase |
| Heating Block | Reactor Position | Sample Description | Co Loading / wt % | Support Number | Wet-Based Mass/g | Co Mass/mg |
|---|---|---|---|---|---|---|
| A | 1 | 10 wt % Co/TiO2—#01 | 10 | 1 | 0.807 | 80 |
| 2 | 10 wt % Co/TiO2—#02 | 10 | 2 | 0.807 | 80 | |
| 3 | 10 wt % Co/TiO2—#03 | 10 | 3 | 0.809 | 80 | |
| 4 | 10 wt % Co/TiO2—#04 | 10 | 4 | 0.808 | 80 | |
| 5 | 10 wt % Co/TiO2—#05 | 10 | 5 | 0.807 | 80 | |
| 6 | 10 wt % Co/TiO2—#07 | 10 | 7 | 0.808 | 80 | |
| 7 | 10 wt % Co/TiO2—#08 | 10 | 8 | 0.809 | 80 | |
| 8 | 10 wt % Co/TiO2—#09 | 10 | 9 | 0.809 | 80 | |
| 9 | 10 wt % Co/TiO2—#10 | 10 | 10 | 0.808 | 80 | |
| 10 | 10 wt % Co/TiO2—#11 | 10 | 11 | 0.807 | 80 | |
| 11 | 20 wt % Co/TiO2—#01 | 20 | 1 | 0.404 | 80 | |
| 12 | 20 wt % Co/TiO2—#05 | 20 | 5 | 0.404 | 80 | |
| 13 | 20 wt % Co/TiO2—#07 | 20 | 7 | 0.405 | 80 | |
| 14 | 20 wt % Co/TiO2—#10 | 20 | 10 | 0.403 | 80 | |
| 15 | 20 wt % Co/TiO2—#11 | 10 | 11 | 0.405 | 80 | |
| 16 | Inert | - | - | - | - | |
| B | 17 | 10 wt % Co/TiO2—#01 | 10 | 1 | 0.405 | 40 |
| 18 | 10 wt % Co/TiO2—#02 | 10 | 2 | 0.404 | 40 | |
| 19 | 10 wt % Co/TiO2—#03 | 10 | 3 | 0.404 | 40 | |
| 20 | 10 wt % Co/TiO2—#04 | 10 | 4 | 0.405 | 40 | |
| 21 | 10 wt % Co/TiO2—#05 | 10 | 5 | 0.405 | 40 | |
| 22 | 10 wt % Co/TiO2—#07 | 10 | 7 | 0.404 | 40 | |
| 23 | 10 wt % Co/TiO2—#08 | 10 | 8 | 0.403 | 40 | |
| 24 | 10 wt % Co/TiO2—#09 | 10 | 9 | 0.403 | 40 | |
| 25 | 10 wt % Co/TiO2—#10 | 10 | 10 | 0.403 | 40 | |
| 26 | 10 wt % Co/TiO2—#11 | 10 | 11 | 0.405 | 40 | |
| 27 | 20 wt % Co/TiO2—#01 | 20 | 1 | 0.203 | 40 | |
| 28 | 20 wt % Co/TiO2—#05 | 20 | 5 | 0.201 | 40 | |
| 29 | 20 wt % Co/TiO2—#07 | 20 | 7 | 0.201 | 40 | |
| 30 | 20 wt % Co/TiO2—#10 | 20 | 10 | 0.203 | 40 | |
| 31 | 20 wt % Co/TiO2—#11 | 20 | 11 | 0.201 | 40 | |
| 32 | Inert | - | - | - | - |
| GHSV/mL/(gCo h) | 20,000 to 80,000 |
| H2/CO ratio | 1 to 2.5 |
| Internal standard Ar/vol % | 10 |
| Temperature/°C | 150 to 233.5 |
| p/bar(g) | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, C.; Kolb, P.; Krupp, D.; Ritter, L.; Haas, A.; Soorholtz, M.; Emmerich Maldonado, T.; Thiede, T.B.; Knobloch, C. Preparation and High-Throughput Testing of TiO2-Supported Co Catalysts for Fischer‒Tropsch Synthesis. Catalysts 2021, 11, 352. https://doi.org/10.3390/catal11030352
Schulz C, Kolb P, Krupp D, Ritter L, Haas A, Soorholtz M, Emmerich Maldonado T, Thiede TB, Knobloch C. Preparation and High-Throughput Testing of TiO2-Supported Co Catalysts for Fischer‒Tropsch Synthesis. Catalysts. 2021; 11(3):352. https://doi.org/10.3390/catal11030352
Chicago/Turabian StyleSchulz, Christian, Peter Kolb, Dennis Krupp, Lars Ritter, Alfred Haas, Mario Soorholtz, Thomas Emmerich Maldonado, Tobias Benjamin Thiede, and Carsten Knobloch. 2021. "Preparation and High-Throughput Testing of TiO2-Supported Co Catalysts for Fischer‒Tropsch Synthesis" Catalysts 11, no. 3: 352. https://doi.org/10.3390/catal11030352